

Abstract
Cyclosporin A (CsA) nephropathy is associated with altered expression of apoptosis regulatory genes such as Fas-ligand and Bcl-2 family members in the glomerular, tubulointerstitial, and vascular compartments. Both hepatocyte growth factor (HGF) and insulin-like growth factor (IGF-I) protect against apoptosis, and HGF specifically up-regulates Bcl-xL, a protein that regulates apoptosis. We investigated whether Bcl-xL and Fas/Fas-ligand were regulated by CsA in cultured podocytes and whether CsA-induced apoptosis was prevented by HGF or IGF-I. A murine podocyte cell line was treated with CsA in the presence or absence of HGF or IGF-I. Apoptosis was quantitated by ELISA and by flow cytometry; Bcl-xL, Fas, and Fas-ligand were measured by Western blotting. Inhibitors of MAP kinase/ERK kinase (MEK)-1 and of phosphatidylinositol 3′-kinase (PI3′-K) were used to determine the signaling pathways involved in Bcl-xL regulation. Apoptosis was induced by CsA in a dose- and time-dependent fashion. CsA also decreased Bcl-xL levels. HGF, but not IGF-I, prevented apoptosis and restored Bcl-xL levels. The regulation of Bcl-xL by HGF was mediated by the PI3′-K but not by the MEK-1 pathway. In summary, we showed that CsA induces apoptosis in podocytes. Apoptosis was prevented by pretreatment with HGF but not IGF-I. Decreased apoptosis appeared to be mediated by regulation of Bcl-xL via the PI3′-K pathway. Our data suggest that the effect of CsA on podocytes may contribute to the glomerular damage and that HGF could provide protection.
Cyclosporin A (CsA) is a widely used immunosuppressive drug. A major limitation in its use is the development of adverse effects such as CsA nephropathy. 1,2 The nephropathy is characterized by tubulointerstitial lesions, arteriopathy, and glomerulosclerosis.
A common finding in kidney biopsies of CsA-treated animals is the presence of apoptosis, and several models of CsA-induced apoptosis have been described. Apoptosis has been shown in tubular and interstitial cells of CsA-treated animals 3 and in tubular cells in vitro. 4,5 In glomeruli, CsA treatment is also associated with altered expression of apoptosis regulatory genes such as Bcl-2, Bax, and Fas-L. 6 In addition, renal tubular cells treated with CsA undergo apoptosis and show increased Fas expression. 4 Although a decrease in apoptosis was observed in the kidneys of CsA-treated animals after nitric oxide blockade or angiotensin II type I receptor antagonism, 3 a rescue factor from CsA-induced apoptosis has not been identified.
Several growth factors have been shown to provide protection against programmed cell death. 7,8 HGF prevented the acute side effects of CsA in mice. 9 Both HGF and IGF-I are modulated by CsA in vivo, 10,11 and their ability to decrease apoptosis has been well established. 12,13 Recently, it has been proposed that HGF protects tubular cells from apoptosis by a dual mechanism, one involving PI3′-kinase/Akt/Bad, and a second one mediated by a later induction of Bcl-xL expression. 14 Bcl-xL, but not other members of the Bcl-2 family, were altered by HGF. It is not known whether the expression of Bcl-xL induced by HGF could be the effect of long-term PI3′-K activation by HGF. Here we examine whether CsA induces apoptosis in cultured podocytes and whether a regulation of Bcl-xL, Fas, and Fas-ligand occurs. Because epithelial cells express c-met, the receptor for HGF, 15 and podocytes bind IGF-I, 16 we study whether HGF and IGF-I prevent apoptosis, and whether Bcl-xL is regulated in a PI3′-K or a MEK-1-dependent manner.
Materials and Methods
Cell Culture
Glomerular epithelial cells derived from normal 4- to 6-week-old female B6SJLF1/J mice were used at passages 23 to 25. These cells have been described in detail previously. 17 Cells were maintained in DMEM/F12 (3:1) medium (Gibco BRL, Rockville, MD) supplemented with 5% fetal bovine serum (Gibco BRL), 1 mmol/L glutamine (Biosource International, Camarillo, CA), 0.075% Na2HCO3 (Gibco BRL), 100 μg/ml penicillin-streptomycin (100 U/ml) (Biosource International), and trace elements (Biosource International). Cells were propagated in fibronectin-coated Nunc flasks.
Apoptosis Detection by ELISA
Cells were plated in 24-well plates. Cyclosporin A (IVAX Corporation, Miami, FL) was diluted in dimethylsulfoxide (DMSO). For dose-dependent experiments, CsA was added at concentrations of 0.1, 0.5, and 1 μg/ml when cells reached confluence. DMSO alone (final concentration 0.1%) was used to treat control cells. Cell lysates were collected after 24 hours of CsA exposure to investigate the presence of apoptosis. Apoptosis was quantitated by ELISA for the determination of histone-associated DNA fragments released into the cytoplasm during the apoptotic process, according to the manufacturer’s suggestions (Cell Death Detection ELISA PLUS, Boehringer Mannheim, Indianapolis, IN). Results are expressed as percent of control. For the time course study of apoptosis, a dose of 0.5 μg/ml was used, and apoptosis was examined after 1, 2, 3, 4, 6, 12, 24, and 48 hours of CsA exposure.
