

Abstract
Glucocorticoid (GC) overexposure in animals has been implicated in hippocampal dysfunctioning and neuronal loss. In major depression, hypercortisolemia, hypothalamic-pituitary-adrenocortical-axis alterations, and reduced hippocampal volumes are commonly observed; hence, hippocampal neurodegeneration is also expected. To study possible GC-related pathology, we investigated hippocampal tissue of 15 major-depressed patients, 16 matched controls, and 9 steroid-treated patients, using in situ-end-labeling for DNA fragmentation and apoptosis, and heat-shock protein 70 and nuclear transcription factor κB immunocytochemistry for damage-related responses. No obvious massive cell loss was observed in any group. In 11 of 15 depressed patients, rare, but convincing apoptosis was found in entorhinal cortex, subiculum, dentate gyrus, CA1, and CA4. Also in three steroid-treated patients, apoptosis was found. Except for several steroid-treated patients, heat-shock protein 70 staining was generally absent, nor was nuclear transcription factor-κB activation found. The detection in 11 of 15 depressed patients, in three steroid-treated, and in one control patient, demonstrates for the first time that apoptosis is involved in steroid-related changes in the human hippocampus. However, in absence of major pyramidal loss, its rare occurrence, that notably was absent from areas at risk for GC damage such as CA3, indicates that apoptosis probably only contributes to a minor extent to the volume changes in depression.
Glucocorticoid hormones (GCs) are important adrenal steroids that affect numerous physiological processes in the brain and body. Their secretion is mediated via the hypothalamic-pituitary-adrenal (HPA) axis and stimulated, for example, by stress through corticotropin-releasing hormone (CRH) in the parvocellular neurons of the paraventricular nucleus (PVN) in the hypothalamus. 1,2 This induces adrenocorticotroph hormone release from the pituitary into the blood, which triggers cortisol release from the adrenal. GC plasma levels are carefully kept within physiological limits through GC-mediated feedback inhibition at specific steroid receptors in the pituitary and PVN. Also the hippocampus, that at least in rat contains high densities of glucocorticoid (GR) and mineralocorticoid (MR) receptors, is sensitive to GC action. 3,4 The hippocampus, among other areas, is furthermore thought to exert an (indirect) tonic inhibitory control on HPA axis activity. 4-7 Disturbance of this negative feedback through hippocampal damage or cell loss could result in a feedforward cascade of cumulative GC exposure and induce pathological steroid overexposure of the brain and hippocampus. This glucocorticoid cascade concept was based on earlier rat studies and has initially been implicated in the pathogenesis of aging and Alzheimer’s disease, in which the extent of hippocampal atrophy correlates well with GC plasma levels and pathology. 8-12,14
In rat, hypercortisolemia, 14 and stress impair memory and learning, as well as electrophysiological responses of the hippocampus. 13-20 Also, they increase neuronal vulnerability to subsequent insults 21-23 and after prolonged GC overexposure, even massive neuronal loss has been reported, mainly in the hippocampal CA3 pyramidal cell layers of the rat and monkey. 10,24,25
In depression, increased HPA axis activity and GC resistance are commonly observed as well, as reflected by the high percentage of dexamethasone nonsuppressors in this population, hypertrophy of the adrenals and pituitary, increased plasma levels of cortisol, and increases in CRH and vasopressin expression in PVN neurons. 2,26-34 Also, decreased hippocampal volumes have been found in depressive patients. 35-37 , 142 –144 On basis of these data, one would expect hippocampal damage in this condition as well. However, the neuropathological correlates of cortisol exposure for the human hippocampus were so far unknown, which is why we set out to study cell death or cell death-related responses in the hippocampus of major depressed patients. Because the adrenal status of the patients in the present study was not known, we further included tissue from nondepressed individuals that had been treated with synthetic steroids.
As GCs may increase susceptibility to apoptosis through calcium- and reactive oxygen species pathways, 38-43 in situ end labeling (ISEL) was applied that identifies fragmented DNA associated with both apoptotic and necrotic cell death, which can be discriminated using morphological criteria. 60,61 Complementary indices of responses to (oxidative) damage and cellular stress were: 1) inducible heat-shock protein-70 (HSP70), that is undetectable in normal brain, but strongly up-regulated in response to insults and cell death 46,47 ; and 2) nuclear transcription factor kappa B (NF-κB), a GC-regulated transcription factor that has been implicated in protection against apoptosis or oxidative stress. 48-51
Materials and Methods
Tissue Selection and Processing
Brain tissue was obtained from the Netherlands Brain Bank (coordinator, Dr. R. Ravid). Fifteen clinically well-defined patients were selected that had suffered from recurrent episodes of major depression or bipolar affective disorder (see Table 1 ▶ for details on the patient data). The absence of neuropathological changes in depressed, steroid-treated patients, and controls was established by neuropathologist Dr. R. A. I. De Vos (Laboratory for Pathology and Microbiology, Enschede, The Netherlands) Prof. Dr. F. C. Stam (Netherlands Brain Bank, Amsterdam), Dr. W. Kamphorst (Free University Amsterdam), or Dr. D. Troost (Academic Medical Center, Amsterdam). The clinical diagnosis of the depressed patients had been established by psychiatrist Dr. W. J. G. Hoogendijk, Amsterdam, on the basis of DSM-IV criteria (see Table 1 ▶ for clinicopathological data and psychiatric history). Six of these 15 depressed patients had been included in previous investigations on the expression of CRH and vasopressin in the hypothalamic PVN (Table 1) ▶ . 2,26,27 A second group of nine patients without any primary neurological or psychiatric disorder but who had been treated for different durations and until death with high doses of synthetic glucocorticoids were included in this study (Table 1) ▶ . The control group consisted of 15 sex- and age-matched controls without any primary neurological or psychiatric disease and without a known history of GC treatment (Table 1) ▶ .
Table 1.
Clinical Data of the Controls, Depressed Patients, and Steroid-Treated Group
| No. | NBB no. | Sex | Age | PMD | Fix | BW | Cause of death | Diseases/medication |
|---|---|---|---|---|---|---|---|---|
| Controls | ||||||||
| 1 | 92-011 | M | 47 | <89:00 | 80 | 1520 | Sepsis, meningitis | Hypertension, antihypertensive medication |
| 2 | 94-035 | M | 49 | 07:40 | 40 | 1404 | CF | Hypertension, antihypertensive medication |
| 3 | 94-118 | M | 49 | 22:20 | 40 | 1254 | Sepsis | None |
| 4 | 96-114 | F | 54 | 08:00 | 35 | 1257 | FR, metastatic breast-CA | Aminogluthetidmide, morphine |
| 5 | 96-010 | M | 63 | 10:20 | 46 | 1250 | Pneumonia | Antibiotics |
| 6 | 96-013 | F | 68 | 10:30 | 39 | 1122 | Hematemesis, metastatic breast CA | Paclitaxel, cisapride |
| 7 | 93-061 | M | 70 | 09:00 | 28 | 1454 | RF | Pneumonia, lobectomy for squamous lung CA |
| 8 | 95-106 | M | 74 | 08:00 | 35 | 1317 | CF | Catecholamines |
| 9 | 94-123 | F | 76 | 09:20 | 42 | 1095 | Respiratory insufficiency | Bronchopneumonia, antibiotics, haloperidole |
| 10 | 90-086 | F | 77 | 44:30 | 343 | 1198 | Postoperative sepsis | Aortic valve surgery, epileptic seizures |
| 11 | 95-093 | M | 78 | 07:00 | 43 | 1440 | Decompensatio cordis, pulmonary embolism | Prostate carcinoma; Renitec, Floxapen, Augmentin, and Capoten |
| 12 | 94-076 | M | 78 | 08:25 | 94 | 1442 | Cardiac arrhythmia, RF | Digoxin, paracetamol |
| 13 | 92-016 | M | 81 | 21:15 | 64 | 1478 | Cardiac/respiratory insufficiency | Pneumonia, antibiotics |
| 14 | 89-045 | M | 82 | <41:00 | 849 | 1284 | Cachexia | Antibiotics, acetylcysteine |
| 15 | 82-018 | M | 85 | 16:15 | 44 | 1400 | Pneumonia | Hydroxurea, allopurinol |
| Mean | 68.7 | 20.8 | 121.5 | 1327 | ||||
| SD | 13.1 | 22.6 | 215.8 | 132 |
| No. | NBB no. | Sex | Age | PMD | Fix | BW | Diagnosis/cause of death |
|---|---|---|---|---|---|---|---|
| Clinical data of the depressed patients | |||||||
| 1 | 93-076 | M | 21 | 33:00 | 75 | 1658 | Depression (overdose of opiates) |
| 2 | 93-090 | M | 39 | 48:00 | 42 | 1220 | Manic depression: RI |
| 3 | 93-146 | M | 51 | 72:00 | 42 | 1390 | Manic depression: RI |
| 4 | 92-003 | F | 55 | 06:45 | 87 | 1320 | Depression, CF |
| 5 | 94-112 | M | 61 | 65:00 | 42 | 1424 | Major Depr., pneumonia |
| 6 | 93-054 | M | 63 | 19:45 | 42 | 1210 | Manic depression, CF |
| 7 | 90-001 | M | 70 | 44:00 | 122 | 1300 | Depression, CF |
| 8 | 94-032 | M | 71 | 41:00 | 42 | 975 | Major Depr., ischemia |
| 9 | 94-094 | M | 71 | 14:00 | 42 | 109 | Major Depr., RI |
| 10 | 94-017 | F | 72 | 41:00 | 42 | 1287 | Major Depr., bronchopneumonia |
| 11 | 94-055 | F | 72 | 29:00 | 42 | 1116 | Major Depr., cardiac arhythmia |
| 12 | 93-021 | F | 73 | 96:00 | 42 | 1032 | Depression, CF |
| 13 | 95-036 | M | 74 | 57:00 | 42 | 1444 | Major Depr., suicide by hanging |
| 14 | 93-115 | M | 79 | 22:00 | 28 | 1530 | Depression, suicide |
| 15 | 91-059 | M | 82 | 48:00 | 406 | 1352 | Depression, RI |
| Mean | 63.6 | 42.4 | 75.8 | 1291 | |||
| SD | 16.3 | 23.6 | 94.5 | 187 |
| No. | NBB no. | Age of onset | No. of psychiatric hospital admissions | Earlier indications for HPA hyperactivity | Previous medication | Diagnosis (DSM-IV) | Last MDE (months before death) |
|---|---|---|---|---|---|---|---|
| Psychiatric history of the depressed patients | |||||||
| 1 | 93-076 | 19 | 0 | Yes 2,26,27 | TCA | MDE (s) | Until death |
| 2 | 93-090 | 28 | 5 | Yes2,26,27 | MAO-I, Li, BZD | BAD (r) | Until death |
| 3 | 93-146 | 41 | 8 | nd | Li, BUT | MDE (R) | 7 |
| 4 | 92-003 | 40 | 2 | Yes2 | SSRI, BUT | MDE (r) | Until death |
