

Abstract
Inactivation of tumor suppressor genes on chromosome 9p is considered a critical event in renal cell carcinoma pathogenesis. Alterations of CDKN2A on 9p21 have been reported in renal cancer cell lines, but their relevance for primary renal carcinomas is unclear. Loss of heterozygosity (LOH) was analyzed by using four polymorphic microsatellites at D9S970 (9p12-9p13), D9S171 (9p13), D9S1748 (9p21), and D9S156 (9p21) in 113 primary conventional clear-cell renal cell carcinomas (CRCCs). Allelic deletion was detected in 21 of 88 informative CRCCs (24%) with the highest rate of LOH being observed at D9S171 on 9p13 (20%). Chromosome 9p LOH was associated with short tumor-specific survival in stage pT3 RCC (P = 0.01). Fluorescence in situ hybridization analysis of 54 CRCCs revealed no homozygous CDKN2A deletions indicating that this mechanism of CDKN2A inactivation is rare in CRCC. Sequencing of 113 CRCCs showed that 13 tumors (12%) had a 24-bp deletion abrogating codons 4 through 11 of CDKN2A. Immunohistochemical CDKN2A expression was absent in normal renal tissue and was only detected in six of 382 CRCCs (1.5%) on a renal tumor microarray. These data suggest that CDKN2A alterations are present in a small subset of CRCCs and a second, yet unknown tumor suppressor gene proximal to the CDKN2A locus, may play a role in CRCC development.
Chromosome 9p is a candidate to harbor an important tumor suppressor gene with relevance for renal carcinoma progression. 9p losses were found in 24 to 36% of conventional clear-cell renal cell carcinomas (CRCCs) by microsatellite analyses or comparative genomic hybridization. 1-4 A shorter recurrence-free survival was observed in CRCCs with 9p losses compared to tumors without 9p loss by comparative genomic hybridization. 3 9p losses were also detected more frequently in renal carcinoma metastases as compared to their primaries, suggesting that alterations in this region contribute to tumor progression. 5
Recent evidence has implicated CDKN2A located at chromosome 9p21 to be frequently aberrant in the germline of members of familial melanoma kindreds, but also in bladder and other solid tumors. 6 CDKN2A encodes a 156-amino acid protein that exclusively associates with CDK4 and CDK6, inhibiting their complexation with D-type cyclins and the consequent phosphorylation of the retinoblastoma protein. 7 This contributes to cell-cycle arrest. Inactivation of CDKN2A has been described as a consequence of homozygous deletion, rearrangement, hypermethylation, or point mutation. 8-10 The involvement of CDKN2A in human cancer is controversial. It has been shown that the rates of homozygous deletions or mutations is smaller in primary tumors than in cell lines. 9,11
The role of CDKN2A in CRCC is uncertain. 1,12 Homozygous deletions of CDKN2A have been detected in up to 56% of kidney cancer cell lines. 13 Hypermethylation of the promoter region of CDKN2A were reported in 23% of RCC cell lines. 10
To further determine the significance of CDKN2A alterations in primary CRCC we performed: 1) sequence analysis of CDKN2A in 113 CRCCs; 2) fluorescence in situ hybridization (FISH) to search for homozygous CDKN2A deletions; 3) immunohistochemical CDKN2A expression analysis using a renal tumor microarray containing 532 renal tumors; and 4) loss of heterozygosity (LOH) analysis using four polymorphic microsatellites on 9p.
Materials and Methods
Tumor Samples for DNA Analysis
All renal tumors from the archive of the Institute for Pathology, University of Basel, were reviewed by one pathologist (HM). One hundred thirteen consecutive nephrectomy specimens from 1985 to 1994 with CRCC 14 were selected for this study. Histological grading and tumor staging were done according to Thoenes and colleagues 15 and International Union Against Cancer (UICC). 16 There were 11 grade 1, 71 grade 2, and 31 grade 3 tumors. Forty tumors were stage pT1, eight were pT2, 64 were pT3, and one was pT4. An extensive tissue sampling to ensure perinephric fat infiltration may account for the relative high proportion of high-stage cases in this tumor set. The mean tumor diameter was 6.6 ± 3.0 cm. There was lymph node metastasis at nephrectomy in seven patients and evidence for hematogenous metastasis (pM1/cM1) in 19 patients.
Overall and tumor-specific survival data were obtained by reviewing the hospital records, by direct communication with the attending physicians, and from the Cancer Registry of Basel. Patients were evaluated from the time of biopsy diagnosis to the last known follow-up. Overall survival was available from 100 patients and tumor-specific survival was available from 83 patients. The mean follow-up time was 48.9 months (median, 48 months; range, 2 to 131 months). Thirty-five patients had a 5-year follow-up. Forty-one patients died within 5 years after surgery. There was a significant association between short tumor-specific survival and presence of metastasis (P < 0.0001), pT stage (P < 0.001), and histological grade (P < 0.05).