For protection experiments, recombinant human IGF-I (Biosource International) or recombinant heterodimeric human HGF-1 (kindly provided by Dr. Ralph Schwall, Genentech, San Bruno, CA, through the Miami VA Geriatric Research, Education, and Clinical Center, Miami, FL) were used. The protective effect of HGF or IGF-I on CsA-induced apoptosis was tested by pretreatment with either 50 or 100 ng/ml of both growth factors. A pretreatment for 1, 12, and 48 hours was performed with HGF or IGF-I (100 ng/ml). Cell layers were collected after 24 hours of CsA exposure.
Apoptosis Detection by Flow Cytometry
Cell death was determined by cell cycle analysis using fluorescence-activated cell sorter flow cytometry (FACS). Cell were plated onto fibronectin-coated 6-well plates and treated for 24 hours with either CsA (0.5 μg/ml) or DMSO (0.1%) as control. The cells were trypsinized and stained with propidium iodide plus RNase. The stained cells were analyzed by FACS (FACScan cytofluorometer, Becton Dickinson and Co., Franklin Lakes, NJ) and a CELL Quest software (Becton Dickinson) was used for analysis of the data. Cell cycle analysis indicates that the cells in G1, S, and G2/M phase were alive whereas the cells in pre-G1 phase (apoptotic phase) were not. The percentage of live or dead cells was calculated as cell number in pre-G1 phase/total cell number × 100.
Bcl-xL, Fas, and Fas-Ligand Analysis by Western Immunoblot
Cells plated onto fibronectin-coated 6-well plates were treated for 1, 12, or 48 hours with HGF or IGF-I (100 ng/ml). For blocking experiments, inhibitors of MEK-1 (PD098059, Calbiochem-Novabiochem Corp., San Diego, CA) and of PI3′-K (LY29400, Calbiochem-Novabiochem) were added 30 minutes before adding the growth factors. After 24 hours of CsA treatment, each well of a 6-well plate was washed twice with cold PBS and 100 μl of cold lysis buffer (20 mmol/L Tris, 140 mmol/L NaCl, 3 mmol/L EDTA, 10 mmol/L NaF, 10 mmol/L Na4P2O7, 2 mmol/L NaVO4, 10% glycerol, pH 7.4, 1% Triton X-100, aprotinin, leupeptin, PMSF) was added for 2 minutes before scraping the cells. Cell lysates were incubated 45 minutes at 4°C and the insoluble material was removed by centrifugation at 20,000 × g for 30 minutes at 4°C. Ten or 50 μg of protein for each sample were loaded on a 15% SDS-PAGE under reducing conditions after being heated for 3 minutes at 100°C. The proteins were electrotransferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL). Blots were blocked for 1 hour at room temperature in Tris-buffered saline (TBS), 5% milk, 0.05% Tween-20. The membrane was incubated overnight at 4°C with a mouse monoclonal antibody against Bcl-xL (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), followed by three washes (total time 35 minutes) with TBS 0.05% Tween 20. For Fas and Fas-ligand detection, rabbit polyclonal antibodies (Santa Cruz Biotechnology) against Fas and Fas-ligand were used at a concentration of 1:500 and processed as for Bcl-xL. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:1000 dilution, Santa Cruz Biotechnology) for 1 hour at room temperature. Peroxidase activity was detected using a chemiluminescence kit (Santa Cruz Biotechnology). Data were analyzed on a Macintosh computer using the public domain NIH Image program (developed at the National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Data Analysis
Each experiment was carried out in duplicate or triplicate and three or four independent experiments were performed. Results are expressed as means ± SD. Results were compared using analysis of variance (ANOVA). When ANOVA showed a statistically significant difference, a group-by-group comparison was performed using a t-test with Tukey’s correction for multiple comparison. Statistical significance was set at P < 0.05.
Results
CsA Induces Apoptosis in Podocytes
CsA exposure induced apoptosis in podocytes in a dose- and time-dependent manner. Apoptosis was detected after 24 hours of treatment with CsA at concentrations ≥ 0.5 μg/ml (Figure 1A) ▶ . Time course experiments using 0.5 μg/ml CsA for 1, 2, 4, 6, and 12 hours showed apoptosis only after 24 hours of exposure (Figure 1B) ▶ . DNA fragmentation was detected at 24 hours, but there was no change in cell number. After 48 hours, both apoptosis and a decrease in cell number were observed. CsA-induced apoptosis was confirmed by FACS analysis. The percentage of cells undergoing apoptosis increased to 10.5 ± 0.1% as compared to 6.1 ± 1.4% in control cells (P < 0.001) (Figure 2) ▶ . In addition, a significant decrease in the percentage of cells in the S phase of the cell cycle was observed (16.41 ± 4.1% versus 26.39 ± 1.5%, CsA-treated cells versus control cells respectively, P < 0.05).