| 5 | 94-112 | 50 | 1 | nd | PHT | MDE (r) | 144 |
| 6 | 93-054 | 14 | 6 | Yes2,26,27 | BUT, PHT, BZD | BAD (R) | Until death |
| 7 | 90-001 | 40 | 3 | Yes2 | SSRI, NL, BZD | MDE (r) | Until death |
| 8 | 94-032 | 53 | 3 | nd | TCA, MAO-I, BZD | MDE (r) | Until death |
| 9 | 94-094 | 65 | 2 | nd | Li, BZD | MDE (R) | 21 |
| 10 | 94-017 | 54 | 1 | nd | AD, BZD | MDE (s) | 36 |
| 11 | 94-055 | 53 | 4 | nd | AD, BZD | MDE (s) | 108 |
| 12 | 93-021 | 71 | 1 | Yes2 | MAO-I, BZD | MDE (s) | Until death |
| 13 | 95-036 | 74 | 1 | nd | SSRI, BUT, BZD | MDE (s) | Until death |
| 14 | 93-115 | nd | 1 | nd | SSRI, BZD | MDE (s) | Until death |
| 15 | 91-059 | nd | 1 | nd | none | MDE (s) | 8 |
| No. | NBB no. | Sex | Age (years) | PMD (hrs) | Fix (days) | BW (g) | Cause of death, summary of the steroid treatment |
|---|---|---|---|---|---|---|---|
| Clinicopathological data of the steroid-treated group | |||||||
| 1 | 86-032 | F | 33 | 41 | 3867 | 1035 | Metastatic lung CA, DEX up to 9 mg/day for >1 month |
| 2 | 83-004 | F | 46 | 5 | 33 | 1360 | Metastatic adrenal carcinoma causing high plasma levels of adrenal steroids; (urine 17-ketosteroid; 4164 μmol/24 hours (normal range: 21-52); 17-OH-corticosteroid; 381 μmol/24 hours (normal range; 10-52) |
| 3 | 95-011 | M | 63 | 6 | 35 | 1352 | Metastatic adeno-CA, PRED 30 mg/day (last 18 days) |
| 4 | 95-051 | M | 65 | 4 | 28 | 1500 | Asthma bronchiale, Beclomethasone inhalation (200 μg last 7 days) |
| 5 | 93-094 | F | 67 | 17 | 79 | 1340 | Metastatic lung-CA, thrombocytopenia, PRED 60 mg/day (last 7 days) |
| 6 | 95-054 | F | 72 | 9 | 47 | 1075 | CF, chronically PRED 5 mg/day up to 30 mg/day last 4 months |
| 7 | 91-120 | F | 73 | 4 | 67 | 975 | Myocardial infarction, SLE with chronic steroid treatment; steroid induced diabetes mellitus |
| 8 | 93-016 | F | 76 | 8 | 284 | 1225 | Metastatic adeno-CA; PRED minimal 60 mg/day (last 8 days) |
| 9 | 95-098 | M | 78 | 4 | 30 | 1259 | Lung emphysema, PRED 40 mg/day >1 month, gradual dose reduction |
| Mean | 63.6 | 10.8 | 496 | 1235 | |||
| SD | 14.9 | 12 | 1266 | 174 |
Abbreviations used; AD, other antidepressant drugs (eg, maprotiline, mianserine); BAD, bipolar affective disorder; BUT, butyrophenone; BW, brain weight (in grams 9 g)); CA, carcinoma; CF, cardiac failure; CX, cortex; SD, standard deviation; Depr., depression; DEX, dexamethasone; BZD, benzodiazepine; F, female; Fix, fixation time (in days in formalin); HPA, hypothalamo-pituitary-adrenocortical; Li, lithium; M, male; MAO-I, monoamineoxidase inhibitor; MDE, major depressive episode (monopolar affective disorder); NBB No., Netherlands Brain Bank number; ND, not determined/not known; PMD: postmorten delay (in hours (hrs)); PRED, prednisone; r, recurrent episodes; RF, renal failure; RI, respiratory insufficiency; s, single episode; SLE, systemic lupus erythematosus; SSRI, selective serotonine reuptake inhibitor; TCA, tricyclic antidepressant.
At autopsy, the hippocampus proper was dissected at a mid-anteroposterior level and fixed in 0.1 mol/L phosphate-buffered 4% formaldehyde (Sigma, St. Louis, MO) solution (pH 7.2) for different periods of time, ranging from 24 to 3,867 days. Tissue was then dehydrated in graded ethanol and embedded in paraffin after which serial sections were cut on a microtome at 10 μm and mounted on Superfrost Plus slides for ISEL and on gelatin-coated slides for immunocytochemistry.
In our parallel study, 71 the same patients were studied for structural and gross morphological changes, using the following markers: 1) conventional Nissl staining for anatomical and structural alterations, 2) glial fibrillary acidic protein for activational changes in astroglia cells, 3) the antibody Alz-50 for early Alzheimer pathological changes, 4) Bodian Silver staining for (pathological) changes in the neuronal network, 5) synaptophysin for synaptic changes, and 6) B-50 for growth-related responses.
ISEL
ISEL was performed as described earlier 44,52,53 with minor adaptations. Tissue sections from controls, depressed, and steroid-treated individuals were always included within the same experiment and processed under identical pretreatment and chromogen developmental conditions. Briefly, 2 to 5 tissue sections per patient or subject, taken at a level of the hippocampus where all main subareas were present, were deparaffinized for 2 × 15 minutes in xylene, hydrated to ethanol 50% and distilled water (DW). Subsequently, sections were preincubated with Proteinase K buffer (10 mmol/L Tris/HCl, 2.6 mmol/L CaCl2, pH 7.5) for 10 minutes, then incubated with a Proteinase K concentration (Sigma Chemical Co., St. Louis, MO) of 10 μg/ml for 15 minutes at room temperature, washed 3 × 5 minutes in DW, and incubated with terminal transferase (TdT) buffer [0.2 mol/L sodium cacodylate, 0.025 mol/L Tris/HCl, and 0.25 mg/ml bovine serum albumin (BSA), pH 6.6] for 15 minutes at room temperature and incubated for 60 minutes at 37°C with a reaction mixture that contained: 0.1 μl TdT (Boehringer Mannheim, Mannheim, Germany)/100 μl reaction mixture, 1.0 μl biotin-16-dUTP (Boehringer Mannheim)/100 μl reaction mixture, and cobalt chloride (25 mmol/L; 5% of the final volume). Incorporation of labeled oligonucleotides was ended by briefly rinsing the sections in DW and in phosphate-buffered saline (PBS) (pH 7.4) for 5 minutes at room temperature. Endogenous peroxidase activity was blocked with 0.3% H2O2 in PBS for 20 minutes at room temperature after which sections were washed 2 × 5 minutes in PBS, preincubated with PBS/1% BSA for 15 minutes, and incubated with peroxidase-conjugated avidin (ABC-elite kit; Vector Laboratories, Burlingame, CA) 1:1,000 in PBS/1% BSA O/N at 4°C. After washing in PBS, sections were incubated with 0.5 mg/ml diaminobenzidine (DAB) (Sigma Chemical Co.) in 0.05 mol/L Tris/HCl (pH 7.5) with 0.02% H2O2 for 10 minutes, washed 2 times in DW and lightly counterstained with methyl green before coverslipping. When ISEL background levels were unacceptably high, as occurred in four cases (94-032, 92-003, 93-115, 93-076), adjacent sections were stained at 50% of the concentration of ISEL enzyme and label as performed before, 53,54 which yielded a strongly improved signal-to-noise ratio, allowing morphological scoring and comparison of the different areas with each other. Positive controls for ISEL were included in every experiment and used to monitor and determine DAB incubation time. They consisted of sections from rat prostate 3 days after castration, a treatment known to induce large amounts of apoptotic cells. 52
HSP70 Immunocytochemistry
HSP70 staining was performed essentially as described before. 55 After deparaffinization, sections were pretreated in a microwave oven (850 W) at full power for 3 × 5 minutes in citrate buffer (pH 6.0), left to cool to room temperature, and then the sections were washed in PBS. Endogenous peroxidase was blocked with 0.2% H2O2-methanol followed by incubation in 10% fetal calf serum in PBS for 10 minutes. The primary antibody was diluted 1 to 200 in PBS/10% fetal calf serum for 1 hour at room temperature and then overnight at 4°C. The monoclonal antibody against HSP 70 (IgG1) (StressGen no. SPA-810; C92; clone C92F3A-5) had originally been isolated from HeLa cells and was purified by ion-exchange chromatography. Its specificity has been proven before. 56 After washing in PBS, sections were incubated for 1 hour in biotinylated anti-mouse antibody (1 to 200; Amersham, Arlington Heights, IL) in PBS/10% fetal calf serum with 3% human serum, followed by a mouse alkaline phosphatase-anti-alkaline phosphatase system (DAKO, Glostrup, Denmark), used 1 to 100 in PBS. The incubation with biotinylated anti-mouse antibody and mouse alkaline phosphatase-anti-alkaline phosphatase were repeated twice (triple alkaline phosphatase-anti-alkaline phosphatase). Fast Red TR salt (F 1500, Sigma) was used as a chromogen. Sections were lightly counterstained with hematoxylin after which sections were mounted in Aquamount (BDH). For a series of sections from the three groups, the first antibody was omitted to control for nonspecific binding of the secondary antibody.
NF-κB Immunocytochemistry
Protocols used were essentially the same as previously described. 57 The polyclonal antibody was raised in rabbit against the p65 subunit (no. sc 109-G; Santa Cruz Biotechnology, Santa Cruz, CA), ie, the activated form of the NF-κB p65-p50 complex. Specificity has been proven before. 49,57 After deparaffinization and rehydration of the sections, the first antibody was applied 1 to 100 in Tris-buffered saline (pH 7.6) overnight at 4°C. Secondary antibody incubation was with biotinylated anti-rabbit 1 to 100, after which the sections were washed again, preincubated in PBS/1% BSA, and incubated in ABC Elite (Vector Laboratories, Inc.) in a 1 to 1,000 dilution in PBS/1% BSA. Chromogen development was performed with 0.5 mg/ml DAB in 0.05 mol/L Tris/HCL (pH 7.5) with 0.02% H2O2 for 10 minutes, followed by washing in distilled water, dehydration, and coverslipping in Entallan (Merck).
In every experiment, hippocampal tissue of an Alzheimer patient [91-51, female 78 years of age, post mortem delay (PMD) 3 hours) with established severe pathology, was included as a positive control (see below and Figure 4D ▶ ).
Figure 4.

NF-κB immunocytochemistry. A: CA4 neurons of depressed patient 92-003 showing a cytoplasmic distribution of staining throughout the area. B: DG neurons of the same patient as in A showing a granular, punctate staining pattern for the p65 subunit only in the cytoplasm. C: Higher magnification of CA1 of control subject 94-035 with prominent staining in the cytoplasm (arrows). D: High magnification of the CA area of an Alzheimer patient that was included as a positive control, showing a similar, granular pattern (arrowhead), but also clear nuclear staining (arrow), indicative of NF-κB activation in selected cells. No such pattern was observed in depressed or steroid-treated patients. Scale bars: 90 μm (A), 38 μm (B), 50 μm (C), and 25 μm (D).
