

Abstract
In this study, we report that infection with Plasmodium yoelii 17XL, a lethal strain of rodent malaria, does not result in death in the DBA/2 strain of mice. In contrast to BALB/c mice, DBA/2 mice developed significantly less parasitemia and never manifested symptoms of cerebral malaria (CM) on infection with this parasite. Moreover, the histological changes evident in the brain of susceptible BALB/c were absent in DBA/2 mice. Interestingly, the resistant DBA/2 mice when treated with recombinant interleukin (IL)-2, were found to develop CM symptoms and the infection became fatal by 6 to 8 days after infection. This condition was associated with an augmented interferon-γ and nitric oxide production. Unexpectedly, IL-10 levels were also elevated in IL-2-treated DBA/2 mice during late stage of infection (at day 6 of infection) whereas the inverse relationship between IL-10 and interferon-γ or nitric oxide was maintained in the early stage of infection (at day 3 after infection). The level of tumor necrosis factor-α production was moderately increased in the late phase of infection in these mice. Histology of brain from IL-2-treated mice demonstrated the presence of parasitized erythrocytes and infiltration of lymphocytes in cerebral vessels, and also displayed some signs of endothelial degeneration. Confocal microscopical studies demonstrated preferential accumulation of γδ T cells in the cerebral vessels of IL-2-treated and -infected mice but not in mice treated with IL-2 alone. The cells recruited in the brain were activated because they demonstrated expression of CD25 (IL-2R) and CD54 (intercellular adhesion molecule 1) molecules. Administration of anti-γδ mAb prevented development of CM in IL-2-treated mice until day 18 after infection whereas mice treated with control antibody showed CM symptoms by day 6 after infection. The information concerning creating pathological sequelae and death in an otherwise resistant mouse strain provides an interesting focus for the burden of pathological attributes on death in an infectious disease.
The pathogenesis of cerebral malaria (CM) has been a subject of considerable interest, and it remains a major cause of death associated with severe Plasmodium falciparum infection in many tropical countries. 1 The early events that lead to human CM are difficult to study experimentally because of ethical constraints and the limitations of postmortem materials. Because of these difficulties, investigators turned to murine models to investigate mechanisms of CM pathogenesis. Considerable effort has been devoted to study the cerebral pathogenesis in the murine models involving Plasmodium berghei ANKA and Plasmodium yoelii 17XL, however, there are many aspects of this pathogenesis that are still poorly understood. 2 Previous studies in the P. berghei ANKA murine model have indicated that CM might be a lymphocyte-mediated disease, in which CD4+, and CD8+ T cells have been postulated to play a role. 3-5 It was recently demonstrated that on treatment with anti-γδ T cell antibody, the susceptible mice failed to develop CM after P. berghei ANKA infection indicating some pathological role of γδ T cells. 6 However, the extent of T cell activation that is required for the development of CM and whether actual T cell infiltration occurs at the site of brain has not been known. Morphological and biochemical studies have so far not recognized a focal or global determinant(s) for CM pathogenesis or a clear relationship between brain lesion and death.
P. yoelii 17XL regularly induces a CM syndrome in susceptible mice that parallels human disease in several respects including cytoadherence of parasitized red blood cells (PRBCs) in the brain and pathological symptoms. 7,8 Whether CM represents an inflammatory pathogenesis was not clarified by studying P. yoelii infection in susceptible mice. It remains debatable whether human CM presents an inflammatory pathogenesis. 9,10 Development of CM in humans infected with P. falciparum has often been accompanied by increased production of interferon (IFN)-γ and tumor necrosis factor (TNF)-α. 11,12 Interleukin (IL)-2 is a potent T cell activator and has the ability to induce production of these pro-inflammatory cytokines. 13,14 Considering that IL-2 is central in the regulation of cell-mediated immune responses, we have hypothesized whether the systemic administration of IL-2 can induce pathological manifestations of CM in a strain of mouse that is otherwise resistant to this syndrome. If it does so, this will be invaluable for analysis of the immune response, particularly of T cell responses and the associated pathology. In this report, we demonstrate that enhanced immunoreactivity induced by IL-2 treatment alters CM-resistant mice to susceptible-like phenotype as revealed by development of symptoms and histological changes characteristic of CM. This condition in resistant DBA/2 mice was associated with increased production of IFN-γ and nitric oxide (NO). Confocal microscopic studies showed preferential recruitment of γδ T-bearing cells in cerebral vessels of IL-2-treated mice manifesting CM symptoms. To our knowledge, this is the first direct evidence of accumulation of γδ T cells in the brain of a host that developed CM. Treatment with anti-γδ T cell mAb arrested the development of CM in IL-2-treated DBA/2 mice. These results suggest that γδ T cells play an important role in the pathways of CM pathogenesis.
Materials and Methods
Parasites and Infection
Female DBA/2 (H-2d) mice, 5 to 6 weeks old, were purchased from the Jackson Laboratory (Bar Harbor, ME) and from Iffa Credo (L’Arbresle, France). All animals were housed in the accredited Animal Research Facility and maintained under the guidelines established by the institution for their use. There are two strains of P. yoelii available, one is lethal and the other is nonlethal. Throughout our present study, we used the lethal (17XL) strain (PYL). The parasite was stored in liquid nitrogen and then used to infect source mice. PRBCs obtained from source mice were suspended in RPMI 1640 and were injected intraperitoneally (5 × 10 5 PRBCs/mouse) in all experimental mice. Parasitemia was monitored by making blood smears and counting Giemsa-stained fields. The survival of mice was assessed until day 30 after infection.
Monoclonal Antibody PRBCs
Antibodies directed against CD3 (mAb 145-2C11), γδ TCR-expressing T cells [mAb GL3, fluorescein isothiocyanate (FITC)-conjugated], αβ TCR-bearing T cells (mAb H57-597, FITC-conjugated), CD25 (mAb 7D4, FITC-conjugated), and CD54 [intercellular adhesion molecule 1 (ICAM-1)] (mAb 3E2, PE-conjugated) were used in this study. All these monoclonal antibodies were obtained from PharMingen (San Diego, CA). Hybridoma secreting anti-γδ TCR mAb (GL3) was obtained from Dr. Pablo Pereira, Institut Pasteur, Paris, France.
Interleukin 2 (IL-2) Treatment
Recombinant IL-2 was a kind gift from Dr. Tom Ciardelli, Department of Toxicology, Dartmouth Medical School. DBA/2 mice were treated with rIL-2 as essentially reported previously. 15 Mice were administered rIL-2, 1 μg in the presence of 1% of normal mouse serum in 200 μl of physiological saline per injection. Administration was via intraperitoneal inoculation three times daily for total of 8 days. It was shown previously that IL-2 was detectable in the serum samples from the mice treated three times a day with the above dose. Two days after the initiation of therapy, mice were infected with P. yoelii 17XL (5 × 10 5 PRBCs/mouse). Age-matched control mice were given similar doses of rIL-2 injections in 1% normal mouse serum but they did not receive any infection.