Figure 1.
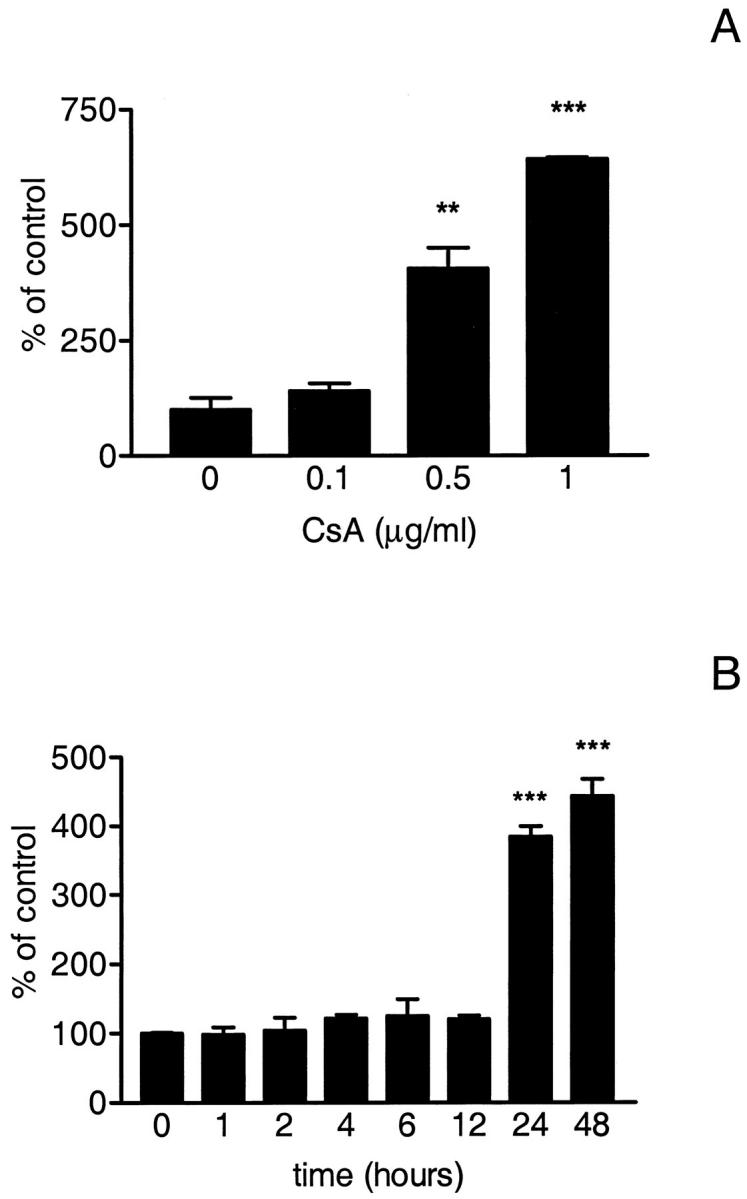
A: Dose-dependent induction of apoptosis by CsA. Cells were treated with increasing doses of 0.1, 0.5, or 1 μg/ml CsA and apoptosis was measured by ELISA and expressed as percentage of control (DMSO 0.1%). The percentage of cells undergoing apoptosis was increased by 0.5 μg/ml CsA (**P < 0.01), and by 1 μg/ml CsA (***P < 0.001). B: Time course of CsA induction of apoptosis. CsA was administered for 1, 2, 4, 6, 12, 24, and 48 hours, and apoptosis was measured by ELISA and expressed as percentage of control (DMSO 0.1%). Apoptosis was increased after at least 24 hours of CsA treatment (***P < 0.001).
Figure 2.

Apoptosis detection by FACS analysis. Representative FACS analysis after propidium iodine staining in control cells (A) and cells treated with 0.5 μg/ml CsA (B). The percentage of cells undergoing apoptosis increased to 10.5 ± 0.1%, as compared to 6.1 ± 1.4% in control cells (P < 0.001).