Evaluation of ISEL, HSP70, and NF-κB
Occurrence and distribution of ISEL-positive apoptotic cells was assessed by one researcher (PJL) unaware of the clinical data of the patients. According to the atlas of Duvernoy, 58 the following anatomical subfields of the human hippocampus were evaluated in great detail in every section at 400× and at 1,000× magnification: the dentate gyrus (polymorphic and granule cell layer); the areas CA1, CA2, CA3, and CA4; and the subiculum, entorhinal cortex, and associated cortical areas. ISEL-positive profiles were scored semiquantitatively per anatomical subarea and assigned to either of the following categories: −, no labeling to two positive profiles present in the subarea of interest; ±, between three to eight positive profiles visible; +, between eight to 15 positive profiles; ++, >15 positive profiles present.
ISEL-positive cells that contained tangle-like morphology, as reported before, 53,59 or when they resembled glia cells, 53,54,91 were scored by T or G, respectively (Table 2) ▶ . When ISEL-positive profiles displayed convincing characteristics of apoptotic cell death, they were marked by A in Table 2 ▶ . As ISEL also detects DNA fragmentation in necrotic or healthy nuclei, but with a different sensitivity, the criteria used for identification of apoptosis included the presence of a brown DAB precipitate, that must be accompanied by an isolated occurrence, strong chromatin or nuclear reorganization or shrinkage, condensation, or the presence of clearly pycnotic nuclei and/or apoptotic bodies (Figure 1, B ▶ -D). 45,60,61 ISEL-positive cells were considered necrotic when none of these apoptotic hallmarks were present, and when no obvious change in size or shape had occurred as compared to neighboring neurons (Figure 2C ▶ , arrow) or when the nuclear or cellular membrane was clearly disrupted.
Table 2.
In Situ End-Labeling (ISEL) Results of the Control Subjects, Depressed Patients, and Steroid-Treated Groups
| Patient no. | CA 1/2 | CA3 | CA4 | DG | Sub | EC | Cx | Remarks | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Controls | ||||||||||||||||
| 1. 92-011 | − | nd | −(2xn) | −(P: +/−) | − | − | − | |||||||||
| 2. 94-035 | − | nd | − | − | − | − | − | |||||||||
| 3. 94-118 | +/− | − | ++ | +− | +/− | − | − | |||||||||
| 4. 96-014 | − | − | − | − | − | +/− | ||||||||||
| 5. 96-010 | −(A1x) | +/− | ++T | +/−P | +/−G | − | − | |||||||||
| 6. 96-013 | + | +/− | + | − | − | − | − | Infiltrate in CA1-subarea | ||||||||
| 7. 93-061 | Nd | − | + | +/− | − | − | − | |||||||||
| 8. 95-106 | − | −R | −R | +/−P | +/−(AG) | − | − | Vessels in alveus positive, remnants of a possible chromatolytic process in CA3+4, some pycnotic cells in the DG | ||||||||
| 9. 94-123 | − | − | − | − | − | − | − | |||||||||
| 10. 90-086 | −R | − | −R | +/−G | − | − | − | Remnants in CA1 and CA4 | ||||||||
| 11. 95-093 | − | − | − | − | nd | nd | − | |||||||||
| 12. 94-076 | − | − | nd | − | − | − | +/− | |||||||||
| 13. 92-016 | − | − | − | − | − | − | − | |||||||||
| 14. 89-045 | − | − | − | − | − | np | np | |||||||||
| 15. 82-018 | +/− | − | +/− | − | +/− | np | − | |||||||||
| Depressed patients | ||||||||||||||||
| 1. 93-076 | − | − | − | nd | nd | nd | − | |||||||||
| 2. 93-090 | −R | − | −1xN, R | +/−A,N | − | − | − | Remnants of a possible chromatolytic process in CA1 and CA4 | ||||||||
| 3. 93-146 | +/−R | − | +/−T,A | −A | − | − | − | Remnants in CA1 | ||||||||
| 4. 92-003 | − | − | +G,N | −P | − | − | − | |||||||||
| 5. 94-112 f | + | +/−R | +A,G,N | +A,P | − | − | − | Remnants in CA3 | ||||||||
| 6. 93-054 | − | − | −A(2x) | +/−A,P | − | − | − | |||||||||
| 7. 90-001 f | −A(1x) | −R | +/− | +/−A(2x) | +/− | − | +/− | Fimbriae, alveus and vessel wall often positive; remnants in CA3 | ||||||||
| 8. 94-032 f | +/− | +/−R | +/−A(1x) | +/− | +/− | +/−A | +/− | Remnants in CA3 | ||||||||
| 9. 94-094 f | +/−A | +/− | +T | +P,A | +/−G | − | − | Positive glia also in alveus; remnants in DG | ||||||||
| 10. 94-017 | + | nd | +(A) | +/−P,A | +/−T | nd | +/−T | |||||||||
| 11. 94-055 | +/− | +/− | ++ | + | +/− | − | +/− | |||||||||
| 12. 93-021 | +/− | +AR | +/−G | +/−P+A | − | − | − | Occasionally vessel wall positive | ||||||||
| 13. 95-036 f | +/−G | +/−NG | +G | G | +/− | − | ||||||||||
| 14. 93-115 | −R | +/− | +/−R | −A(2x) | − | − | − | Some remnants in CA4/CA1 | ||||||||
| 15. 91-059 f | − | +/− | +/−R | +/−(A) | − | − | − | Remnants in CA4 | ||||||||
| Steroid-treated group | ||||||||||||||||
| 1. 86-032 | − | − | − | − | − | − | − | |||||||||
| 2. 83-004 f | − | −1xN | −2xN,G | +/−A(1x) | − | − | −A(1x) | Apoptotic cell in DG. | ||||||||
| 3. 95-011 f | −A(1x) | − | +/−A(2x) | +/−A(1x) | − | − | − | Obvious size variation in DG as well as some pycnotic cells in DG tip; only one ISEL-positive | ||||||||
| 4. 95-051 | − | +/− | +/−G | − | − | +/−A | +/− | Some ISEL positive cells in alveus; also positive vessels in cortex | ||||||||
| 5. 93-094 | − | − | − | − | − | − | − | |||||||||
| 6. 95-054 f | − | − | +/−N,A | +/−N,G | − | − | +/−T | Many pycnotic cells in DG; infiltrate/bleeding in sulcus/stratum moleculare | ||||||||
| 7. 91-120 | +/− | +/− | +G | +/− | − | − | − | |||||||||
| 8. 93-016 f | −R | +/−P,GR | +/−R | +/− | − | − | − | Remnants in CA4, CA3 and CA1 | ||||||||
| 9. 95-098 | − | − | − | − | − | − | − | Occasionally vessel wall positive in CA4 and Sub | ||||||||
Abbreviations used for ISEL scoring: P, small, seemingly pycnotic cells; ISEL-negative; N, ISEL-positive cell with necrotic phenotype; A, ISEL-positive cell with a convincing, clearly apoptotic phenotype (see text for details). G; ISEL-positive cell displaying glia-like morphology. T; ISEL-positive cell displaying a tangle-like appearance. R or remnants: granular staining pattern, suggestive of remnants of chromatolysis; nd, not determined. ISEL positive profiles were scored semiquantitatively per anatomical subarea and assigned to either of the following categories; −: no labeling to 2 positive profiles present in the subarea of interest, +/−: between 3–8 positive profiles visible, +: between 8–15 positive profiles, ++: more than 15 positive profiles present. f, examples shown in the figures.
Figure 1.
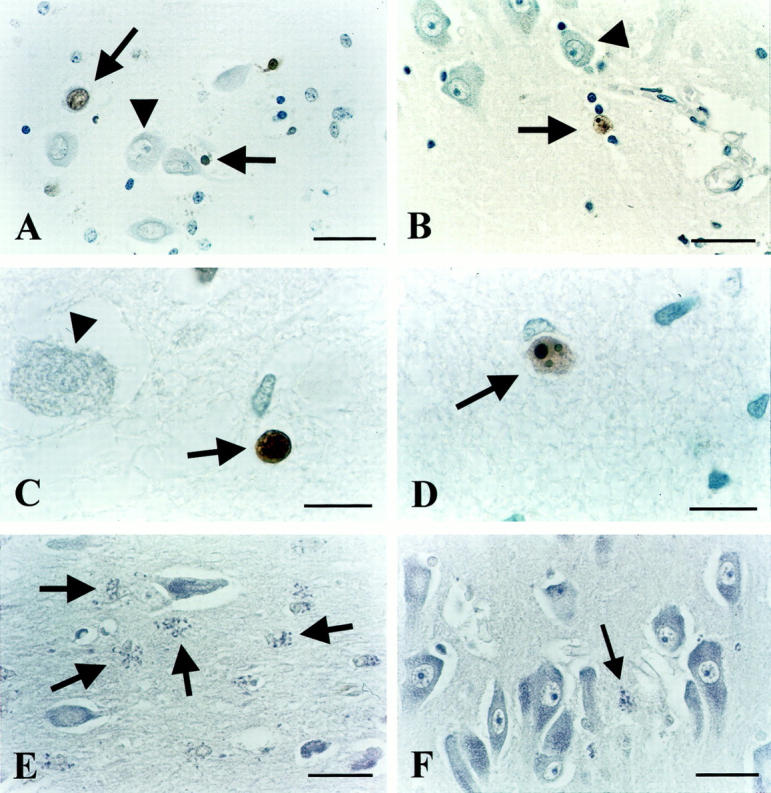
ISEL results. A: Positive labeling in the CA4 area of depressed patient 94-112, showing necrotic morphology (upper arrow) as indicated by the comparable size as an intact, neighboring neuron (arrowhead) without chromatin re-organization or apoptotic bodies visible. Also seen is a labeled apoptotic cell as evidenced by its pycnotic appearance, strong condensation, and brown DAB precipitate (horizontal arrow). B: ISEL-positive neuron (arrow) just outside the CA1 cell layer of depressed patient 90-001 with clear apoptotic morphology, ie, a reduced size as compared to unstained, healthy-looking neurons (triangle), and apoptotic bodies clearly visible. C: ISEL-positive, apoptotic cell (arrow) with a pycnotic, condensed appearance adjacent to a nonstained large cell (arrowhead). CA1 of depressed patient 94-094. D: Apoptotic neuron (arrow) in the subiculum of depressed patient 94-032 with three clear apoptotic bodies visible. E: Frequent, granular morphology (arrows) suggestive of chromatolytic processes, adjacent to normal looking neurons in CA3 of depressed patient 90-001. F: Normal-appearing neurons in CA1 of control subject 94-123. Also, one granular, chromatolytic-like structure is visible (arrow). Scale bars: 34 μm (A, B, E, and F) and 15 μm (C and D).
Figure 2.