Histopathological Analysis
The brain of each mouse was removed immediately after anesthesia and placed in 10% buffered neutral formaldehyde (Polyscience Inc., Warrington, PA). Paraffin-embedded brain tissues were sectioned and stained with hematoxylin and eosin. These are random sections of the cerebrum. The slides were examined and then photographed.
T Cell Preparation and Culture
Mice were killed on days 3 and 6 after infection, spleens were removed and gently dissociated into single-cell suspensions. Red blood cells were removed by using lysing buffer (Sigma Chemical Co., St. Louis, MO). Cell suspensions were passed through nylon wool columns to enrich for T cells. These cells were ∼90% T cells. Bulk cultures were set up in 24-well plates with 2 × 10 6 cells/well, and stimulated with cross-linked anti-CD3 mAb as described previously. 15 For anti-CD3 mAb (145–2C11 mAb; PharMingen,) driven activation, culture-plate wells were precoated with goat anti-hamster IgG (14 μg of anti-hamster IgG) (Jackson Immunology Research Laboratories, West Grove, PA) overnight at 4°C. After washing, the wells were incubated with 5 μg/ml of anti-CD3 mAb at 37°C for several hours.
Enzyme-Linked Immunosorbent Assay Quantitation of Cytokine in Splenic T-Cell Culture Supernatants
Supernatants from cultures of cross-linked anti-CD3 mAb stimulated splenocytes from the various groups of mice were collected at 24 and 48 hours after the beginning of culture. The bulk cultures were set up in 24-well plates (Nunc, Rochester, NY) with 2 × 10 6 cells/well, in a final volume of 1 ml of RPMI 1640 medium supplemented with fetal calf serum, HEPES, l-glutamine, 2 mercaptoethanol, and gentamicin. In preliminary experiments, a 48-hour incubation time was found to be optimum for cytokine and nitrite productions. Cytokine enzyme-linked immunosorbent assays were conducted using paired capture and biotinylated detection antibodies from R&D Systems (Minneapolis, MN) following the manufacturer’s recommendations. The cytokine level was calculated by reference to standard units provided by the manufacturer. The results were presented as pg/ml.
Assay for Nitrite Production
Nitrite production in 48-hour culture supernatants was measured using Griess reagent. 16 Briefly, 0.05 ml of supernatant was mixed with 0.05 ml of Griess reagent (0.5% sulfanilamide and 0.05% N-1-naphthylenedimine hydrochloride in 2.5% H3PO4) then incubated for 10 minutes at room temperature and the absorbency was read at 570 nm by using an automated plate reader. The nitrite concentration was calculated from a NaNO2 (Sigma Chemical Co.) standard curve. The results were presented as μmol/L.
Immunofluorescence for T Cell Visualization in Brains by Confocal Microscopy
The blocks of brain tissue were trimmed, and 30- to 70-μm sections were cut using a vibratome (V1000, TPI; Energy Beam Sciences, Agawam, MA). Sections were maintained in ice-cold phosphate-buffered saline (PBS) throughout processing. To phenotype the immune cells present in the brain, immunofluorescent staining of tissue sections was conducted according to the method described previously. 17 Briefly, immediately after cutting, for direct staining, 2 μg/100 μl each of FITC-, PE-labeled Abs in PBS/1% bovine serum albumin/0.1% azide (PBA)-containing mouse Ig (6 mg/ml to block nonspecific binding) were added to sections in 96-well plates and incubated overnight at 4°C in the dark with continuous gentle agitation. Unbound Ab was removed from the sections by aspiration followed by four 20-minute washes in PBA. Washed sections were then fixed overnight in the same buffer containing 1% paraformaldehyde. Stained sections were wet-mounted in anti-fade (Molecular Probes, Eugene, OR), sealed with nail varnish, and stored at 4°C in the dark for up to 10 days before confocal imaging. Unstained and fluorescein isotype controls were used to control for autofluorescence and nonspecific Ab binding, respectively.
In Vivo Administration of Anti-γδ TCR mAb
Protein G-purified anti-γδ TCR mAb was injected intraperitoneally at a dose of 500 μg as described previously. 6 Groups (n = 7) of IL-2-treated DBA/2 mice were used to assess the effect of depletion of γδ T cells on development of CM. One group was administered with anti-γδ TCR mAb on days −1 and +2 after infection, and another group received injections on days +3 and +6 after infection. Control groups were administered with purified hamster IgG (Jackson Immunology Research Laboratories) diluted with PBS in the manner as in experimental groups. The efficacy of γδ T-cell depletion was determined by flow cytometry of spleen lymphocytes. The percentage of γδ T cells was <0.02% after anti-γδ mAb treatment.
Statistical Significance
Probability significance was determined by two-tailed Student’s t-test, assuming equal variances. Statistical significance was set at P of <0.05 for all comparisons except murine survival after immunization. Fisher’s exact test was used for murine survival experiments.
Results
DBA/2 Mice after Infection with P. yoelii 17XL (a Lethal Strain) Did Not Develop CM Pathology or Succumb to Infection
In a previous study, we showed that BALB/c mice infected with P. yoelii 17XL developed symptoms of CM and died by days 6 to 8 after infection. 18 In this study, BALB/c and DBA/2 mice were given different doses of blood-stage infection with P. yoelii 17XL and we then evaluated parasitemia and survival in these two strains of mice. Whatever inoculum was given the level of parasitemia was almost the same in both BALB/c and DBA/2 mice at day 4 after infection but it increased rapidly and became significantly higher in BALB/c mice than in DBA/2 mice on day 6 of infection (P < 0.005). When infection was given with a lower dose (0.2 × 10 5 PRBCs), the peak of parasitemia was slightly delayed in BALB/c mice compared to that after infection with a higher dose (1 × 10 5 PRBCs) (Figure 1) ▶ . The results presented in Figure 2 ▶ clearly demonstrate that whatever dose of inoculum ranging from 0.2 × 10 5 to 1 × 10 7 was given, all of DBA/2 mice survived the infection whereas all of the BALB/c mice died by day 6 to 8 after infection. Both DBA/2 and BALB/c mice were infected with 5 × 10 5 PRBCs, and then brain sections were obtained from them on day 6 of infection. The results of brain histology showed the presence of parasitized erythrocytes adjacent to endothelial wall, infiltration of lymphocytes, and signs of endothelial cell activation in susceptible BALB/c (Figure 3A) ▶ but not in DBA/2 mice (Figure 3B) ▶ .
Figure 1.

Levels of parasitemia in BALB/c and DBA/2 mice (n = 5/group) after infection with P. yoelii (17XL strain). Mice were infected intraperitoneally with different doses of PRBCs: 0.2 × 10 5 (a), 1.0 × 10 5 (b), 5.0 × 10 5 (c), 1.0 × 10 7 (d). This experiment was repeated three times.
Figure 2.