DNA Preparation
Formalin-fixed, paraffin-embedded tumor tissue from each of the 113 patients was selected on the basis of hematoxylin and eosin (H&E)-stained sections to ensure a minimum of 75% tumor cells in the samples. Twenty to 30 mg of normal and tumor tissue was scraped away from the paraffin blocks using a scalpel. Areas of normal tissue were defined using H&E tissue sections. Deparaffinizing of the tissues and DNA extraction were performed according to the QIAmp Tissue Kit protocol (Qiagen, Basel, Switzerland).
Microsatellite Analysis
Analysis of allelic deletions was performed using primers for microsatellites D9S970 (9p12-9p13), D9S171 (9p13), D9S1748 (9p21), and D9S156 (9p21). Primer sequences were obtained from the Genome Data Base. The locus of D9S1748 lies upstream of exon 1 of CDKN2A. 17 Primers were labeled with T4-polynucleotide kinase (Catalys, Wallisellen, Switzerland) and γ32P-ATP (Amersham/Pharmacia, Zurich, Switzerland) for microsatellite analysis. Polymerase chain reaction (PCR) amplification was performed in a total of 15 μl containing 50 to 100 ng DNA, 1× Taq buffer (Qiagen), 200 μmol/L dNTPs, 3 pmol of each primer, 0.4 pmol 32P-labeled primer, and 1 U Taq DNA polymerase (Qiagen). An initial denaturation step of 95°C for 3 minutes followed by 35 cycles each of 95°C for 30 seconds, 50°C to 55°C for 30 seconds, and 72°C for 1 minute with a final extension step of 72°C for 5 minutes comprised the PCR profile. Products were separated by electrophoresis in denaturing 6% polyacrylamide gels followed by autoradiography as previously described. 18 For informative cases, allelic loss was scored if the radiographic signal of one allele was >50% reduced in the tumor DNA as compared with the corresponding normal allele.
FISH for Detection of CDKN2A Deletions
FISH was performed as previously described. 19 One section from each tissue block was stained with H&E to ensure the presence of at least 90% tumor cells and nuclei of formalin-fixed tissue blocks were dissociated and dropped onto slides. Two-color FISH was performed using a 180-kb Spectrum Orange-labeled 9p21 probe (Vysis, Downer’s Grove, IL), spanning the minimal homozygously deleted region that includes CDKN2A and excludes CDKN2B as described by Cairns and colleagues, 1 together with a corresponding Spectrum Green-labeled centromeric 9 α satellite probe (CEP 9, Vysis). Slide pretreatment, hybridization, and washing procedures were performed as previously described. 19 The hybridization mixture contained 3 μl of each of the probes and Cot-1 DNA (1 mg/ml). Slides were counterstained with 0.2 μmol/L 4,6-diamidino-2-phenylindole. At least 100 nuclei were selected for scoring using 4,6-diamidino-2-phenylindole staining. A loss of one CDKN2A allele was defined as presence of less 9p21 than centromere 9 signals in >40% of nuclei. A tumor was considered monosomic for chromosome 9 if >50% of the nuclei showed only one signal for (chromosome) 9p21 and CEP9. A homozygous deletion of 9p21 was considered, if >50% of the nuclei showed centromere 9 signals without 9p21 signals.
Sequence Analysis
Exon 1 and exon 2 of CDKN2A were amplified with two primer sets (Table 1) ▶ , each resulting in two overlapping fragments. PCR samples for the first PCR contained ∼100 ng of genomic DNA, 10 pmol of each primer, 1× Taq buffer (Qiagen), 200 μmol/L of each nucleotide (dATP, dCTP, dGTP, dTTP) and 1 U Taq polymerase (Qiagen) in a 20 μl total volume. PCR conditions for the step-down PCR were: 95°C for 3 minutes; 10 cycles of 95°C for 40 seconds, 62.5°C to 64.6°C for 40 seconds, 72°C for 1 minute; 25 cycles of 95°C for 40 seconds, 55.5°C to 57°C for 30 seconds, 72°C for 1 minute; 72°C for 5 minutes. PCR conditions for exon 3 were: 95°C for 3 minutes; 35 cycles of 95°C for 40 seconds, 55°C for 30 seconds, 72°C for 1 minute; 72°C for 5 minutes.
Table 1.