ISEL results (continued from Figure 1 ▶ ). A: Isolated, ISEL-positive apoptotic cell with a clearly condensed size (arrow) in an otherwise ISEL-negative DG area of depressed patient 93-146. B: ISEL-positive apoptotic cell (arrow) in the DG of steroid-treated patient 95-11. C: Apoptotic cell (arrowhead) with clear membrane blebbing as well as a single apoptotic body visible, adjacent to an isolated ISEL-positive nucleus (large arrow) that appears necrotic, with a comparable size that seems even a little swollen, as compared to neighboring DG cells. Both cells are located on the inner border between the otherwise ISEL-negative DG and CA4. Depressed patient 93-090. D: Similar to C, an apoptotic cell close to a necrotic one (arrow). Clearly apoptotic bodies are indicated by arrowheads. Also, the enhanced levels of DNA fragmentation in some of the other cells is visible. Depressed patient 94-17. E: Apoptotic cell at the inner border of the DG of steroid-treated patient 83-004. Arrowheads indicate apoptotic bodies. F: Prominent ISEL labeling of a cell displaying glia morphology with DAB precipitate present throughout its protrusions. Steroid-treated patient 93-021. Scale bars: 42 μm (A), 25 μm (B), 16 μm (C–E), and 10 μm (F).
For HSP70 immunocytochemistry, staining was classified as neuropil staining (N) or cellular staining (C) with intensity being scored as follows: −, no staining present; ±, weak to moderate staining present; or +, strong, prominent staining observed. In addition, punctate granular deposits in the neuropil were marked with S.
Activation of NF-κB is histologically reflected by translocation of the p65 subunit from the cytoplasm to the nucleus. 49,50,57,62 For this reason, attention was paid to the subcellular distribution of the DAB precipitate and recorded whether it was found in the cytoplasm or nucleus.
Results
Gross histological examination of the methyl green counterstained ISEL sections did not reveal any obvious anatomical or cellular differences in hippocampal morphology or organization between the groups studied. Notably, no indications for overt, massive neuronal loss in any of the cell layers of the hippocampus could be observed. Also, no abnormally organized, heterotopically displaced, or irregularly oriented hippocampal or cortical neurons were seen. In eight out of 15 depressed patients, one steroid-treated patient, and two out of 15 controls, methyl green counterstaining revealed an ISEL-negative, granular staining pattern without a distinct neuronal membrane, that varied between patients in frequency and distribution (Table 2) ▶ . This pattern was suggestive of remnants of chromatolytic processes and was often found in CA4, CA3, and CA1 (see Table 2 ▶ , scored with R; and Figure 1, E and F ▶ ).
ISEL
Patterns of DNA fragmentation between different sections of the same patient were comparable. In general, slightly more ISEL-positive cells as well as occasionally, moderate increases in nuclear staining intensity were found in the depressed group (Table 2) ▶ , with the CA4 and DG area frequently affected (14 out of 15 patients scored ± or higher in these areas, compared to five out of 15 controls; see Table 2 ▶ ). Although less strongly, CA1 and subiculum were also affected, whereas CA3 was moderately positive (scored “+”) only one time. The DG displayed isolated, ISEL-positive cells at a low frequency in several depressed patients (12 patients scored ± or higher) and in five controls. Several of the ISEL-positive cells displayed a necrotic phenotype, as judged from their comparable size as neighboring, nonstained neurons, and the absence of a shrunken or condensed, pycnotic nucleus (Figure 2, B and E ▶ , arrow). Also, ISEL-positive glia-cells were occasionally observed in controls (95-106 and 90-086), depressed individuals (94-112, 93-021, and 95-036), and steroid-treated patients (93-16 and 91-120). These cells were present in both neuron-dense and neuron-sparse areas and clearly displayed positively labeled protrusions (Figure 2F) ▶ . No labeling was observed if the TdT enzyme was omitted (results not shown).
In addition to necrotic and glia-like morphology, ISEL revealed positive cells with a clear and convincing apoptotic morphology at a very low incidence of approximately one cell per hippocampal section, in 11 depressed patients, one control, and three steroid-treated patients. Apoptotic cells in depressed patients were found in the DG (10 patients), CA4 (five patients), CA1/2 (two patients), CA3 (one patient), and in the entorhinal cortex (one patient). Based on their location these cells were presumed to be neuronal. Furthermore, in three steroid-treated patients, in addition to necrotic cells in other hippocampal areas (see Table 2 ▶ for details), isolated, ISEL-positive apoptotic cells were found in cortex (one patient), entorhinal cortex (one patient), the granule cell layer of the DG (two patients), and also in CA1 (one patient) and in CA4 (two patients).
HSP70 Immunoreactivity
As depicted in Table 3 ▶ , several controls and depressed patients showed very little, if any iHSP70 signal or only a very weak neuropil or cellular staining. Cytoplasmatic staining was observed in some neurons, eg, in five depressed patients (93-54, 93-146, 94-32, 94-112, and 94-094) and in one control (94-118) moderate but clear cytoplasmatic HSP70 immunoreactivity was found in the CA3 or CA4 area (scored as “+”) (Figure 3A) ▶ . Some others displayed faint neuropil staining (eg, controls 94-118 and 96-013 and depressed patient 94-032 and 93-146). The most prominent cellular staining was found in the steroid-treated patients in CA4 and CA3 (patients 83-004, 95-011, 95-051, 93-094, and 95-054). Furthermore, in the DG, a clear cellular-staining pattern was observed in only one depressed (94-017) and in two steroid-treated patients (95-011, 95-054). Small punctate deposits were observed over the neuropil of three controls, five depressed patients and one steroid-treated patient (indicated with S in Table 3 ▶ ). This pattern seems specific as it was absent in control sections of the same patients in which the first antibody was omitted (not shown). In those steroid-treated individuals that showed HSP70 immunoreactivity, the hippocampus was often preferentially affected as the surrounding cortical tissue of the same tissue section was almost devoid of any staining.
Table 3.
Heat-Shock Protein (HSP) 70 Results of the Control Subjects, Depressed Patients, and Steroid-Treated Group
| Patient no. | CA1/2 | CA3 | CA4 | DG | Sub | EC | Cx | Remarks | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Controls | ||||||||||||||||
| 1. 92-011 | − | − | − | −P | − | nd | nd | |||||||||
| 2. 94-035 | − | nd | +/−C | − | − | − | +II/III | |||||||||
| 3. 94-0118f | +/−N | +NC,S | +NC | −N | +/−N | +/−N | +II/III C | |||||||||
| 4. 96-014 | +/−(1x) | +/−C | − | − | − | nd | − | |||||||||
| 5. 96-10 | − | − | − | − | − | − | − | |||||||||
| 6. 96-013 | − | +/−N | +/−N | − | − | +/−C | +/−N | |||||||||
| 7. 93-061 | nd | − | − | − | − | − | +/−N | |||||||||
| 8. 95-106 | +/−N | −NC | −S, R\ {Tc}− | − | − | − | − | |||||||||
| 9. 94-123 | − | +/−C | +/−N | nd | − | − | − | |||||||||
| 10. 90-086 | − | − | − | − | − | − | − | |||||||||
| 11. 95-093 | − | +/−NC | +/−C | −P | nd | nd | II/III: +/−C | |||||||||
| 12. 94-076 | − | − | − | −P | − | − | − | Infiltrates/bleeding in sulcus | ||||||||
| 13. 92-016 | − | +/−C | +/−C, S | − | − | − | − | |||||||||
| 14. 89-045 | − | − | − | −P | − | − | − | |||||||||
| 15. 82-018 | − | − | − | − | − | − | − | |||||||||
| Depressed patients | ||||||||||||||||
| 1. 93-076 | nd | − | − | − | nd | nd | − | |||||||||
| 2. 93-090 | − | − | − | −P | − | − | − | |||||||||
| 3. 93-146 | +/−N | +/−N | +C | +/−N | +/−N | +/−N | +/−N | |||||||||
| 4. 92-003 | − | − | − | +/−P | +/−N | +/−A | − | |||||||||
| 5. 94-112f | +/−N,C | +N,C | +N | +/−N,P | +/−N | − | −S | Nuclear staining of some individual DG cells | ||||||||
| 6. 93-054 | +/−C,N | +/−C | +C,S | −P | +/−N | − | +/−C,S | |||||||||
| 7. 90-001 | − | +/−C | − | − | − | − | +/−(2x) | |||||||||
| 8. 94-032 | +/−N | +C,S | +/−C,N | −P | +/−NC | − | +/−II−VG | |||||||||
| 9. 94-094f | +/−N | +C | +/−C | − | +/−N | − | +/− | |||||||||
| 10. 94-017 | − | − | +/−C | +C | +/−N | nd | +/−N | |||||||||
| 11. 94-055 | − | +/−N | +/−C | +/−N | − | − | +II−V,S | |||||||||
| 12. 93-021 | −(N) | +/−C | +/−C | −P | − | − | − | |||||||||
| 13. 95-036 | − | +/−C | +/−C | −P | +/− | − | +/−C,S | |||||||||
| 14. 93-115 | Nd | − | − | − | − | − | +II−V,S | |||||||||
| 15. 91-059 | − | − | −R | − | − | − | − | |||||||||
| Steroid-treated group | ||||||||||||||||
| 1. 86-032 | − | − | − | +/−P | − | − | − | |||||||||
| 2. 83-004 | − | +C | ++C | − | − | − | − | |||||||||
| 3. 95-011f | − | +C | ++C | ++C | − | − | − | |||||||||
| 4. 95-051 | +/−N | +/−C | ++C | − | +/−N | +/−N | +/− | |||||||||
| 5. 93-094 | +/−N,C | ++C | ++C | −P | +C | +/− | − | |||||||||
| 6. 95-054f | +/−N | +/−C | ++C | +C,S | ++C | − | II/III: +/−C,S | Infiltration/bleeding in stratum moleculare; positive cells in DG | ||||||||
| 7. 91-120 | − | − | − | − | − | − | − | |||||||||
| 8. 93-016 | − | − | +C | − | − | − | − | |||||||||
| 9. 95-098 | − | − | − | − | − | − | − | |||||||||
Abbreviations used for HSP scoring: N, neuropil staining; C, cellular, cytoplasmic staining pattern; S, small punctate staining of neuropil; P, presence of small, condensed, pycnotic-like cells in the DG granule layer; 2x; denotes the presence of eg, two positive cells in a subarea that is further devoid of staining; A, cellular staining pattern resembling an apoptotic cell. nd: not determined or not present; II, III or II-V relates to cortical layer II and III, and II through LV respectively.
Figure 3.

iHSP70 immunocytochemistry. A: Clear cellular labeling of the pyramidal CA1, CA2, CA3, and CA4 layers of 71-year-old depressed patient 94-094. Arrowheads indicate DG granule cell layer. The asterisk marks the CA3 area. No immuno-cytochemical staining is present in the DG. B: Predominant cellular (top arrowhead) and neuropil staining (between bottom arrowhead and the asterisk) in the CA1-3 area of control subject 94-118. C: Higher magnification of the DG of a 63-year-old steroid-treated patient 95-11 that shows some individual neurons showing clearly enhanced staining (arrows). D: CA1 area of depressed patient 94-112 showing prominent cellular staining (arrows) in a subpopulation of neurons in this area whereas cells without this cytoplasmic staining are also observed in the same area. E: Weak cytoplasmic staining of CA1 neurons of steroid-treated patient 95-054. F: Prominent cellular staining of CA4 neurons in steroid-treated patient 95-11. Scale bars: 400 μm (A and B), 42 μm (C), 80 μm (D), and 40 μm (E and F).