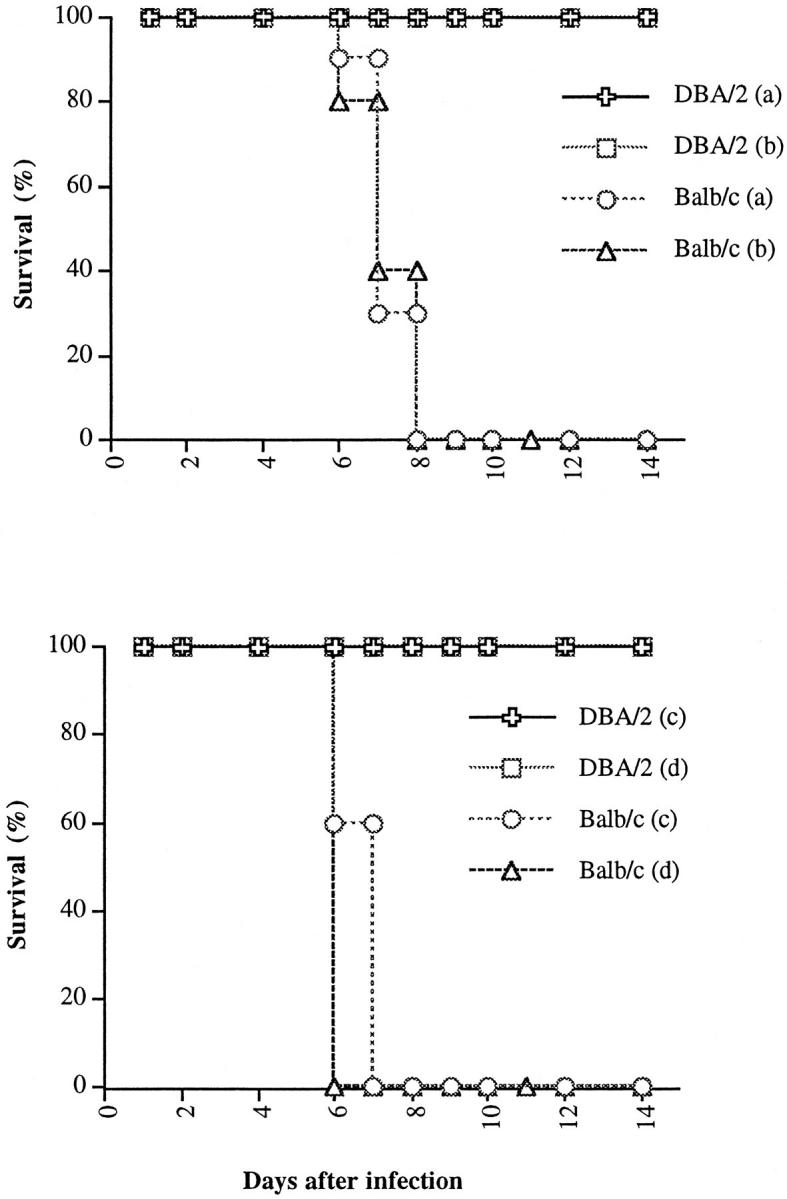
Survival of BALB/c and DBA/2 mice (n = 10/group) given infection with P. yoelii (17XL strain). Both strains of mice received the same dose of PRBCs: 0.2 × 10 5 (a), 1.0 × 10 5 (b), 5.0 × 10 5 (c), 1.0 × 10 7 (d). DBA/2 infected mice survived until day 30, end of observation period (not shown).
Figure 3.
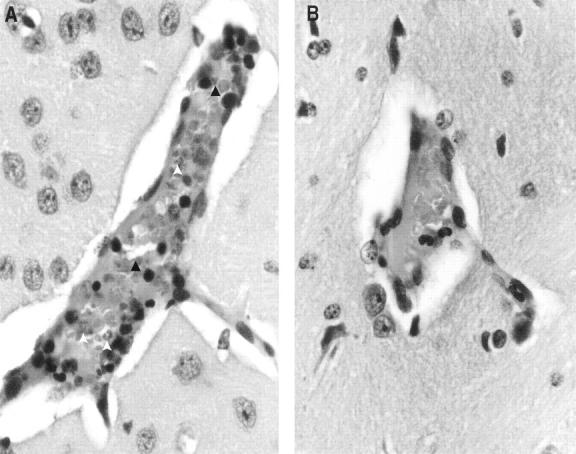
Histology of the brain (original magnification, 20 × 1.6) shows accumulation of parasitized erythrocytes (open arrow) and infiltration of lymphocytes (dark arrow) within cerebral vessels of BALB/c (A) but not in DBA/2 (B) mice after infection with P. yoelii (17XL strain).
IL-2 Treatment Alters CM-Resistant Mice to Susceptible-Like Phenotype
To determine whether CM pathology is related to an enhanced T cell activation, resistant DBA/2 mice were administered with rIL-2. After the treatment, all of the DBA/2 mice that were infected with P. yoelii 17XL (TI) developed symptoms of CM by day 6 after infection. The symptoms include: appearance of ruffled hair, convulsions, seizure, ataxia, and coma, whereas none of the nontreated but infected mice (IO) showed any of these CM signs. All TI animals succumbed to infection by 6 to 8 days after infection whereas none of the IO mice died by the same time period (Figure 4) ▶ . The regimen of IL-2 treatment was not toxic for DBA/2 mice because all of the IL-2-treated but noninfected mice (TO) were in good health and survived until the end of observation period (Figure 4) ▶ . There was no significant difference in parasitemia between treated and untreated mice at day 3 after infection. The IL-2-treated moribund mice at day 6 after infection showed slightly higher parasitemia than nontreated infected mice (Figure 5) ▶ .
Figure 4.

Survival of P. yoelii 17XL-infected and IL-2-treated mice. DBA/2 mice treated with IL-2 and infected with P. yoelii (17XL strain; n = 10); mice treated with IL-2 but not given infection (n = 10); or mice received infection alone (n = 10) (C). DBA/2 mice treated/no infection or mice given infection only survived until day 35, the end of observation period (not shown).
Figure 5.

Effect of IL-2 treatment on parasitemia during the course of infection with P. yoelii (17XL strain) in DBA/2 mice (n = 5/group). The parasitemia in IL-2-treated and -infected moribund DBA/2 mice was slightly increased compared to mice given infection alone at day 6 after infection. Evaluation of parasitemia could not be continued in treated and infected mice beyond day 6 of infection as they died by this time period, whereas parasitemia was shown in surviving mice given infection alone until day 30 of infection. This experiment was repeated three times.