HGF, but Not IGF-I, Prevented CsA-Induced Apoptosis
Pretreatment with HGF for 1, 12, and 48 hours before CsA exposure reduced apoptosis to control levels (Figure 3A) ▶ . The percentage of cells undergoing CsA-mediated apoptosis increased to 173.9 ± 13.1% (P < 0.05 as compared to 100 ± 34.4% control) an effect which was reversed by HGF pretreatment for 1 hour (95.7 ± 20.6% versus 173.9 ± 13.1%, P < 0.01), 12 hours (73.3 ± 10.2% versus 173.9 ± 13.1%, P < 0.01) and 48 hours (69 ± 11.4% versus 173.9 ± 13.1%, P < 0.05). The percentage of cells undergoing apoptosis was decreased by pretreatment with 50 ng/ml HGF (140.7 ± 14.76% versus 186.6 ± 8.00%, HGF + CsA versus CsA alone, respectively), and was reduced to control levels by 100 ng/ml HGF (85.98 ± 6.47% versus 186.6 ± 8.00%, HGF + CsA versus CsA alone, respectively) (Figure 3B) ▶ . When HGF and CsA were administered simultaneously, there was no decrease in apoptosis.
Figure 3.
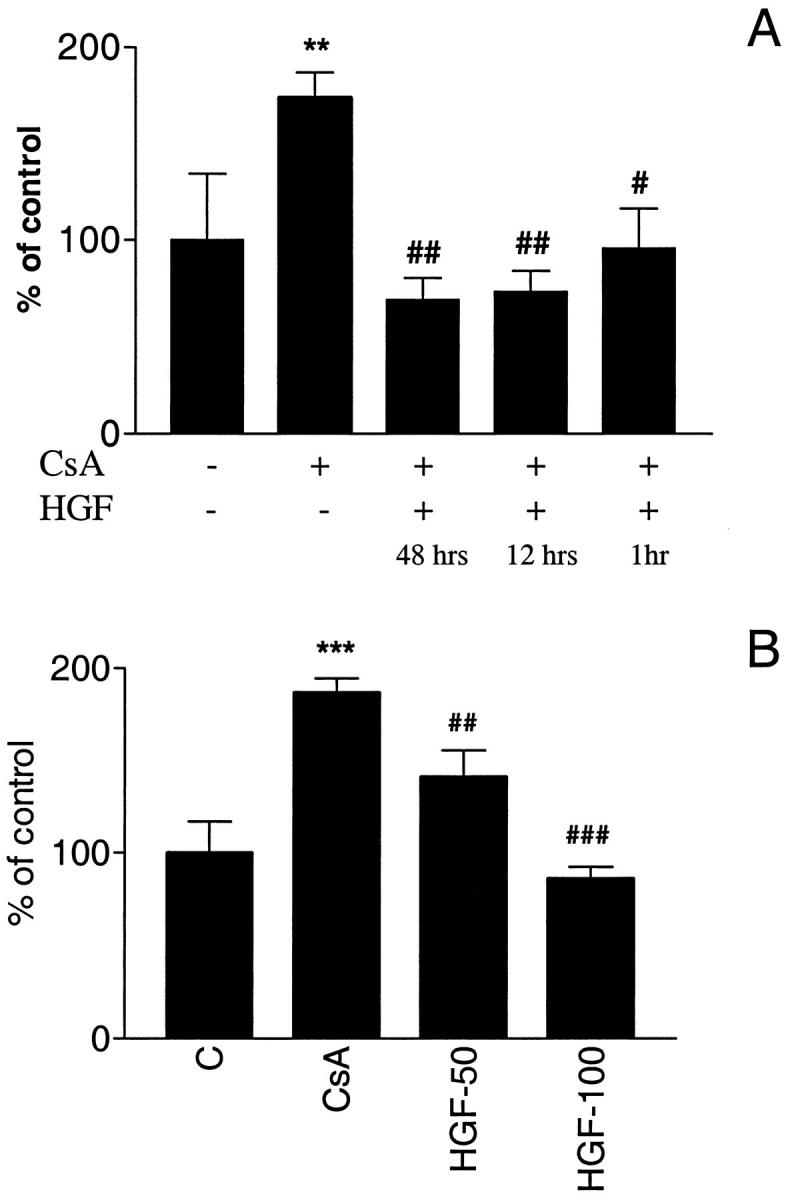
Effect of HGF on CsA-induced apoptosis. A: HGF pretreatment (100 ng/ml) for 48, 12, and 1 hour before CsA exposure (0.5 μg/ml) prevented CsA-induced apoptosis. **P < 0.01 (CsA treated cells versus non-treated cells). ##P < 0.01 (HGF-pretreatment for 48 and 12 hours cells versus no pretreatment). #P < 0.05 (1 hour HGF pretreatment versus no pretreatment). B: The protective effect of HGF is dose-dependent. ***P < 0.001 (CsA treated cells versus control cells). ##P < 0.01 (50 ng/ml HGF pretreatment versus no pretreatment). #P < 0.05 (100 ng/ml HGF pretreatment versus no pretreatment).
IGF-I pretreatment for 1, 12, or 48 hours did not affect podocyte apoptosis after CsA exposure (Figure 4A) ▶ at any of the doses investigated (Figure 4B) ▶ .
Figure 4.