NF-κB
For NF-κB p65 immunocytochemistry, no clear differences were observed between the groups or between the anatomical subareas studied. A granular and punctate staining pattern was obtained, that was restricted to the cytoplasm in almost all neurons of the hippocampal pyramidal and granule cell layers (Figure 4, A ▶ -C). No nuclear staining that would have reflected NF-κB activation could be found. The positive control sections of a confirmed severe Alzheimer’s disease patient (NBB section number 91-51, female 78 years of age, PMD 3 hours) clearly did show occasional nuclear translocation as reflected by the nuclear localization of the signal (Figure 4D) ▶ .
Discussion
Numerous clinical and preclinical studies have now shown that hyperactivity and disturbance of HPA axis function are implicated in the pathogenesis of depression, 2,26-37,63-68 whereas other factors are also involved. 65,69,70 In the present and in a related study on the same patients, 71 we investigated for the first time the anatomical consequences of GC overexposure for neuronal viability in the human hippocampus. Although initially proposed to cause hippocampal pathology in, for example, aging and Alzheimer’s disease, later studies have further developed the glucocorticoid cascade concept. 72-75,82,84,96,139 Deleterious effects of chronic GC exposure are now thought to range from initial functional deficits 16,76-78 via significant, but transient and still reversible, atrophy of the CA3 dendritic trees, 79 to increased vulnerability to (metabolic) insults 23,80,81 and eventually, cell loss in primarily CA3 under extreme or prolonged stress conditions. Although some debate exists concerning the type involved, GC-induced cell death is generally believed to be apoptotic. 43,72,82
If GC overexposure would also cause neurodegenerative changes in the human hippocampus, one would predict increased levels of pathology, obvious cell loss, as reported after chronic stress or GC treatment, 24,25 and possibly even visible changes in the structural integrity of the CA3 pyramidal field in depression. All patients in our group were established to have suffered from severe depression for a prolonged period, but no significant structural, synaptic, or Alzheimer-like alterations could be detected using Alz-50, glial fibrillary acidic protein, Nissl, or Bodian Silver stain, nor with synaptophysin or B-50, markers for synaptic density and plasticity in tissue from the same patients that are presented in a separate paper. 71 The observed moderate level of DNA fragmentation in some depressed cases is consistent with enhanced oxidative damage, as has been proposed to occur after GC overexposure. 83,84 Whether such damage has actually induced apoptosis, is difficult to establish in the present tissue. The low numbers of apoptotic cells agree with the rapid time kinetics 85 and consequently low chance of detecting ongoing cell death in thin tissue sections. 86-88 The fact that not all patients showed apoptosis could relate to the fact that not all of them died in a depressive period, whereas the presence of both enhanced DNA fragmentation and apoptosis suggests an enhanced neuronal vulnerability in depression (Table 2) ▶ .
It should be noted that the amount of depression-related hippocampal cell loss may be too small to be judged correctly with the present techniques. Although no major changes could be detected with the markers mentioned above, 71 establishing the exact extent of neuronal loss awaits a detailed stereological disector survey of the hippocampus of major depressed patients that are, for example, preferably not on antidepressant medication, which is so far difficult to find. However, the prominent gross morphological changes in previous animal experiments, that were already apparent at low-power morphological examination of conventionally stained sections, 10,24,25 were clearly absent in the present patients, as is evident from both the present and our parallel study in which additional structural markers were applied. 71 In the absence of such extensive cell loss, or neuropathology and, importantly, no obvious loss in areas at risk for GC endangerment such as CA3, our joint observations 71 do not support the notion that corticosteroid overexposure causes major permanent structural damage to the human hippocampus. 10,11,24,25
A methodological factor that might, in theory, have influenced our ISEL results, is PMD (Table 1) ▶ , that, when prolonged, was initially claimed to yield false-positive ISEL results. 89,90 Other studies, however, could not establish such a relationship studying PMDs of up to 65 hours, 44,53-55,91 whereas differences in methodology and fixation also contributed to these differences. 92,93 In the current study, no obvious relation with PMD was found either, with, for example, very little ISEL staining in patient 93-090 despite a PMD of 48 hours (Table 2) ▶ . Neither is it very likely that the consistently found increases in number of ISEL-positive cells in, for example, Alzheimer’s disease as reported by several independent groups now, are all because of PMD, that clearly differed between these studies. 88,93 Furthermore, although differences in ISEL distribution in the present study were scored in a semiquantitative manner, the detection of apoptosis is not influenced by PMD because it is based on specific, additional morphological criteria (Figure 1, B and D ▶ , and Figure 2, C–E ▶ ). The presence of ISEL-negative granular staining, however, that is suggestive of remnants of a chromatolytic process and was found in CA1, CA3, and CA4 of some patients (Table 2 ▶ and Figure 1, E and F ▶ ), may relate to PMD, because it was preferably observed in depressed cases with long PMD values (eg, 93-090, 93-146, 94-112), and in control 90-086, with a long PMD of 44 hours. Only one steroid-treated patient with a PMD less than 8 hours also showed this staining pattern, but to a considerably lesser extent, for which no explanation can be given at present.
Recent in vivo MRI studies suggest a correlation between hippocampal or brain atrophy, memory deficits, and cumulative GC exposure during, for example, aging and depression, 35-37,67,94-99 although also exceptions have been reported. 142,143 However, such studies do not provide conclusive evidence for permanent changes, such as cell loss. Hippocampal volume reductions in Cushing’s disease, were reversible after a decrease or cessation of the steroid exposure. 99 This agrees with the general clinical experience with depressive or Cushing’s patients, in which treatment or operation can relieve the depressive symptoms, several of the HPA alterations, and even the hippocampal atrophy as recently demonstrated. 33,34,99,101-104 Consistent with this, the CA3 atrophy in rat and tree shrew hippocampus after chronic stress or GC excess, disappeared once the treatment was stopped or antidepressant treatment commenced. 79,100 Hence, reversible and adaptive, rather than neurotoxic phenomena are expected in this subarea. 72 Furthermore, because CA3 in man constitutes only a relatively small part of the hippocampus proper, it awaits further study whether GC-induced volume changes in this specific subarea indeed contribute significantly to the atrophy of the entire hippocampus that is already detectable at the MRI level.
It is interesting to note that unlike older studies, 10,24,25 applying shrinkage-sensitive density measures, several recent studies have used modern stereological methods for unbiased neuronal counting. 105,106 Such studies on the hippocampus of chronically stressed tree shrews, stressed or GC-exposed rats, and GC-treated (aged) primates, 107-111 all failed to find major reductions in neuron number in hippocampal subareas, which agrees with our present observations using structural markers on the hippocampus of depressed patients presented elsewhere. 71 In addition, analysis of apoptosis in the chronically stressed tree shrew revealed differential changes in different subareas, rather than an increase in CA3, that was expected, in stressed as compared to control animals. 137 Although cortisol application is clearly a different condition than stress per se, the extent of the stressor also differs strongly from the initial rat studies, in which rather extreme, physical stressors were applied. 10,24,25 Although some older studies that used different quantification methods, suggest otherwise, 140-141 studies based on unbiased stereological analysis even question the presence of a relationship between hippocampal neuron number and learning deficits, suggesting that the structural correlates of memory and cognition are more likely to be found in parameters other than neuron number. 112-115
In this respect, primary alterations in GR or MR affinity, function, or number can create a relative insensitivity of hippocampal neurons to GC excess. 4,103,116,117 Studies on GR kinetics in depression, have unfortunately been inconsistent so far, whereas quantification and localization of GR and MR protein or mRNA levels in human brain await further methodological development, also on other HPA feedback areas. 65,118,119 So far, however, GR protein levels in primate and human hippocampus seem rather low (P. J. Lucassen, E. Fuchs, and D. F. Swaab, unpublished observations). 120 Also, medication is important, as it can affect MR, GR, or serotonin receptor levels and even neuronal viability. 65,101,103,116,121-123 Although almost all patients were on an antidepressant or neuroleptic drug treatment at the time of death, we could not establish a relation between medication type and either of the present markers studied. Even if some antidepressants are neurotoxic in vitro, 124 safety testing in vivo should have revealed such a toxic potential, which has, to our knowledge, not been reported.
Regarding the discrepancy between hippocampal volume reductions in the absence of obvious changes in neuron number, a possibility could be a shift in water content. Not only is the brain volume reduction occurring during high-dose GC treatment or in Cushing’s syndrome, reversible after a decrease in or cessation of steroid exposure, a recent study by Starkman and co-workers 99 also convincingly demonstrated the reversibility of GC-induced hippocampal volume changes and the close correlation between the extent of hypercortisolemia and brain volume reduction, that are likely because of changes in water content or balance. 104,138 Another possibility are GC effects on glia cells, that not only possess GRs, but are sensitive to steroid action, 125 and can even undergo apoptosis after exposure to oxidative stress. 126 Interestingly, recent stereological analysis of hippocampal subareas of rats subjected to stress or GC treatment, revealed no changes in neuron number, whereas volume reductions were found, notably, in neuron-sparse subareas of the hippocampus, that contain mainly glia. 107,108,114 At least for astroglia, no obvious changes could be found in our parallel study, 71 which is in line with recent data showing that glial fibrillary acidic protein is resistant to down-regulation by endogenous glucocorticoids 125 and hence suggests a role for other glia types.
A limitation of our current study is that no plasma cortisol levels were available from the depressed patients. However, aside from the well-documented clinical confirmation, six of 15 depressed patients had been included in earlier studies from our group, in which a clear activation of the HPA axis was found, as evidenced by increases in the numbers of CRH-expressing neurons, in the fraction that showed co-localization with vasopressin and by enhanced levels of CRH mRNA in the PVN. 2,26,27 In addition, duration of depression was found to be the strongest predictor for hippocampal atrophy, 37 which was long lasting in the present cohort (Table 1) ▶ .
HSPs are part of a family of ATP-binding proteins that function as molecular chaperones and form complexes, for example, with the GR, to assist in protein folding. The inducible form of HSP70, as currently studied, is hardly detectable under normal conditions, but is strongly induced after various forms of injury, including excitatory insults, ischemia, or cell death. 47,127,128 Although still matter of debate whether iHSP70 plays a protective role, it is considered a general response to cellular stress. A recent study in which iHSP70 was overexpressed indeed showed improved neuronal survival after ischemia and kainic acid injections, which supports a protective role. 47,127,128 Although in most controls, only weak cellular staining was observed, a few depressed patients (eg, CA4 of 93-146, 94-032, and 94-094) showed cellular iHSP70 staining in some neurons. However, the most prominent cellular staining was found in CA4, and to a lesser extent, CA3 area of the prednisone- or beclomethasone-treated patients 93-004, 95-011, 95-051, 93-094 and 95-054, which suggests that if up-regulation of iHSP70 has a protective role, it seems to be more strongly induced by synthetic steroids, rather then by endogenous corticosteroids or the depressive state.