IL-2 Treatment Induced Augmented Production of Pro-inflammatory Cytokine in Moribund Mice with CM
The extent of T cell activation after administration of rIL-2 was assessed by measuring cytokine production by T-cell enriched populations obtained from spleens. Splenic cells were enriched for T lymphocytes by passage on nylon-wool columns and then were stimulated with cross-linked anti-CD3 mAb. This TCR-mediated activation has allowed potential generation of cytokines by mouse T cells. A significantly lower IFN-γ production was found at day 3 after infection in TI mice than in TO, IO, or in normal mice (P < 0.005) (Figure 6A) ▶ . The diminished production of IFN-γ at an early stage of infection (on day 3 of infection) in TI animals was accompanied by an enhanced production of IL-10, an immunoregulatory cytokine (Figure 6A) ▶ . The level of IFN-γ was significantly augmented in TI mice at day 6 after infection compared to that in the early stage of infection (P < 0.005) (Figure 6B) ▶ . There was no significant difference in the level of IFN-γ between IO and TO mice on day 6 of infection (Figure 6B) ▶ . Our findings showed significantly higher IL-10 production in the early stage of infection in TI mice than in IO or TO mice (P < 0.005). No such difference in IL-10 production was seen between these groups in the late stage of infection (P > 05). The level of TNF-α production was lower in TI mice (532 ± 57 pg/ml) than in IO animals (820 ± 170 pg/ml) during the early stage of infection whereas this became higher in TI mice (772 ± 57 pg/ml) than in IO animals (508 ± 94 pg/ml) in the late stage of infection. There was an increase in the production of TNF-α in TI mice in the late stage of infection compared to that in the early stage. Although these differences in TNF-α levels are significant (P < 0.05), they are not extremely as significant as seen in the case of IFN-γ production. These results are summarized in Figure 6 ▶ .
Figure 6.
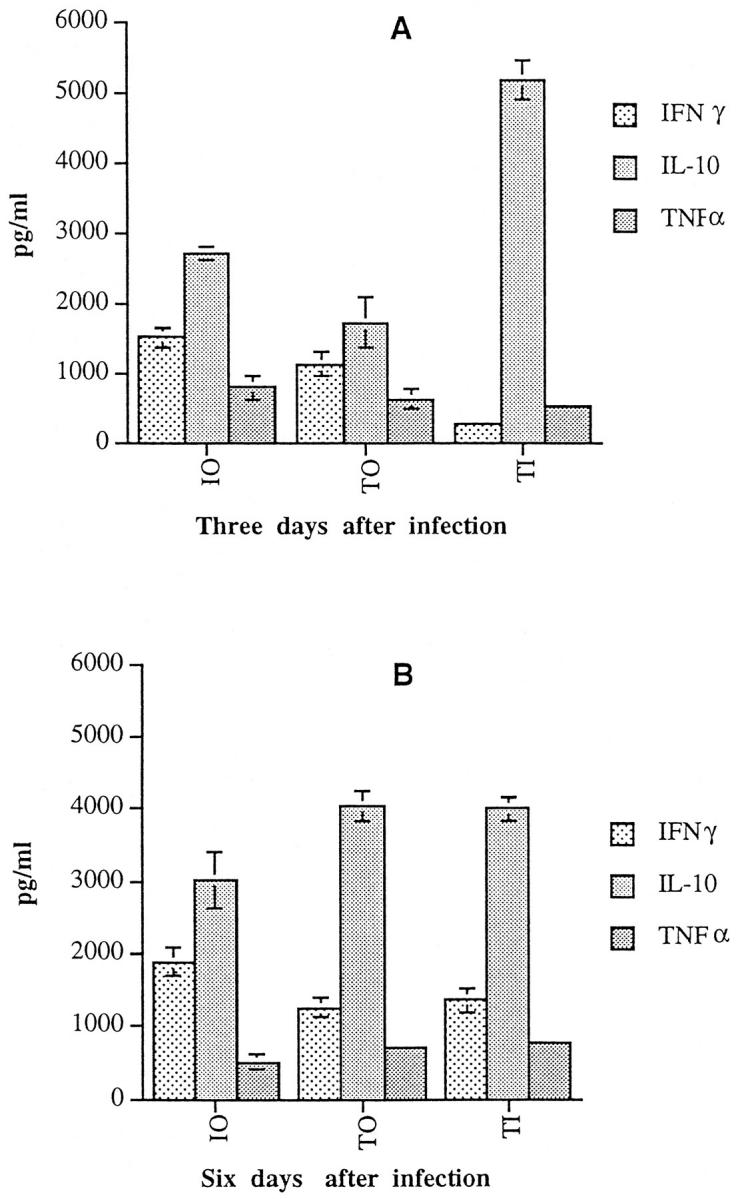
Cytokine production in DBA/2 mice treated with IL-2 and infected with P. yoelii (17XL strain). Mice (n = 3) were either given IL-2 treatment only or treated with IL-2 and given infection with P. yoelii. Spleen cells were harvested from mice infected for 3 or 6 days and enriched for T cells by passage on nylon-wool column and bulk cultures were set up with 2 × 10 6 cells/well. Cells were stimulated for 48 hours with cross-linked anti-CD3 mAb and the cytokine production in culture supernatants was assessed. The production of IFN-γ, IL-10, and TNF-α by normal mouse cells stimulated with cross-linked anti-CD3 mAb was 966 ± 321 pg/ml, 1,500 ± 102 pg/ml, and 456 ± 58 pg/ml, respectively. The production of IFN-γ, TNF-α, or IL-10 in the absence of stimulation (ie, medium alone) in culture supernatants of cells from treated or untreated mice with or without infection was <200 pg/ml.
Effect of IL-2 Treatment on Nitrite Production
The production of NO by T-cell enriched splenic cell populations after stimulation with cross-linked anti-CD3 mAb was assayed. Nitrite production in TI mice was significantly reduced on day 3 of infection compared to that in TO animals (P < 0.05). The reduced production of nitrite in TI was associated with higher production of IL-10 at this stage. When the IL-2 treatment continued and the infection progressed to the late stage (day 6 of infection), the level of NO production augmented in TI mice and became significantly higher than in TO mice or in normal mice (P < 0.005). These results are presented in Figure 7 ▶ .
Figure 7.

Effect of IL-2 treatment on nitrite production in DBA/2 mice after infection with blood stages of P. yoelii (17XL strain). Mice (n = 3) either received IL-2 treatment only or were treated with IL-2 and given infection with P. yoelii. Spleen cells were harvested at days 3 and 6 after infection. They were then enriched for T cells by nylon-wool purification and were stimulated with cross-linked anti-CD3 mAb. Nitrite levels were measured in 48-hour culture supernatants. Cells were unstimulated (solid bars) or stimulated with anti-CD3 mAb (hatched bars).
Histological Changes in the Brain of IL-2-Treated Mice
Histological analysis revealed the changes in the brain in IL-2-treated DBA/2 mice at day 6 after infection that closely resembled those described in susceptible BALB/c mice. These include the accumulation of PRBCs adjacent to endothelial wall, signs of activation of endothelial cells by enlargement of their nuclei, weakness in the integrity of blood-brain barrier, and signs of hemorrhage. In addition, we observed infiltration of lymphocytes within cerebral vessels of IL-2-treated and infected mice (Figure 8D) ▶ . In contrast, no such histological alterations were seen in the brains of normal mice (Figure 8A) ▶ , in mice that were given IL-2 only (Figure 8B) ▶ , or in mice that received infection but no IL-2 treatment (Figure 8C) ▶ .
Figure 8.