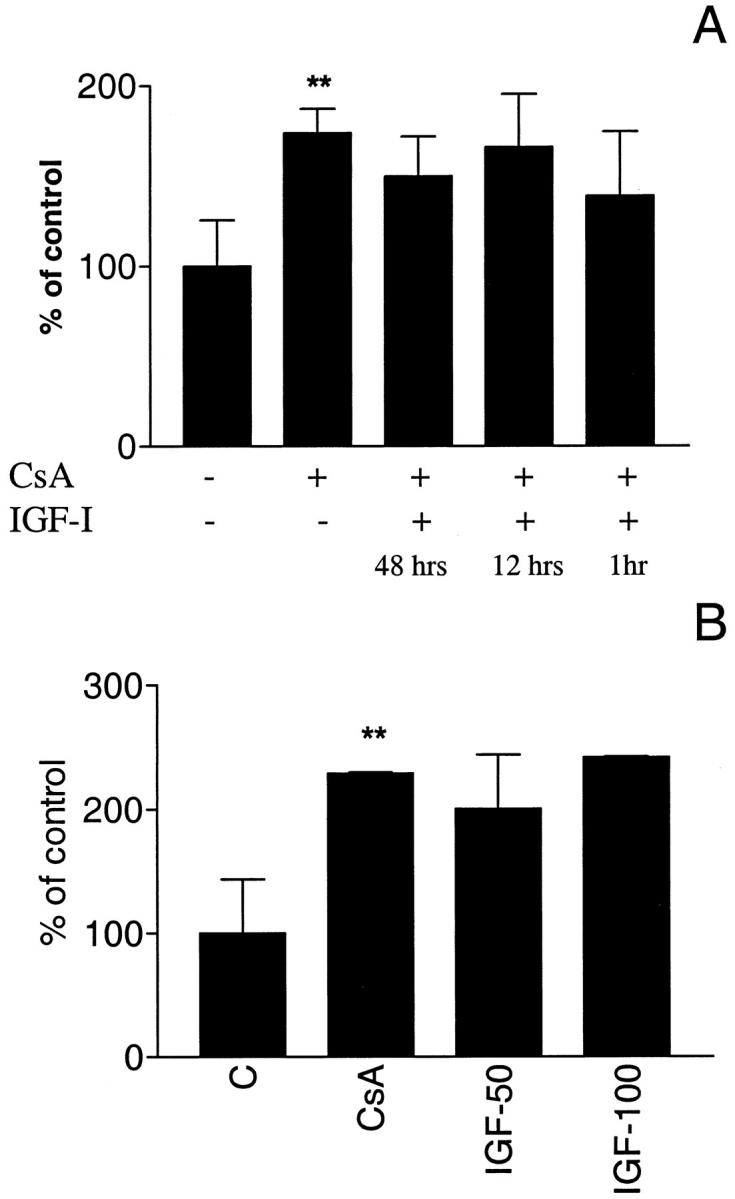
Effect of IGF-I on CsA-induced apoptosis. A: IGF-I pretreatment (100 ng/ml) for 48, 12 and 1 hour before CsA exposure (0.5 μg/ml) did not prevent CsA-induced apoptosis. **P < 0.01 (CsA-treated cells versus control cells). B: IGF-I pretreatment does not protect from CsA-induced apoptosis at any of the doses investigated (50 and 100 ng/ml) **P < 0.01 (IGF-I pretreatment versus no pretreatment).
The addition of HGF or IGF-I for 48 hours led to a 1.27 ± 0.03-fold or a 1.46 ± 0.11-fold increase in cell number, respectively.
Effect of CsA and HGF or IGF-I on Bcl-xL, Fas and Fas-Ligand Production
CsA exposure (0.5 μg/ml for 48 hours) did not change Fas or Fas-ligand production (Figure 5) ▶ , but reduced Bcl-xL production to 40.4 ± 15.7% of control (100 ± 3.7%) levels (P < 0.05) (Figure 6A) ▶ . However, in the presence of HGF pretreatment, the reductions in Bcl-xL levels were inhibited (72.88 ± 27.2% at 1 hour, 69.9 ± 12.4% at 12 hours, and 92.8 ± 8.2% at 48 hours, respectively) (Figure 6A) ▶ . In contrast, IGF-I pretreatment for 1, 12, or 48 hours did not affect the CsA-induced Bcl-xL down-regulation (Figure 6B) ▶ .
Figure 5.

Effect of CsA on Fas and Fas-ligand. CsA treatment (0.5 μg/ml) for 24 hours did not affect Fas and Fas-ligand production.
Figure 6.