NF-κB is an important transcription factor regulated by GCs. 48 Increased GC exposure, through the induction and accumulation of reactive oxygen species, might induce NF-κB activation, which has furthermore been implicated in both the induction of and protection against neurotoxicity and apoptosis. 51,84,129,130 Activated NF-κB has been found in dopaminergic neurons in Parkinson’s and also in Alzheimer’s disease, as reflected by enhanced nuclear levels in hippocampal neurons (Figure 4D) ▶ . 49,50,129,131 NF-κB activation, as would have been evidenced by translocation of the p65 subunit to the nucleus, was absent in the present study, suggesting that apparently, no protective response against oxygen radicals or apoptosis was initiated. This is consistent with a study that failed to find an association between NF-κB activation and apoptosis in amyotrophic lateral sclerosis 57 and suggests that other mechanisms are involved.
As dexamethasone poorly passes the blood-brain barrier, 132,133 synthetic steroid treatment may induce HPA feedback inhibition only at the level of the pituitary without reaching the hypothalamus or hippocampus. Consequently, this may deplete hippocampal MRs and GRs from their endogenous GC ligand, and induce a condition of chemical adrenalectomy, which is expected to yield apoptosis in the DG. 133-135 An earlier study on the same steroid-treated patients already demonstrated that the doses used strongly suppressed hypothalamic CRH and vasopressin expression, 136 which suggests that HPA feedback inhibition on a hypothalamic level has occurred. Although such a central effect agrees with the presence of ISEL-positive cells in the DG of two of the steroid-treated patients (Table 2 ▶ and Figure 2 ▶ ), a conclusive statement on whether chemical adrenalectomy in human is primarily induced by synthetic steroid treatment awaits further research on the blood-brain barrier kinetics of the specific steroids, and on the temporal profile of GC depletion in man.
In conclusion, in our joint studies, 71 no massive structural hippocampal changes could be detected in depression. Although small effects undetectable by the present methods cannot be excluded and await detailed morphometrical analysis, they could be significant enough to already affect feedback disturbance or hippocampal functioning. Moderate increases in DNA fragmentation as well as clear evidence for apoptotic cell death were observed for the first time in depression, but the latter at a very low frequency and not in areas predicted to be at GC risk, which rather points to an increased vulnerability in this condition. As several recent animal studies also failed to show massive cell loss after severe stress or GC excess, actual hippocampal neuronal loss seems to be induced only by very extreme GC excess conditions. It hence becomes unlikely whether such conditions are indeed relevant for the situation in human depression.
Acknowledgments
We thank Dr. A. Migheli, University of Turino, Italy, for his generous gift of NF-κB antibody; Dr. R. Ravid (coordinator), the Netherlands Brain Bank; Dr. R. De Vos, Department of Neuropathology, University of Nijmegen, for provision of the well-documented human brain tissue; and Mr. S. Van Mechelen, University of Amsterdam, for expert photographic assistance.
Footnotes
Address reprint requests to P. J. Lucassen, Institute for Neurobiology, Faculty of Science, University of Amsterdam, Kruislaan 320, 1098 SM, Amsterdam, The Netherlands. E-mail: lucassen@bio.uva.nl.
Supported by the Netherlands Organization For Scientific Research (NWO) (to P. J. L.), by the Internationale Stichting Alzheimer Onderzoek (SAO) (to E. R. D. K. and P. J. L.), and by the Platform Alternatieven Dierproeven (to D. F. S.).
References
- 1.Raadsheer FC, Sluiter AA, Ravid R, Tilders FJ, Swaab DF: Localization of corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the human hypothalamus; age-dependent colocalization with vasopressin. Brain Res 1993, 615:50-62 [DOI] [PubMed] [Google Scholar]
- 2.Raadsheer FC, van Heerikhuize JJ, Lucassen PJ, Hoogendijk WJ, Tilders FJ, Swaab DF: Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus of patients with Alzheimer’s disease and depression. Am J Psychiatry 1995, 152:1372-1376 [DOI] [PubMed] [Google Scholar]
- 3.Reul JM, de Kloet ER: Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 1985, 117:2505-2511 [DOI] [PubMed] [Google Scholar]
- 4.De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M: Brain corticosteroid receptor balance in health and disease. Endocr Rev 1998, 19:269-301 [DOI] [PubMed] [Google Scholar]
- 5.Herman JP, Cullinan WE, Morano MI, Akil H, Watson SJ: Contribution of the ventral subiculum to inhibitory regulation of the hypothalamo-pituitary-adrenocortical axis. J Neuroendocrinol 1995, 7:475-482 [DOI] [PubMed] [Google Scholar]
- 6.Herman JP, Prewitt CM, Cullinan WE: Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stress axis. Crit Rev Neurobiol 1996, 10:371-394 [DOI] [PubMed] [Google Scholar]
- 7.Herman JP, Cullinan WE: Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 1997, 20:78-84 [DOI] [PubMed] [Google Scholar]
- 8.Bobinski M, Wegiel J, Wisniewski HM, Tarnawski M, Reisberg B, Mlodzik B, de Leon MJ, Miller DC: Atrophy of hippocampal formation subdivisions correlates with stage and duration of Alzheimer disease. Dementia 1995, 6:205-210 [DOI] [PubMed] [Google Scholar]
- 9.De Leon MJ, George AE, Golomb J, Tarshish C, Convit A, Kluger A, De Santi S, McRae T, Ferris SH, Reisberg B, Ince C, Rusinek H, Bobinski M, Quinn B, Miller DC, Wisniewski HM: Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging 1997, 18:1-11 [DOI] [PubMed] [Google Scholar]
- 10.Sapolsky RM, Krey LC, McEwen BS: Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci 1985, 5:1222-1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sapolsky RM, Krey LC, McEwen BS: The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev 1986, 7:284-301 [DOI] [PubMed] [Google Scholar]
- 12.Weiner MF, Vobach S, Olsson K, Svetlik D, Risser RC: Cortisol secretion and Alzheimer’s disease progression. Biol Psychiatry 1997, 42:1030-1038 [DOI] [PubMed] [Google Scholar]
- 13.Conrad CD, Galea LA, Kuroda Y, McEwen BS: Chronic stress impairs rat spatial memory on the Y maze, and this effect is blocked by tianeptine pretreatment. Behav Neurosci 1996, 110:1321-1334 [DOI] [PubMed] [Google Scholar]
- 14.Landfield PW, Waymire JC, Lynch G: Hippocampal aging and adrenocorticoids: quantitative correlations. Science 1978, 202:1098-1102 [DOI] [PubMed] [Google Scholar]
- 15.Landfield PW, Baskin RK, Pitler TA: Brain aging correlates: retardation by hormonal-pharmacological treatments. Science 1981, 214:581-584 [DOI] [PubMed] [Google Scholar]
- 16.Luine V, Villegas M, Martinez C, McEwen BS: Repeated stress causes reversible impairments of spatial memory performance. Brain Res 1994, 639:167-170 [DOI] [PubMed] [Google Scholar]
- 17.Lupien SJ, McEwen BS: The acute effects of corticosteroids on cognition: integration of animal and human model studies. Brain Res Brain Res Rev 1997, 24:1-27 [DOI] [PubMed] [Google Scholar]
- 18.Issa AM, Rowe W, Gauthier S, Meaney MJ: Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J Neurosci 1990, 10:3247-3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joels M: Steroid hormones and excitability in the mammalian brain. Front Neuroendocrinol 1997, 18:2-48 [DOI] [PubMed] [Google Scholar]
- 20.Kerr DS, Campbell LW, Applegate MD, Brodish A, Landfield PW: Chronic stress-induced acceleration of electrophysiologic and morphometric biomarkers of hippocampal aging. J Neurosci 1991, 11:1316-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapolsky RM: A mechanism for glucocorticoid toxicity in the hippocampus: increased neuronal vulnerability to metabolic insults. J Neurosci 1985, 5:1228-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein-Behrens BA, Elliott EM, Miller CA, Schilling JW, Newcombe R, Sapolsky RM: Glucocorticoids exacerbate kainic acid-induced extracellular accumulation of excitatory amino acids in the rat hippocampus. J Neurochem 1992, 58:1730-1735 [DOI] [PubMed] [Google Scholar]
- 23.Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R: Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci 1994, 14:5373-5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sapolsky RM, Uno H, Rebert CS, Finch CE: Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci 1990, 10:2897-2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uno H, Tarara R, Else JG, Suleman MA, Sapolsky RM: Hippocampal damage associated with prolonged and fatal stress in primates. J Neurosci 1989, 9:1705-1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF: Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 1994, 60:436-444 [DOI] [PubMed] [Google Scholar]
- 27.Purba JS, Hoogendijk WJ, Hofman MA, Swaab DF: Increased number of vasopressin- and oxytocin-expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Arch Gen Psychiatry 1996, 53:137-143 [DOI] [PubMed] [Google Scholar]
- 28.Nemeroff CB, Krishnan KR, Reed D, Leder R, Beam C, Dunnick NR: Adrenal gland enlargement in major depression. A computed tomographic study. Arch Gen Psychiatry 1992, 49:384-387 [DOI] [PubMed] [Google Scholar]
- 29.Nemeroff CB: The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry 1996, 1:336-342 [PubMed] [Google Scholar]
- 30.Holsboer F: The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res 1999, 33:181-214 [DOI] [PubMed] [Google Scholar]
- 31.Holsboer F, Lauer CJ, Schreiber W, Krieg JC: Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology 1995, 62:340-347 [DOI] [PubMed] [Google Scholar]
- 32.Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, Standhardt H, Lammers CH, Heuser I: Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab 1997, 82:234-238 [DOI] [PubMed] [Google Scholar]
- 33.Rubin RT, Phillips JJ, Sadow TF, McCracken JT: Adrenal gland volume in major depression. Increase during the depressive episode and decrease with successful treatment. Arch Gen Psychiatry 1995, 52:213-218 [DOI] [PubMed] [Google Scholar]
- 34.Heuser I: Anna-Monika-Prize paper. The hypothalamic-pituitary-adrenal system in depression. Pharmacopsychiatry 1998, 31:10-13 [DOI] [PubMed] [Google Scholar]
- 35.Sapolsky RM: Why stress is bad for your brain. Science 1996, 273:749-750 [DOI] [PubMed] [Google Scholar]