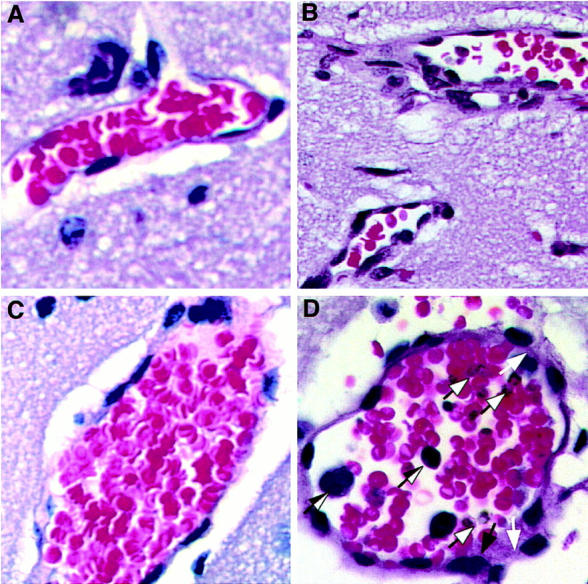
Histology of the brain from DBA/2 mice treated with IL-2 and given infection with P. yoelii (strain 17XL). Sections of brain vessel (original magnification, 40 × 1.6) from normal mouse (A); from mouse that received no treatment but was infected for 6 days (B); from mouse receiving treatment only (C); from treated and infected mouse at day 6 after infection (D) showing presence of several parasitized erythrocytes in the vessel (dark-bordered arrow), infiltration of lymphocytes (one-side dark arrow), endothelial cell activation with enlarged nuclei (dark arrow) and areas of vessel disruption (clear arrow).
IL-2 Treatment Evokes Accumulation of γδ T Cells within Cerebral Vessels of DBA/2 Mice Infected with P. yoelli 17XL
Confocal microscopy was performed with brain specimens obtained from TI mice to characterize the nature of the cellular infiltrates. Infiltration of lymphocytes was not observed in the brain of TI mice at day 3 after infection in histological analysis (data not shown). For this reason, brain samples were obtained only from the moribund TI mice at day 6 after infection. Thin sections from brains were prepared and these sections were incubated with fluorescent conjugated antibodies to αβ or γδ TCRs. When analyzed by confocal microscopy, we could observe recruitment of a few T cells belonging to αβ lineage in the brains of TI mice (Figure 9D) ▶ . Interestingly, the T cells that were predominantly accumulated in the brains of mice with CM were seen to be γδ-bearing T cells (Figure 9E) ▶ . Further, we were able to detect the expression of CD25 (IL-2R)-positive cells in the brains of TI but not in TO mice (data not shown). This finding suggested that T cells recruited in the brains were in a state of activation. Another important activation marker, CD54 (ICAM-1) molecules were detected on the surface of recruited cells (Figure 9F) ▶ .
Figure 9.

Confocal microscopy performed on vibratome sections of the brain from mice that received IL-2 treatment and were infected with P. yoelii (strain 17XL) for 6 days. Sections of brain from a mouse given IL-2 treatment but no infection, and stained with anti-αβ TCR antibody (A), with anti-γδ TCR antibody (B), or with anti-CD54 (ICAM-1) antibody (C); no marked staining was seen in any of the cases. Sections of brain from a mouse given IL-2 treatment and infection, and incubated with anti-αβ TCR antibody (D), with anti-γδ TCR antibody (E), or with anti-CD54 antibody (F); a number of cells with positive staining was seen in each cases, notably with anti-γδ TCR antibody. Laser power, PMT gains, and confocal thresholds were set using FITC-IgG isotype controls and kept constant throughout the experiment.
Effects of in Vivo Administration of Anti-γδ mAb on CM Development in DBA/2 Mice
Having observed the preferential recruitment of γδ T cells within cerebral vessels of TI mice, we wanted to investigate the in vivo role of γδ T cells in CM pathology and subsequent mortality. Because of the nonavailability of γδ T-cell-deficient mice with DBA/2 background, we studied the effect of depletion of γδ T cells by antibody treatment on the outcome of infection in IL-2-treated mice. None of the anti-γδ mAb-treated TI mice developed CM symptoms at day 6 after infection whereas all of the control mice demonstrated signs of CM on day 6 of infection. Five of the seven anti-γδ mAb-treated TI mice ultimately developed CM symptoms and died by days 18 to 21 after infection. The remaining two mice stayed healthy until the last day of observation. In a separate experiment, TI mice that received anti-γδ T cell antibody on days −1 and +2 after infection developed CM symptoms and died by days 6 to 8 after infection. Only one out of seven mice survived the infection in this group. All of the control TI mice included in both of these experiments became moribund with signs of CM by day 6 after infection and succumbed to infection by day 8 after infection. The results of these experiments are presented in Table 1 ▶ .
Table 1.
Effects of Anti-γδ mAb Administration on Development of Cerebral Malaria in IL-2-Treated and Infected DBA/2 Mice (n = 7/Group)
| Experiment | Treatment | Cerebral malaria* |
|---|---|---|
| Experiment 1† | TcR γδ mAb (on days+3 and+6 after infection) | 0 /7 |
| Isotype match control | 7 /7 | |
| Experiment 2‡ | TcR γδ mAb (on days−1 and+2 after infection) | 6 /7 |
| Isotype match control | 7 /7 |
*Cerebral malaria symptoms were examined at day 6 after infection.
†Mice were injected with anti-γδ on days +3 and +6 after infection.
‡Mice were given injections with anti-γδ mAb on days −1 and +2 after infection.
Discussion
BALB/c mice, when infected with P. yoelii 17XL (a lethal strain), readily develop CM symptoms by days 6 to 8 after infection and succumb to infection. 18 In this study, we show that DBA/2 mice inoculated with the same strain of parasite survived infection and never developed signs of CM. CM syndrome in DBA/2 mice could not be induced even by increasing the inoculum by 500 times. The level of parasitemia in this mouse strain was never as high as in BALB/c mice even with a large inoculum. Further, histological examination of brain revealed a lack of parasitized erythrocytes within vessels and no observed infiltration of lymphocytes within cerebral vessels in DBA/2 mice, in comparison to susceptible BALB/c mice that showed parasitized erythrocytes and perivascular lymphocytic infiltrates. These findings clearly illustrate that DBA/2 strain of mice is resistant to the pathological hallmarks of CM.
It is still unclear whether human CM presents an immune-mediated disease. 9,10 In the current study, we have considered the possibility that large doses of IL-2 administration may enhance cell-mediated immune responses in resistant DBA/2 mice, and this in turn may stimulate pathways for the development of CM pathology from infection with P. yoelii 17XL. Indeed, treatment with IL-2 led to the development of CM symptoms by day 6 after infection in DBA/2 mice (TI), which subsequently died on days 6 to 8 of infection. The regimen of IL-2 treatment was not toxic for DBA/2 mice because mice treated in the same way and not given infection (TO) showed no signs of disease until the end of observation period. These findings clearly demonstrated that treatment with IL-2, a potent activator of T cells, induced the CM-resistant mice to demonstrate a susceptible phenotype and compromised their ability to survive a P. yoelii 17XL infection. Our results suggest that development of CM, at least in IL-2-treated DBA/2 mice, is independent of level of parasitemia because parasitemia was increased only slightly in treated, moribund mice compared to untreated, infected mice (IO). It seems that overall parasitemia remains unpredictable as a marker of fatal infection in malaria. In an earlier study we showed that after IL-2 treatment ∼70% of BALB/c mice infected with a nonlethal strain of P. yoelii (17XNL strain) died, although parasitemia was markedly decreased in those mice. 15 Recently Landau and her colleagues, 19 by controlling parasitemia through subcurative drug treatment, have concluded that pathological sequelae are related not only to acute infections with high parasite levels, but also are consequent to chronic infection with low-grade parasitemias.