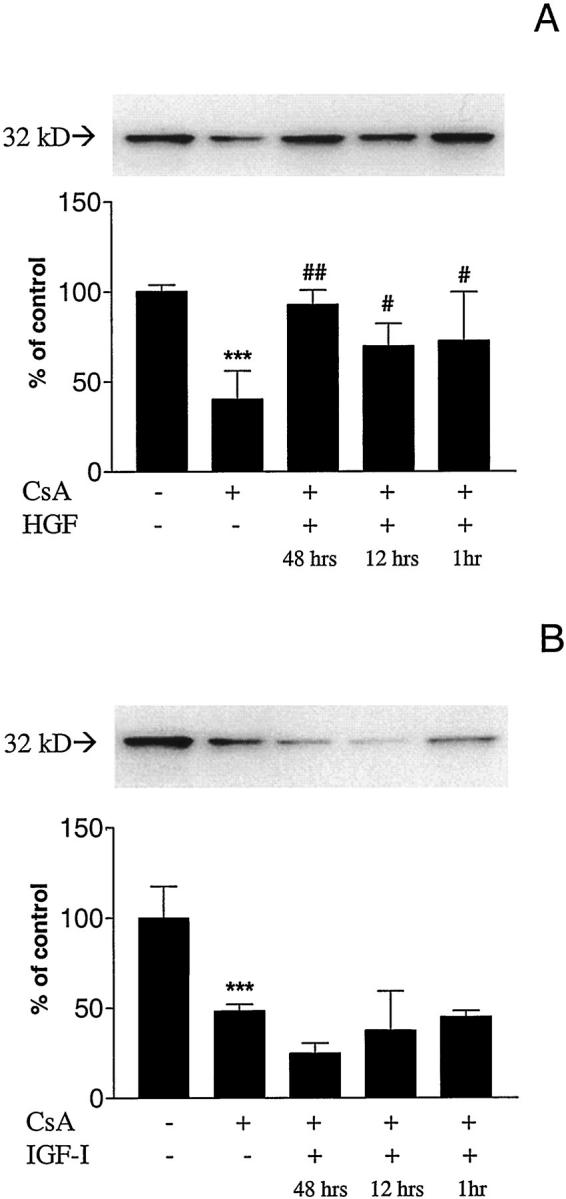
Effect of CsA with and without HGF or IGF-I pretreatment on Bcl-xL. A: CsA treatment (0.5 μg/ml) for 24 hours decreased Bcl-xL protein levels (***P < 0.001). HGF pretreatment for 48 hours (##P < 0.01) and for 12 and 1 hour (#P < 0.05) preserved Bcl-xL levels, compared to non-pretreated cells. B: CsA treatment (0.5 μg/ml) for 24 hours decreased Bcl-xL protein levels (***P < 0.001). IGF-I pretreatment for 48, 12, or 1 hour did not preserve Bcl-xL levels, compared to non pretreated cells.
Role of PI3′-K and MEK-1 in Bcl-xL Up-Regulation by HGF
PI3′-kinase inhibition by LY29400 completely abolished Bcl-xL up-regulation by HGF in CsA-treated podocytes, although the inhibitors of MEK-1 (PD098059) did not have this effect. In particular, a 50 ± 9.6% reduction in Bcl-xL production in CsA-treated samples receiving HGF pretreatment was observed when PI3′-kinase pathway was inhibited by LY29400 pretreatment (Figure 7) ▶ , which was not significantly different compared to CsA-treated samples without HGF pretreatment.
Figure 7.

PI3′-K and MEK-1 pathways in Bcl-xL levels after HGF pretreatment. When 4 μmol/L LY29400 (PI3′-K inhibitor) or PD098059 (MEK-1 inhibitor) were added before HGF stimulation, LY29400, but not PD098059, blunted the HGF effect on Bcl-xL. ♦♦P < 0.01, LY29400 + HGF + CsA-treated cells versus HGF + CsA-treated cells. **P < 0.01, CsA treated cells versus non-pretreated cells. ##P < 0.01, HGF + CsA and HGF + PD098059 + CsA versus non-pretreated cells.
Discussion
CsA nephrotoxicity has been shown to be associated with apoptosis, 3 and the ability of CsA to induce apoptosis is cell type-specific. CsA induces apoptosis in tubular renal cells, 4,5 glioma cells, 18 and hepatocytes, 19 but protects endothelial cells 20 and myeloid leukemia cells 21 from cell death. In this in vitro study of podocytes, we found that CsA treatment was associated with a dose- and time-dependent increase in apoptosis. Interestingly, an increase in DNA fragments in CsA-treated cells was detectable before a decrease in cell number occurred, suggesting that DNA fragmentation is an early event in the apoptotic process in podocytes in vitro. Among the several growth factors which reduce apoptosis, we studied HGF and IGF-I because both have been implicated in CsA-induced nephropathy. 10,11 Only one previous study investigated the role of IGF-I and HGF on serum starvation-induced apoptosis in human umbilical vein endothelial cells. 22 They found that HGF protected against apoptosis, whereas IGF-I did not. We showed that HGF, but not IGF-I, protected against CsA-induced apoptosis, suggesting that different pathways are activated by these growth factors in podocytes. Although pretreatment with HGF was effective, the simultaneous administration of HGF and CsA was not able to prevent apoptosis. This suggests that the CsA-induced apoptosis occurs faster than the induction of protective genes by HGF.