- 36.Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW: Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996, 93:3908-3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheline YI, Sanghavi M, Mintun MA, Gado MH: Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 1999, 19:5034-5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliott EM, Sapolsky RM: Corticosterone impairs hippocampal neuronal calcium regulation—possible mediating mechanisms. Brain Res 1993, 602:84-90 [DOI] [PubMed] [Google Scholar]
- 39.McConkey DJ, Orrenius S: The role of calcium in the regulation of apoptosis. Biochem Biophys Res Commun 1997, 239:357-366 [DOI] [PubMed] [Google Scholar]
- 40.McIntosh LJ, Sapolsky RM: Glucocorticoids may enhance oxygen radical-mediated neurotoxicity. Neurotoxicology 1996, 17:873-882 [PubMed] [Google Scholar]
- 41.McIntosh LJ, Cortopassi KM, Sapolsky RM: Glucocorticoids may alter antioxidant enzyme capacity in the brain: kainic acid studies. Brain Res 1998, 791:215-222 [DOI] [PubMed] [Google Scholar]
- 42.McIntosh LJ, Sapolsky RM: Glucocorticoids increase the accumulation of reactive oxygen species and enhance adriamycin-induced toxicity in neuronal culture. Exp Neurol 1996, 141:201-206 [DOI] [PubMed] [Google Scholar]
- 43.Reagan LP, McEwen BS: Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat 1997, 13:149-167 [DOI] [PubMed] [Google Scholar]
- 44.Lucassen PJ, Chung WC, Vermeulen JP, Van Lookeren Campagne M, Van Dierendonck JH, Swaab DF: Microwave-enhanced in situ end-labeling of fragmented DNA: parametric studies in relation to postmortem delay and fixation of rat and human brain. J Histochem Cytochem 1995, 43:1163-1171 [DOI] [PubMed] [Google Scholar]
- 45.Kerr JF, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26:239-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hasegawa K, Litt L, Espanol MT, Sharp FR, Chan PH: Expression of c-fos and hsp70 mRNA in neonatal rat cerebrocortical slices during NMDA-induced necrosis and apoptosis. Brain Res 1998, 785:262-278 [DOI] [PubMed] [Google Scholar]
- 47.Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK: The neuroprotective potential of heat shock protein 70 (HSP70). Mol Med Today 1999, 5:525-531 [DOI] [PubMed] [Google Scholar]
- 48.Unlap T, Jope RS: Inhibition of NFκB DNA binding activity by glucocorticoids in rat brain. Neurosci Lett 1995, 198:41-44 [DOI] [PubMed] [Google Scholar]
- 49.Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC: Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci USA 1997, 94:7531-7536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C: Inhibition of NF-κB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA 1999, 96:9409-9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lezoualc’h F, Sagara Y, Holsboer F, Behl C: High constitutive NF-κB activity mediates resistance to oxidative stress in neuronal cells. J Neurosci 1998, 18:3224-3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH: A new method to detect apoptosis in paraffin sections: in situ end-labeling of fragmented DNA. J Histochem Cytochem 1993, 41:7-12 [DOI] [PubMed] [Google Scholar]
- 53.Lucassen PJ, Chung WC, Kamphorst W, Swaab DF: DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer disease in the absence of apoptotic morphology. J Neuropathol Exp Neurol 1997, 56:887-900 [DOI] [PubMed] [Google Scholar]
- 54.Migheli A, Cavalla P, Marino S, Schiffer D: A study of apoptosis in normal and pathologic nervous tissue after in situ end-labeling of DNA strand breaks. J Neuropathol Exp Neurol 1994, 53:606-616 [DOI] [PubMed] [Google Scholar]
- 55.Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H: Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 1998, 57:456-464 [DOI] [PubMed] [Google Scholar]
- 56.Milarski KL, Morimoto RI: Mutational analysis of the human HSP70 protein: distinct domains for nucleolar localization and adenosine triphosphate binding. J Cell Biol 1989, 109:1947-1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Migheli A, Piva R, Atzori C, Troost D, Schiffer D: c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 1997, 56:1314-1322 [DOI] [PubMed] [Google Scholar]
- 58.Duvernoy HM: The Human Hippocampus, an Atlas of Applied Anatomy. 1988. Bergmann Verlag, Munchen
- 59.Sheng JG, Mrak RE, Griffin WS: Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J Neuropathol Exp Neurol 1998, 57:323-328 [DOI] [PubMed] [Google Scholar]
- 60.Cohen JJ: Apoptosis. Immunol Today 1993, 14:126-130 [DOI] [PubMed] [Google Scholar]
- 61.Wyllie AH: Apoptosis: an overview. Br Med Bull 1997, 53:451-465 [DOI] [PubMed] [Google Scholar]
- 62.Heck S, Lezoualc’h F, Engert S, Behl C: Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor κB. J Biol Chem 1999, 274:9828-9835 [DOI] [PubMed] [Google Scholar]
- 63.Holsboer F, Barden N: Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev 1996, 17:187-205 [DOI] [PubMed] [Google Scholar]
- 64.Nemeroff CB: The neurobiology of depression. Sci Am 1998, 278:42-49 [DOI] [PubMed] [Google Scholar]
- 65.Lopez JF, Chalmers DT, Little KY, Watson SJ: A. E. Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry 1998, 43:547-573 [DOI] [PubMed] [Google Scholar]
- 66.Deuschle M, Weber B, Colla M, Depner M, Heuser I: Effects of major depression, aging and gender upon calculated diurnal free plasma cortisol concentrations: a re-evaluation study. Stress 1998, 2:281-287 [DOI] [PubMed] [Google Scholar]
- 67.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS: Hippocampal volume reduction in major depression. Am J Psychiatry 2000, 157:115-118 [DOI] [PubMed] [Google Scholar]
- 68.Krishnan KR, Doraiswamy PM, Figiel GS, Husain MM, Shah SA, Na C, Boyko OB, McDonald WM, Nemeroff CB, Ellinwood EH, Jr: Hippocampal abnormalities in depression. J Neuropsychiatry Clin Neurosci 1991, 3:387-391 [DOI] [PubMed] [Google Scholar]
- 69.Meltzer CC, Smith G, DeKosky ST, Pollock BG, Mathis CA, Moore RY, Kupfer DJ, Reynolds CF, III: Serotonin in aging, late-life depression, and Alzheimer’s disease: the emerging role of functional imaging [see comments]. Neuropsychopharmacology 1998, 18:407-430 [DOI] [PubMed] [Google Scholar]
- 70.Owens MJ, Nemeroff CB: Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem 1994, 40:288-295 [PubMed] [Google Scholar]
- 71.Müller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJG, Holsboer F, Swaab DF: The human hippocampus in major depression or following administration of glucocorticoids: no evidence for major structural alterations. Society for Neuroscience Annual Meeting 1998, abstract 386.14
- 72.McEwen BS: Stress and hippocampal plasticity. Annu Rev Neurosci 1999, 22:105-122 [DOI] [PubMed] [Google Scholar]
- 73.McEwen BS: Stress and the aging hippocampus. Front Neuroendocrinol 1999, 20:49-70 [DOI] [PubMed] [Google Scholar]
- 74.Seckl JR, Olsson T: Glucocorticoid hypersecretion and the age-impaired hippocampus: cause or effect? J Endocrinol 1995, 145:201-211 [DOI] [PubMed] [Google Scholar]
- 75.Sapolsky RM: Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol 1999, 34:721-732 [DOI] [PubMed] [Google Scholar]
- 76.Lupien SJ, Gaudreau S, Tchiteya BM, Maheu F, Sharma S, Nair NP, Hauger RL, McEwen BS, Meaney MJ: Stress-induced declarative memory impairment in healthy elderly subjects: relationship to cortisol reactivity. J Clin Endocrinol Metab 1997, 82:2070-2075 [DOI] [PubMed] [Google Scholar]
- 77.McEwen BS, Sapolsky RM: Stress and cognitive function. Curr Opin Neurobiol 1995, 5:205-216 [DOI] [PubMed] [Google Scholar]
- 78.Newcomer JW, Selke G, Melson AK, Hershey T, Craft S, Richards K, Alderson AL: Decreased memory performance in healthy humans induced by stress-level cortisol treatment. Arch Gen Psychiatry 1999, 56:527-533 [DOI] [PubMed] [Google Scholar]
- 79.Magarinos AM, McEwen BS, Flugge G, Fuchs E: Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 1996, 16:3534-3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lawrence MS, Sapolsky RM: Glucocorticoids accelerate ATP loss following metabolic insults in cultured hippocampal neurons. Brain Res 1994, 646:303-306 [DOI] [PubMed] [Google Scholar]
- 81.Sapolsky RM: Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J Neurosci 1986, 6:2240-2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sapolsky RM: Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress 1996, 1:1-19 [DOI] [PubMed] [Google Scholar]
- 83.McIntosh LJ, Hong KE, Sapolsky RM: Glucocorticoids may alter antioxidant enzyme capacity in the brain: baseline studies. Brain Res 1998, 791:209-214 [DOI] [PubMed] [Google Scholar]
- 84.Behl C: Effects of glucocorticoids on oxidative stress-induced hippocampal cell death: implications for the pathogenesis of Alzheimer’s disease. Exp Gerontol 1998, 33:689-696 [DOI] [PubMed] [Google Scholar]
- 85.Hu Z, Yuri K, Ozawa H, Lu H, Kawata M: The in vivo time course for elimination of adrenalectomy-induced apoptotic profiles from the granule cell layer of the rat hippocampus. J Neurosci 1997, 17:3981-3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Perry G, Nunomura A, Smith MA: A suicide note from Alzheimer disease neurons? Nat Med 1998, 4:897-898 [DOI] [PubMed] [Google Scholar]
- 87.Perry G, Nunomura A, Lucassen P, Lassmann H, Smith MA: Apoptosis and Alzheimer’s disease. Science 1998, 282:1268-1269 [DOI] [PubMed] [Google Scholar]
- 88.Lucassen PJ: Presenilins and DNA damage; a link through amyloid ? J Alzheimer’s Dis 2000, 2:61–67 [DOI] [PubMed]
- 89.Anderson AJ, Su JH, Cotman CW: DNA damage and apoptosis in Alzheimer’s disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci 1996, 16:1710-1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petito CK, Roberts B: Effect of postmortem interval on in situ end-labeling of DNA oligonucleosomes. J Neuropathol Exp Neurol 1995, 54:761-765 [DOI] [PubMed] [Google Scholar]
- 91.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM: Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol 1995, 89:35-41 [DOI] [PubMed] [Google Scholar]
- 92.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, Negoescu A: TUNEL apoptotic cell detection in tissue sections: critical evaluation and improvement. J Histochem Cytochem 1998, 46:327-334 [DOI] [PubMed] [Google Scholar]
- 93.Lucassen PJ, Negoescu A, Labat-Moleur F, Van Lookeren Campagne M: Microwave-enhanced in situ end labeling; pitfalls and limitations. Biotechniques; Microwave Applications for Pathology. Eaton Publishing 2000, pp 71–91