T-cell enriched populations from the spleen, which is an active lymphoid organ in mice and an important site for PRBC clearance, 20 were stimulated with cross-linked anti-CD3 mAb. Measurement of cytokine production by this TCR-mediated activation has allowed us to evaluate the extent of T cell activation after administration of rIL-2. Our results demonstrated that the production of IFN-γ was increased in moribund TI mice with CM symptoms during the late stage of infection compared to that in the early stage of infection. Further, early IFN-γ response in TI mice was markedly diminished compared to that in IO mice that had survived the infection. These results correlate with previous reports by others and by us that show a weak IFN-γ response in the initial stage followed by an increased expression of IFN-γ in the late stage of infection that led to fatal infections. 2,18 It has been shown that stimulation of strong IFN-γ response in the initial stage of infection with P. yoelii seems to defend the host against the development of CM in susceptible mice. 2,18,21 These findings emphasize that the timing and level of IFN-γ production is critically important in determining whether the effects would be beneficial or harmful. It has not yet been clearly established whether NO plays a role in the development of CM. Some authors have suggested that NO might be implicated in CM pathogenesis whereas others have failed to show a relationship between them. 22-24 Data presented in this study clearly show heightened NO production was associated with CM development in IL-2-treated mice. Patients undergoing IL-2 therapy have been shown to express increased NO synthesis from l-arginine. 25 IL-2 treatment of DBA/2 mice may influence a similar NO synthesis pathway, which may affect neurotransmission in the brain. 26 The effects of IL-2 treatment on production of IL-10 by primed T cells are not clear. However, it seems from our results that the well-recognized inverse relationship between IL-10 and IFN-γ, 27,28 or between IL-10 and NO 29 is maintained when the animals received IL-2 treatment for a short duration (3 days of infection and 2 days before infection).
The balance of these factors became unstable when the immune cells were exposed to IL-2 stimulation for a longer time (6 days of infection and 2 days before infection). It is of note that simultaneous elevated serum levels of IL-10 and IFN-γ have previously been reported in patients with acute P. falciparum malaria. 30 It is possible that the inhibitory effect induced by IL-10 on IFN-γ or on NO may be dependent on their respective levels and/or other regulatory mechanisms that are involved after extended IL-2-mediated stimulation. 31
We have demonstrated in this study that there was an augmentation in TNF-α production in TI mice in the late stage compared to that in early stage of infection, which indicated that TNF-α response remained stimulated in moribund TI mice that developed CM. The differences in TNF-α production are significant but less marked than to changes in IFN-γ levels observed among different groups. It may be noted that in the current study we measured cytokine production by T-cell enriched populations after TCR-mediated stimulation and thus primarily excluded production of TNF-α by cells from the monocyte/macrophage lineage. The moderately elevated TNF-α response that we observed in TI mice may, along with other factors, participate in the development of CM. TNF-α may not be the sole factor responsible for CM. Recently Shear et al 32 described that TNF-α levels were not significantly different between infections with lethal and nonlethal strains of P. yoelii. Treatment of patients with a mAb against TNF-α did not diminish the incidence of CM. 33 Also, therapy with pentoxyfylline, an inhibitor of TNF-α, did not improve the condition of patients in a controlled study of P. falciparum infection. 34 However, another study reported reduction of both TNF-α and duration of coma in CM in children after treatment. 35 Production of cytokine in TO mice is likely to involve a cytokine cascade initiated by the administration of IL-2 and is not dependent on antigen stimulation because TO mice were not exposed to infection and thus not primed with P. yoelii blood-stage antigens.
In this study, histological analysis of brains from TI mice demonstrated several anomalies, which include activation of endothelial cells by enlargement of their nuclei, infiltration of erythrocytes through the blood-brain barrier, and subsequent edema and hemorrhage. This condition was associated with accumulation of PRBCs and infiltration of lymphocytes in the perivascular space of cerebral venules. These changes clearly correlated with the development of clinical CM in TI mice. Some of the infiltrating lymphocytes were seen to advance toward the endothelial wall. Examination of multiple brain sections from TI mice did not show the presence of lymphocytes in the parenchyma. Sequestration of recruited T lymphocytes to activated microvascular endothelial cells may well occur via receptor-ligand interaction. Weakness in the blood-brain barrier has been reported in P. falciparum CM patients but transmigration of leukocytes in brain parenchyma was not clearly demonstrated. 36 No infiltration of lymphocytes and parasitized erythrocytes or endothelial damage was seen in the brains of mice that were infected but received no IL-2 treatment, or in mice that were given IL-2 only. The histological changes seen in the brain of IL-2-treated and infected DBA/2 mice closely resemble those described in susceptible mice (see Figure 3 ▶ in this study and Yoelii and Hargreaves 7 and Kaul et al 8 ).
A critical issue concerning pathology might be the preferential recruitment of some activated T cells in certain microvascular beds, particularly in the brain. The results of our confocal microscopical studies showed infiltration of only a few αβ T cells in cerebral vessels. The T cells accumulated in the brain vessels of mice with CM were predominantly γδ-bearing T cells. Recruitment of γδ T cells was not observed in the brain of mice given IL-2 only. As expected, IL-2 treatment evoked a cascade of cytokine response that was not sufficient to induce recruitment of γδ T cells in cerebral vessels of TO mice in the absence of infection. It has been postulated that only memory T cells can enter and be retained in the central nervous system, and only if the relevant antigens are present. 37 These results clearly demonstrated that recruitment of γδ T cells in the brain of TI mice was infection-induced, and retention of γδ T cells may be dependent on the local presence of antigens from accumulated PRBCs. Some of the activation markers such as CD25 (IL-2R) and CD54 (ICAM-1) molecules were detected on the surface of recruited cells. 38,39 This indicated that T cells recruited in cerebral vessels of TI mice were in a state of activation. ICAM-1 can be expressed on endothelial cells, but the cells that showed positive staining with anti-ICAM-1 antibody looked morphologically more like T cells than endothelial cells. The ICAM-1 molecules expressed on activated T cells can induce aggregation, adherence, and sequestration of T lymphocytes in the cerebral venule. 40 Moreover, accumulated γδ T cells that are in a state of activation can induce local production of IFN-γ and TNF-α, 41,42 and also perhaps generate NO. 43 These mediators will induce/up-regulate the expression of receptors at the brain endothelium where accumulated PRBCs and T lymphocytes could sequester, and this will lead to endothelial degeneration.