The mechanisms for CsA-induced apoptosis and for HGF and IGF-I protection are still a matter of investigation. A recent study suggested that CsA-induced apoptosis in the kidney could be mediated by a down-regulation of Bax/Bcl-2 as well as by an up-regulation of Fas-L. 6 Immunofluorescence experiments conducted in this latter study showed that Bcl-2 and Fas-L were altered in the glomeruli as well as in the interstitial and vascular compartment of CsA-treated rats, suggesting that several pathways were probably involved. Although the ability of podocytes to express Fas and Fas-ligand is not known, 23 we also tested if CsA regulates Fas/Fas-ligand. In contrast to what has been previously shown in renal tubular cells, 4 we did not find changes in either Fas or Fas-ligand production in CsA-treated podocytes.
Although evidence exists for a PI3′-kinase and MEK-1 dependent regulation of apoptosis by IGF-I, 24 less is known about the means by which HGF exerts a protective effect. A recent study suggests that both a rapid posttranslational PI3-K/AKT activation and a later Bcl-xL de novo gene expression could be involved in the HGF protection from apoptosis of human proximal tubular epithelial HKC cells. 12 This specific regulation of Bcl-xL among various apoptosis regulatory proteins by HGF led us to hypothesize that Bcl-xL was involved in podocyte apoptosis, and that its levels were maintained by HGF when given before CsA treatment. Thus, we investigated whether CsA-induced apoptosis in podocytes was accompanied by an alteration in the levels of Bcl-xL. We found that CsA-induced apoptosis was associated with a down-regulation of Bcl-xL, and that HGF protects podocytes from CsA-induced apoptosis, probably by regulation of Bcl-xL in a PI3′-K dependent but MEK-1 independent manner. In addition, this effect was specific for HGF, because IGF-I did not protect from apoptosis in podocytes and did not modulate Bcl-xL production. However, the intracellular signaling pathways of both growth factors were functional in podocytes, as shown by the preservation of their effect on cell proliferation.
In conclusion, we found that CsA can induce apoptosis in podocytes in vitro and that this phenomenon could be completely prevented by HGF, but not IGF-I, pretreatment. In addition, we propose that the preservation of Bcl-xL levels by HGF could be responsible for its protection from CsA-induced apoptosis in podocytes. Our data suggest that a direct effect of CsA on podocytes may contribute to the glomerular damage observed in some patients, and that HGF could have a protective effect. Further studies will be needed to assess the clinical relevance of HGF in CsA-induced nephropathy, as well as in other glomerular diseases, such as rapidly progressive glomerulonephritis, HIV nephropathy, or lupus nephritis, where podocyte apoptosis is observed and could play a role in disease initiation and progression. Finally, the ability of CsA to induce apoptosis in podocytes raises some concerns about the widespread use of this drug in glomerulopathy with nephrotic syndrome resistant to conventional therapy, 25 and should be further investigated.
Footnotes
Address reprint requests to Dr. Liliane J. Striker, Renal Cell Biology Laboratory, University of Miami School of Medicine, P. O. Box 016960 (R126), Miami, FL 33101. E-mail: lstriker@med.miami.edu.
References
- 1.Shihab FS: Cyclosporine nephropathy: pathophysiology and clinical impact. Semin Nephrol 1996, 16:536-547 [PubMed] [Google Scholar]
- 2.Myers BD, Ross J, Newton L, Luetscher J, Perlroth M: Cyclosporine-associated chronic nephropathy. N Engl J Med 1984, 311:699-705 [DOI] [PubMed] [Google Scholar]
- 3.Thomas SE, Andoh TF, Pichler RH, Shankland SJ, Couser WG, Bennett WM, Johnson RJ: Accelerated apoptosis characterizes cyclosporine-associated interstitial fibrosis. Kidney Int 1998, 53:897-908 [DOI] [PubMed] [Google Scholar]
- 4.Healy E, Dempsey M, Lally C, Ryan MP: Apoptosis and necrosis: mechanisms of cell death induced by cyclosporine A in a renal proximal tubular cell line. Kidney Int 1998, 54:1955-1966 [DOI] [PubMed] [Google Scholar]
- 5.Ortiz A, Lorz C, Catalan M, Coca S, Egido J: Cyclosporine A induces apoptosis in murine tubular epithelial cells: role of caspases. Kidney Int Suppl 1998, 68:S25-S29 [DOI] [PubMed] [Google Scholar]