- 94.Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ: Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci 1998, 1:69-73 [DOI] [PubMed] [Google Scholar]
- 95.Lupien SJ, Nair NP, Briere S, Maheu F, Tu MT, Lemay M, McEwen BS, Meaney MJ: Increased cortisol levels and impaired cognition in human aging: implication for depression and dementia in later life. Rev Neurosci 1999, 10:117-139 [DOI] [PubMed] [Google Scholar]
- 96.McEwen BS, de Leon MJ, Lupien SJ, Meaney MJ: Corticosteroids, the aging brain and cognition. Trends Endocrinol Metab 1999, 10:92-96 [DOI] [PubMed] [Google Scholar]
- 97.Dahabra S, Ashton CH, Bahrainian M, Britton PG, Ferrier IN, McAllister VA, Marsh VR, Moore PB: Structural and functional abnormalities in elderly patients clinically recovered from early- and late-onset depression. Biol Psychiatry 1998, 44:34-46 [DOI] [PubMed] [Google Scholar]
- 98.Convit A, de Leon MJ, Tarshish C, De Santi S, Kluger A, Rusinek H, George AE: Hippocampal volume losses in minimally impaired elderly. Lancet 1995, 345:266. [DOI] [PubMed] [Google Scholar]
- 99.Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE: Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing’s disease. Biol Psychiatry 1999, 46:1595-1602 [DOI] [PubMed] [Google Scholar]
- 100.Magarinos AM, Deslandes A, McEwen BS: Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur J Pharmacol 1999, 371:113-122 [DOI] [PubMed] [Google Scholar]
- 101.Yau JL, Olsson T, Morris RG, Meaney MJ, Seckl JR: Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: relationship with spatial learning in young and aged rats. Neuroscience 1995, 66:571-581 [DOI] [PubMed] [Google Scholar]
- 102.Rowe W, Steverman A, Walker M, Sharma S, Barden N, Seckl JR, Meaney MJ: Antidepressants restore hypothalamic-pituitary-adrenal feedback function in aged, cognitively-impaired rats. Neurobiol Aging 1997, 18:527-533 [DOI] [PubMed] [Google Scholar]
- 103.Barden N: Regulation of corticosteroid receptor gene expression in depression and antidepressant action. J Psychiatry Neurosci 1999, 24:25-39 [PMC free article] [PubMed] [Google Scholar]
- 104.Starkman MN, Schteingart DE, Schork MA: Cushing’s syndrome after treatment: changes in cortisol and ACTH levels, and amelioration of the depressive syndrome. Psychiatry Res 1986, 19:177-188 [DOI] [PubMed] [Google Scholar]
- 105.Swaab DF, Uylings HB: Comments on review by Coleman and Flood, ‘Neuron Numbers and Dendritic Extent in Normal Aging and Alzheimer’s Disease.’ Density measures: parameters to avoid. Neurobiol Aging 1987, 8:574-576 [DOI] [PubMed] [Google Scholar]
- 106.West MJ: Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 1999, 22:51-61 [DOI] [PubMed] [Google Scholar]
- 107.Sousa N, Madeira MD, Paula-Barbosa MM: Effects of corticosterone treatment and rehabilitation on the hippocampal formation of neonatal and adult rats. An unbiased stereological study. Brain Res 1998, 794:199-210 [DOI] [PubMed] [Google Scholar]
- 108.Sousa N, Paula-Barbosa MM, Almeida OF: Ligand and subfield specificity of corticoid-induced neuronal loss in the rat hippocampal formation. Neuroscience 1999, 89:1079-1087 [DOI] [PubMed] [Google Scholar]
- 109.Leverenz JB, Wilkinson CW, Wamble M, Corbin S, Grabber JE, Raskind MA, Peskind ER: Effect of chronic high-dose exogenous cortisol on hippocampal neuronal number in aged nonhuman primates. J Neurosci 1999, 19:2356-2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vollmann-Honsdorf GK, Flugge G, Fuchs E: Chronic psychosocial stress does not affect the number of pyramidal neurons in tree shrew hippocampus. Neurosci Lett 1997, 233:121-124 [DOI] [PubMed] [Google Scholar]
- 111.Bodnoff SR, Humphreys AG, Lehman JC, Diamond DM, Rose GM, Meaney MJ: Enduring effects of chronic corticosterone treatment on spatial learning, synaptic plasticity, and hippocampal neuropathology in young and mid-aged rats. J Neurosci 1995, 15:61-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Geinisman Y, Disterhoft JF, Gundersen HJ, McEchron MD, Persina IS, Power JM, van der Zee EA, West MJ: Remodeling of hippocampal synapses after hippocampus-dependent associative learning. J Comp Neurol 2000, 417:49-59 [PubMed] [Google Scholar]
- 113.Rapp PR, Gallagher M: Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci USA 1996, 93:9926-9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rapp PR, Stack EC, Gallagher M: Morphometric studies of the aged hippocampus: I. Volumetric analysis in behaviorally characterized rats. J Comp Neurol 1999, 403:459-470 [PubMed] [Google Scholar]
- 115.Rasmussen T, Schliemann T, Sorensen JC, Zimmer J, West MJ: Memory impaired aged rats: no loss of principal hippocampal and subicular neurons. Neurobiol Aging 1996, 17:143-147 [DOI] [PubMed] [Google Scholar]
- 116.Barden N: Modulation of glucocorticoid receptor gene expression by antidepressant drugs. Pharmacopsychiatry 1996, 29:12-22 [DOI] [PubMed] [Google Scholar]
- 117.Karanth S, Linthorst AC, Stalla GK, Barden N, Holsboer F, Reul JM: Hypothalamic-pituitary-adrenocortical axis changes in a transgenic mouse with impaired glucocorticoid receptor function. Endocrinology 1997, 138:3476-3485 [DOI] [PubMed] [Google Scholar]
- 118.Seckl JR, Dickson KL, Yates C, Fink G: Distribution of glucocorticoid and mineralocorticoid receptor messenger RNA expression in human postmortem hippocampus. Brain Res 1991, 561:332-337 [DOI] [PubMed] [Google Scholar]
- 119.Pariante CM, Nemeroff CB, Miller AH: Glucocorticoid receptors in depression. Isr J Med Sci 1995, 31:705-712 [PubMed] [Google Scholar]
- 120.Sanchez MM, Young LJ, Plotsky PM, Insel TR: Distribution of corticosteroid receptors in the rhesus brain: relative absence of glucocorticoid receptors in the hippocampal formation. J Neurosci 2000, 20:4657-4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yau JL, Kelly PA, Olsson T, Noble J, Seckl JR: Chronic amitriptyline administration increases serotonin transporter binding sites in the hippocampus of aged rats. Neurosci Lett 1999, 261:183-185 [DOI] [PubMed] [Google Scholar]
- 122.Yau JL, Kelly PA, Seckl JR: Increased glucocorticoid receptor gene expression in the rat hippocampus following combined serotonergic and medial septal cholinergic lesions. Brain Res Mol Brain Res 1994, 27:174-178 [DOI] [PubMed] [Google Scholar]
- 123.Seckl JR, Fink G: Antidepressants increase glucocorticoid and mineralocorticoid receptor mRNA expression in rat hippocampus in vivo. Neuroendocrinology 1992, 55:621-626 [DOI] [PubMed] [Google Scholar]
- 124.Post A, Crochemore C, Uhr M, Holsboer F, Behl C: Differential induction of NF-kappa B activity and neural cell death by antidepressants in vitro. Eur J Neurosci 2000, 12:4331-4337 [DOI] [PubMed] [Google Scholar]
- 125.Nichols NR: Glial responses to steroids as markers of brain aging. J Neurobiol 1999, 40:585-601 [PubMed] [Google Scholar]
- 126.Kitamura Y, Taniguchi T, Shimohama S: Apoptotic cell death in neurons and glial cells: implications for Alzheimer’s disease. Jpn J Pharmacol 1999, 79:1-5 [DOI] [PubMed] [Google Scholar]
- 127.Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, Onley D, Ho DY, Sapolsky RM, Steinberg GK: Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol 1998, 44:584-591 [DOI] [PubMed] [Google Scholar]
- 128.Sharp FR: Stress genes protect brain. Ann Neurol 1998, 44:581-583 [DOI] [PubMed] [Google Scholar]
- 129.Lezoualc’h F, Behl C: Transcription factor NF-κB: friend or foe of neurons? Mol Psychiatry 1998, 3:15-20 [DOI] [PubMed] [Google Scholar]
- 130.Behl C, Lezoualc’h F, Trapp T, Widmann M, Skutella T, Holsboer F: Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 1997, 138:101-106 [DOI] [PubMed] [Google Scholar]
- 131.Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C: Transcription factor NF-κB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA 1997, 94:2642-2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Meijer OC, de Lange EC, Breimer DD, de Boer AG, Workel JO, de Kloet ER: Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology 1998, 139:1789-1793 [DOI] [PubMed] [Google Scholar]
- 133.De Kloet ER: Why dexamethasone poorly penetrates in brain. Stress 1997, 2:13-20 [DOI] [PubMed] [Google Scholar]
- 134.Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF: Exacerbation of apoptosis in the dentate gyrus of the aged rat by dexamethasone and the protective role of corticosterone. Exp Neurol 1996, 140(1):43-52 [DOI] [PubMed] [Google Scholar]
- 135.Sloviter RS, Sollas AL, Dean E, Neubort S: Adrenalectomy-induced granule cell degeneration in the rat hippocampal dentate gyrus: characterization of an in vivo model of controlled neuronal death. J Comp Neurol 1993, 330:324-336 [DOI] [PubMed] [Google Scholar]
- 136.Erkut ZA, Pool C, Swaab DF: Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J Clin Endocrinol Metab 1998, 83:2066-2073 [DOI] [PubMed] [Google Scholar]
- 137.Lucassen PJ, Vollmann-Honsdorf GK, Gleisberg M, Czeh B, De Kloet ER, Fuchs E: Chronic psychosocial stress differentially affects apoptosis in hippocampal subareas and cortex of the adult tree shrew. Eur J Neurosci (in press) [DOI] [PubMed]
- 138.Lagenstein I, Willig RP, Kuhne D: Cranial computed tomography (CCT) findings in children treated with ACTH and dexamethasone: first results. Neuropädiatrie 1979, 10:370–384 [DOI] [PubMed]
- 139.Lucassen PJ, De Kloet ER: Glucocorticoids and the aging brain; cause or consequence? Hof P Mobbs C eds. Functional Neurobiology of Aging. 2001, :pp 883-905 Academic Press, San Diego [Google Scholar]
- 140.Sass KJ, Buchanan CP, Kraemer S, Westerveld M, Kim JH, Spencer DD: Verbal memory impairment resulting from hippocampal neuron loss among epileptic patients with structural lesions. Neurology 1995, 45:2154-2158 [DOI] [PubMed] [Google Scholar]
- 141.Arbel I, Kadar T, Silbermann M, Levy A: The effects of long-term corticosterone administration on hippocampal morphology and cognitive performance of middle-aged rats. Brain Res 1994, 657:227-235 [DOI] [PubMed] [Google Scholar]
- 142.Vakili K, Pillay SS, Lafer B, Fava M, Renshaw PF, Bonello-Cintron CM, Yurgelun-Todd DA: Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biol Psychiatry 2000, 47:1087-1090 [DOI] [PubMed] [Google Scholar]
- 143.Mervaala E, Fohr J, Kononen M, Valkonen-Korhonen M, Vainio P, Partanen K, Partanen J, Tiihonen J, Viinamaki H, Karjalainen AK, Lehtonen J: Quantitative MRI of the hippocampus and amygdala in severe depression. Psychol Med 2000, 30:117-125 [DOI] [PubMed] [Google Scholar]