The evidence of an important role of γδ T cells in the induction of CM pathology in TI mice was bolstered by reversal of CM in these mice by treatment with anti-γδ antibody. Approximately 70% of TI mice treated with anti-γδ mAb succumbed to infection by days 18 to 20 after infection. This may be explained either by the pathological activities of other cell types in the absence of γδ T cells or by the restoration of γδ T cell expansion later in the infection, because anti-γδ treatment terminated at day 6 after infection in our experiments. Of note, treatment was inefficient when first given 1 day before infection and stopped at day 2 after infection. This observation suggests a biphasic mechanism of action of γδ T cells (early protective/late pathological) in this murine malaria model. It has recently been shown that mice depleted of γδ T cells by antibody failed to develop CM after infection with P. berghei ANKA, however, mice deficient in γδ TCR genes developed CM pathology. 6 The biphasic activity of γδ T cells may explain the failure to inhibit CM pathology in γδ-deficient KO mice. It may be noted that several authors have reported a protective role of γδ T cells in malaria infections. 44-46 An early infection-induced response of natural killer cells and γδ T cells, as a part of innate immunity, may develop followed by, eventually, an adaptive immune response. But abundant response of γδ T cells late in malarial infection could be detrimental for hosts, as has recently been described in influenza. 47
The results of the present study suggest a potential mechanism of CM pathogenesis and support the hypothesis that CM is an immune-mediated disease. Our findings show that the genetic resistance in mice to CM syndrome may be altered by provoking enhanced immunoreactivity in the hosts. Our results provide strong evidence that preferential accumulation of γδ T cells in the brain is crucial for induction of the pathogenesis of CM.
Acknowledgments
We thank Drs. Hanna Shear (Albert Einstein College of Medicine, New York) and Pablo Pereira (Institut Pasteur, Paris, France) for providing P. yoelii parasite stock and hybridoma secreting anti-γδ TCR antibody, respectively; Dr. Tom Ciardelli (Dartmouth Medical School, Hanover) for generously providing rIL-2; Drs. Jane Collins and Grant Yeaman (Dartmouth Medical School, Lebanon) for help and technical advice; Mr. Kenneth Orndorff (Dartmouth Medical School, Lebanon) and Mr. Claude Alesia (Faculté de Médecine, Université de la Mediterranee, Marseille) for their expert help with confocal microscopy and photoshop; and Dr. Bill Hickey (Dartmouth Medical School, Lebanon) for helpful discussion and his encouragement.
Footnotes
Address reprint requests to A. Haque, Ph.D., Chercheur au CNRS, Immunologie et Génétique des Maladies Parasitaires, INSERM U399, Faculté de Médecine, Université de la Mediterranee, 27 Bvd. Jean Moulin, 13385 Marseille, France. E-mail: h.azizul@medecine.univ-mrs.fr.
Supported by institutional funding to Unité de Recherche 399, INSERM.
References
- 1.Oaks SC, Mitchell VS, Pearson GW, Carpenter CJ (Eds): Malaria: Obstacles and Opportunities. Washington, DC, National Academy Press, 1991, pp 56–65 [PubMed]
- 2.Taylor-Robinson AW, Smith EC: A role for cytokines in potentiation of malaria vaccines through immunological modulation of blood stage infection. Immunol Rev 1999, 171:105-124 [DOI] [PubMed] [Google Scholar]
- 3.Grau GE, Piguet PF, Engers HD, Louis JA, Vassalli P, Lambert PH: L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J Immunol 1986, 137:2348-2354 [PubMed] [Google Scholar]
- 4.Yanez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC: Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol 1996, 157:1620-1624 [PubMed] [Google Scholar]
- 5.Hermsen C, van de Wiel T, Mommers E, Sauerwein R, Eling W: Depletion of CD4+ or CD8+ T-cells prevents Plasmodium berghei induced cerebral malaria in end-stage disease. Parasitology 1997, 114:7-12 [DOI] [PubMed] [Google Scholar]
- 6.Yanez DM, Batchelder J, van der Heyde HC, Manning DD, Weidanz WP: Gamma delta T-cell function in pathogenesis of cerebral malaria in mice infected with Plasmodium berghei ANKA. Infect Immun 1999, 67:446-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoelii M, Hargreaves BJ: Brain capillary blockage produced by a virulent strain of rodent malaria. Science 1974, 184:572-574 [DOI] [PubMed] [Google Scholar]
- 8.Kaul DK, Nagel RL, Llena JF, Shear HL: Cerebral malaria in mice: demonstration of cytoadherence of infected red blood cells and microrheologic correlates. Am J Trop Med Hyg 1994, 50:512-521 [DOI] [PubMed] [Google Scholar]
- 9.Miller LH, Good MF, Milon G: Malaria pathogenesis. Science 1994, 264:1878-1883 [DOI] [PubMed] [Google Scholar]
- 10.Turner GD, Morrison H, Jones M, Davis TM, Looareesuwan S, Buley ID, Gatter KC, Newbold CI, Pukritayakamee S, Nagachinta B, White NJ, Berendt AR: An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol 1994, 145:1057-1069 [PMC free article] [PubMed] [Google Scholar]
- 11.Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert PH: Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med 1989, 320:1586-1591 [DOI] [PubMed] [Google Scholar]
- 12.McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D: Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 1994, 371:508-510 [DOI] [PubMed] [Google Scholar]
- 13.Minami Y, Kono T, Miyazaki T, Taniguchi T: The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol 1993, 11:245-268 [DOI] [PubMed] [Google Scholar]
- 14.Engert A, Sausville EA, Vitteta ES: Clinical Applications of Immunotoxins, Frankel AE eds. 1997, vol 2.:pp 13-33 Springer, Berlin [Google Scholar]
- 15.Lucas B, Kasper LH, Smith K, Haque A: In vivo treatment with interleukin 2 reduces parasitemia and restores IFN-γ gene expression and T cell proliferation during acute murine malaria. C R Acad Sci Paris Sciences de la Vie/Life Sciences 1996, 319:705-710 [PubMed] [Google Scholar]
- 16.Green LC, Wagner DA, Glogowski J, Skepper PL, Sishnok JS, Tannenbaum SR: Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem 1982, 126:131-138 [DOI] [PubMed] [Google Scholar]
- 17.Yeaman GR, Collins JE, Currie JK, Guyre PM, Wira CR, Fanger MW: IFN-γ is produced by polymorphonuclear neutrophils in human uterine endometrium and by cultured peripheral blood polymorphonuclear neutrophils. J Immunol 1998, 160:5145-5153 [PubMed] [Google Scholar]
- 18.Haque A, Graille M, Kasper LH, Haque S: Immunization with heat-killed Toxoplasma gondii stimulates an early IFN-gamma response and induces protection against virulent murine malaria. Vaccine 1999, 17:2604-2611 [DOI] [PubMed] [Google Scholar]