- 6.Shihab FS, Andoh TF, Tanner AM, Yi H, Bennett WM: Expression of apoptosis regulatory genes in chronic cyclosporine nephrotoxicity favors apoptosis. Kidney Int 1999, 56:2147-2159 [DOI] [PubMed] [Google Scholar]
- 7.Butt AJ, Firth SM, Baxter RC: The IGF axis and programmed cell death. Immunol Cell Biol 1999, 77:256-262 [DOI] [PubMed] [Google Scholar]
- 8.Mene P, Amore A: Apoptosis: potential role in renal diseases. Nephrol Dial Transplant 1998, 13:1936-1943 [DOI] [PubMed] [Google Scholar]
- 9.Amaike H, Matsumoto K, Oka T, Nakamura T: Preventive effect of hepatocyte growth factor on acute side effects of cyclosporin A in mice. Cytokine 1996, 8:387-394 [DOI] [PubMed] [Google Scholar]
- 10.Kasai S, Yoshimura R, Sugimura K, Ohyama A, Harimoto K, Kishimoto T, Yoshimura N: Study of hepatocyte growth factor in cyclosporine-induced nephropathy. Transplant Proc 1997, 29:1724-1725 [DOI] [PubMed] [Google Scholar]
- 11.Johnson DW, Saunders HJ, Johnson FJ, Huq SO, Field MJ, Pollock CA: Cyclosporin exerts a direct fibrogenic effect on human tubulointerstitial cells: roles of insulin-like growth factor I, transforming growth factor beta1, and platelet-derived growth factor. J Pharmacol Exp Ther 1999, 289:535-542 [PubMed] [Google Scholar]
- 12.Liu Y, Sun AM, Dworkin LD: Hepatocyte growth factor protects renal epithelial cells from apoptotic cell death. Biochem Biophys Res Commun 1998, 246:821-826 [DOI] [PubMed] [Google Scholar]
- 13.Isgaard J, Tivesten A: The role of growth hormone and insulin-like growth factor I in the regulation of apoptosis. Growth Horm IGF Res 1999, 9 (suppl A):125-128 [DOI] [PubMed] [Google Scholar]
- 14.Liu Y: Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol 1999, 277:F624-F633 [DOI] [PubMed] [Google Scholar]
- 15.Yo Y, Morishita R, Nakamura S, Tomita N, Yamamoto K, Moriguchi A, Matsumoto K, Nakamura T, Higaki J, Ogihara T: Potential role of hepatocyte growth factor in the maintenance of renal structure: anti-apoptotic action of HGF on epithelial cells. Kidney Int 1998, 54:1128-1138 [DOI] [PubMed] [Google Scholar]
- 16.Conti FG, Elliot SJ, Striker LJ, Striker GE: Binding of insulin-like growth factor-I by glomerular endothelial and epithelial cells: further evidence for IGF-I action in the renal glomerulus. Biochem Biophys Res Commun 1989, 163:952-958 [DOI] [PubMed] [Google Scholar]
- 17.MacKay K, Striker LJ, Stauffer JW, Doi T, Agodoa LY, Striker GE: Transforming growth factor-beta: murine glomerular receptors and responses of isolated glomerular cells. J Clin Invest 1989, 83:1160-1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pyrzynska B, Mosieniak G, Kaminska B: Changes of the trans-activating potential of AP-1 transcription factor during cyclosporin A-induced apoptosis of glioma cells are mediated by phosphorylation and alterations of AP-1 composition. J Neurochem 2000, 74:42-51 [DOI] [PubMed] [Google Scholar]
- 19.Roman ID, Rodriguez-Henche N, Fueyo JA, Zueco JA, Menor Prieto JC, Guijarro LG: Cyclosporin A induces apoptosis in rat hepatocytes in culture. Arch Toxicol 1998, 72:559-565 [DOI] [PubMed] [Google Scholar]
- 20.Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S: Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation 1998, 98:1153-1157 [DOI] [PubMed] [Google Scholar]
- 21.Lotem J, Sachs L: Different mechanisms for suppression of apoptosis by cytokines and calcium mobilizing compounds. Proc Natl Acad Sci USA 1998, 95:4601-4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinoshita M, Shimokado K: Autocrine FGF-2 is responsible for the cell density-dependent susceptibility to apoptosis of HUVEC: a role of a calpain inhibitor-sensitive mechanism. Arterioscler Thromb Vasc Biol 1999, 19:2323-2329 [DOI] [PubMed] [Google Scholar]
- 23.Ortiz A, Lorz C, Egido J: The Fas ligand/Fas system in renal injury. Nephrol Dial Transplant 1999, 14:1831-1834 [DOI] [PubMed] [Google Scholar]
- 24.Navarro P, Valverde AM, Benito M, Lorenzo M: Insulin/IGF-I rescues immortalized brown adipocytes from apoptosis down-regulating Bcl-xS expression, in a PI 3-kinase- and MAP kinase-dependent manner. Exp Cell Res 1998, 243:213-221 [DOI] [PubMed] [Google Scholar]
- 25.Klein M, Radhakrishnan J, Appel G: Cyclosporine treatment of glomerular diseases. Annu Rev Med 1999, 50:1-15 [DOI] [PubMed] [Google Scholar]