- 19.Vuong PN, Richard F, Snounou G, Coquelin F, Renia L, Gonnet F, Chabaud AG, Landau I: Development of reversible lesions in the brain, heart and kidney following acute and chronic murine malaria infection. Parasitology 1999, 119:543-553 [DOI] [PubMed] [Google Scholar]
- 20.Weiss L: The spleen in malaria: the role of barrier cells. Immunol Lett 1990, 25:165-172 [DOI] [PubMed] [Google Scholar]
- 21.Shear HL, Srinivasan R, Nolan T, Ng C: Role of IFN-gamma in lethal and nonlethal malaria in susceptible and resistant murine hosts. J Immunol 1989, 143:2038-2044 [PubMed] [Google Scholar]
- 22.Clark IA, Rockett KA, Cowden WB: Possible central role of nitric oxide in conditions clinically similar to cerebral malaria. Lancet 1992, 340:894-896 [DOI] [PubMed] [Google Scholar]
- 23.Al Yaman FM, Mokela D, Genton B, Rockett KA, Alpers MP, Clark IA: Association between serum levels of reactive nitrogen intermediates and coma in children with cerebral malaria in Papua New Guinea. Trans R Soc Trop Med Hyg 1996, 90:270-273 [DOI] [PubMed] [Google Scholar]
- 24.Anstey NM, Weinberg JB, Hassanali MY, Mwaikambo ED, Manyenga D, Misukonis MA, Arnelle DR, Hollis D, McDonald MI, Granger DL: Nitric oxide in Tanzanian children with malaria: inverse relationship between malaria severity and nitric oxide production/nitric oxide synthase type 2 expression. J Exp Med 1996, 184:557-567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hibbs JB, Jr, Westenfelder C, Taintor R, Vavrin Z, Kablitz C, Baranowski RL, Ward JH, Menlove RL, McMurry MP, Kushner JP, Samlowski WE: Evidence for cytokine-inducible nitric oxide synthesis from L-arginine in patients receiving interleukin-2 therapy. J Clin Invest 1992, 89:867-877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bredt DS, Snyder SH: Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem 1994, 63:175-195 [DOI] [PubMed] [Google Scholar]
- 27.Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O’Garra A: IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol 1991, 146:3444-3451 [PubMed] [Google Scholar]
- 28.Moore KW, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann TR: Interleukin-10. Annu Rev Immunol 1993, 11:165-190 [DOI] [PubMed] [Google Scholar]
- 29.Cunha FQ, Moncada S, Liew FY: Interleukin 10 (IL-10) inhibits the induction of nitric oxide synthase by interferon-γ in murine macrophages. Biochem Biophys Res Commun 1992, 186:1155-1159 [DOI] [PubMed] [Google Scholar]
- 30.Wenisch C, Parschalk B, Narzt E, Looareesuwan S, Graninger W: Elevated serum levels of IL-10 and IFN-gamma in patients with acute Plasmodium falciparum malaria. Clin Immunol Immunopathol 1995, 74:115-117 [DOI] [PubMed] [Google Scholar]
- 31.Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, Larner AC, Finbloom DS: Interleukin-10 inhibits expression of both interferon α- and interferon γ-induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 1999, 93:1456-1463 [PubMed] [Google Scholar]
- 32.Shear HL, Marino MW, Wanidworanun C, Berman JW, Nagel RL: Correlation of increased expression of intercellular adhesion molecule-1, but not high levels of tumor necrosis factor-alpha with lethality of Plasmodium yoelii 17XL, a rodent model of cerebral malaria. Am J Trop Med Hyg 1998, 59:852-858 [DOI] [PubMed] [Google Scholar]
- 33.van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, Memming H, Frenkel J, Enwere G, Bennett S, Kwiatkowski D, Greenwood BM: The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis 1996, 174:1091-1097 [DOI] [PubMed] [Google Scholar]
- 34.Moling O, Mian P: Use of pentoxyfelline in the treatment of cerebral malaria. Clin Infect Dis 1994, 19:558-559 [DOI] [PubMed] [Google Scholar]
- 35.Di Perri G, Di Guasparri I, Monteiro GB, Bonora S, Henning C, Cassatella M, Micciolo R, Vento S, Dusi S, Bassetti D, Concia E: Pentoxyfelline as a supportive agent in the treatment of cerebral malaria in children. J Infect Dis 1995, 171:1317-1322 [DOI] [PubMed] [Google Scholar]
- 36.Brown H, Hien TT, Day N, Mai NT, Chuong LV, Chau TT, Loc PP, Phu NH, Bethell D, Farrar J, Gatter K, White N, Turner G: Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol 1999, 25:331-340 [DOI] [PubMed] [Google Scholar]
- 37.Hickey WF, Hsu BL, Kimura H: T lymphocyte entry into the central nervous system. J Neurosci Res 1991, 28:254-260 [DOI] [PubMed] [Google Scholar]
- 38.Rusterholz C, Henrioud PC, Nabholz M: Interleukin-2 (IL-2) regulates the accessibility of the IL-2-responsive enhancer in the IL-2 receptor alpha gene to transcription factors. Mol Cell Biol 1999, 19:2681-2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Springer TA, Dustin ML, Kishimoto TK, Marlin SD: The lymphocyte function associated LFA-1, CD2, and LFA-3 molecules: cell adhesion receptors of the immune system. Annu Rev Immunol 1987, 5:223-252 [DOI] [PubMed] [Google Scholar]
- 40.Chao CC, Sandor M, Dailey OM: Expression and regulation of adhesion molecules by γδ T cells from lymphoid tissues and intestinal epithelium. Eur J Immunol 1994, 24:3180-3187 [DOI] [PubMed] [Google Scholar]
- 41.Christmas SE, Meager A: Production of interferon-gamma and tumour necrosis factor-alpha by human T-cell clones expressing different forms of the gamma delta receptor. Immunology 1990, 1:486-492 [PMC free article] [PubMed] [Google Scholar]
- 42.Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H: Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature 1995, 373:255-257 [DOI] [PubMed] [Google Scholar]
- 43.Jones-Carson J, Vazquez-Torres A, van der Heyde HC, Warner T, Wagner RD, Balish E: Gamma delta T cell-induced nitric oxide production enhances resistance to mucosal candidiasis. Nat Med 1995, 1:552-557 [DOI] [PubMed] [Google Scholar]
- 44.van der Heyde HC, Elloso MM, Chang WL, Kaplan M, Manning DD, Weidanz WP: Gamma delta T cells function in cell-mediated immunity to acute blood-stage Plasmodium chabaudi adami malaria. J Immunol 1995, 154:3985-3990 [PubMed] [Google Scholar]
- 45.Seixas EM, Langhorne J: Gamma delta T cells contribute to control of chronic parasitemia in Plasmodium chabaudi infections in mice. J Immunol 1999, 162:2837-2841 [PubMed] [Google Scholar]
- 46.McKenna KC, Tsuji M, Sarzotti M, Sacci JB, Jr, Witney AA, Azad AF: Gamma delta T cells are a component of early immunity against preerythrocytic malaria parasites. Infect Immun 2000, 68:2224-2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carding SR, Allan W, Kyes S, Hayday A, Bottomly K, Doherty PC: Late dominance of the inflammatory process in murine influenza by γδ T cells in vivo. J Exp Med 1990, 172:1225-1231 [DOI] [PMC free article] [PubMed] [Google Scholar]