PCR Primers for CDKN2A Mutation Analysis
| First PCR | |
|---|---|
| Exon 1 | Forward 5′-CGG AGA GGG GGA GAG CAG-3′ |
| Reverse 5′-GAC CGT AAC TAT TCG GTG CGT T-3′ | |
| Forward 5′-CAG CAT GGA GCC TTC GGC TGA-3′ | |
| Reverse 5′-GCG CTA CCT GAT TCC AAT TC-3′ | |
| Exon 2 | Forward 5′-GCT CTA CAC AAG CTT CCT TTC C-3′ |
| Reverse 5′-CAG CTC CTC AGC CAG GTC C-3′ | |
| Forward 5′-CTG GAC ACG CTG GTG GTG CT-3′ | |
| Reverse 5′-GGG CTG AAC TTT CTG TGC TGG-3′ | |
| Exon 3 | Forward 5′-GTG CCA CAC ATC TTT GAC CTC A-3′ |
| Reverse 5′-CGG TGA CTG ATG ATC TAA GT-3′ | |
| Second PCR | |
| Exon 1 | Forward 5′-M13*-AGA GGG GGA GAG CAG GCA-3′ |
| Reverse 5′-CCA GCA GCG CCC GCA CCT C-3′ | |
| Forward 5′-GAG CCT TCG GCT GAC TGG CTG-3′ | |
| Reverse 5′-M13-CAA ACT TCG TCC TCC AGA GT-3′ | |
| Exon 2 | Forward 5′-M13-AAG CTT CCT TTC CGT CAT GCC-3′ |
| Reverse 5′-GCC AGG TCC ACG GGC AGA C-3′ | |
| Forward 5′-M13-GAC ACG CTG GTG GTG CTG CA-3′ | |
| Reverse 5′-TCT GAG CTT TGG AAG CTC TC-3′ | |
| Exon 3 | Forward 5′-GAC CTC AGG TTT CTA ACG CCT-3′ |
| Reverse 5′-CGG TGA CTG ATG ATC TAA GT-3′ |
*Primers were tailed with M13 universal (−21) and M13 reverse (−29), respectively.
The products of the first PCR were analyzed on a 1.2% agarose gel. One μl of the supernatant of excised, snap-frozen, and re-thawed bands were taken for the second PCR. The samples for the second PCR contained 10 pmol of each primer, 1× Taq buffer, 200 μmol/L of each nucleotide and 1 U of Taq polymerase (Qiagen). PCR conditions were: 95°C for 3 minutes, 30 cycles of 95°C for 30 seconds, 55°C to 65°C for 30 seconds, 72°C for 30 seconds; 72°C for 5 minutes.
IRD800-labeled primers (MWG-Biotech, Ebersberg, Germany) were used to direct cycle-sequencing of the PCR products. Cycle sequencing was done according to the protocol of the manufacturer’s instructions (MWG-Biotech). Sequence products were analyzed on a LICOR-DNA sequencer (Model 4000). Tumor DNA that showed a sequence change was subjected to a second PCR and sequence analysis. In those cases the matched normal DNA was also examined.
Immunohistochemistry for CDKN2A Expression on a Renal Tumor Tissue Microarray
To evaluate the prevalence of CDKN2A expression in renal tumors, a renal tumor microarray was used containing tumor specimens from 532 renal tumors and tissue from six normal kidneys. The construction of the tumor microarray was previously described. 20,21 There were 383 conventional (clear cell), 57 papillary, 23 chromophobe, three collecting duct RCCs, and 17 oncocytomas. The presence of tumor tissue on the arrayed samples was verified on one H&E-stained section. Sections (5-μm thick) were cut for immunohistochemistry. Standard indirect immunoperoxidase procedures were used for immunohistochemistry (ABC-Elite; Vector Laboratories, Burlingame, CA) on the tumor microarray as previously described. 21 A well-characterized antibody to CDKN2A 7 was used for detection of CDKN2A expression (1:25, Ab-1; Oncogene Research Products, Cambridge, MA) after pronase pretreatment. Tumors were considered positive if an unequivocal nuclear positivity was seen in tumor cells. Cases of prostate and bladder carcinoma with nuclear CDKN2A expression served as external positive control.
Statistics
Contingency table analysis was used to analyze the relationship between allelic deletion, grade, stage, and presence of metastasis. Overall survival rates were plotted using the Kaplan-Meier method. Statistical differences between tumors with and without 9p deletions were determined with the log-rank test. The proportional hazard model was used to test for independent prognostic information.
Results
Allelic Deletions
Eighty-eight normal/tumor pairs could be interpreted for LOH analysis on chromosome 9p. Twenty-one of 88 CRCCs (24%) showed allelic deletion with at least one microsatellite. Most deletions were detected at D9S171 on 9p13. Twelve of 59 informative tumors (20%) displayed allelic loss in this region. In contrast, only seven of 73 informative cases (10%) showed LOH at D9S1748 (9p21), eight of 68 (9%) demonstrated allelic deletion at D9S156 (9p21), and five of 68 (7%) displayed LOH at D9S970 (9p12-9p13). Allelic deletion at only one of the microsatellite loci examined were found in six tumors (10%) at D9S171, followed by three tumors at D9S1748 and D9S156 (4% each), respectively, and two tumors at D9S970 (Figure 1, A and B) ▶ .
Figure 1.
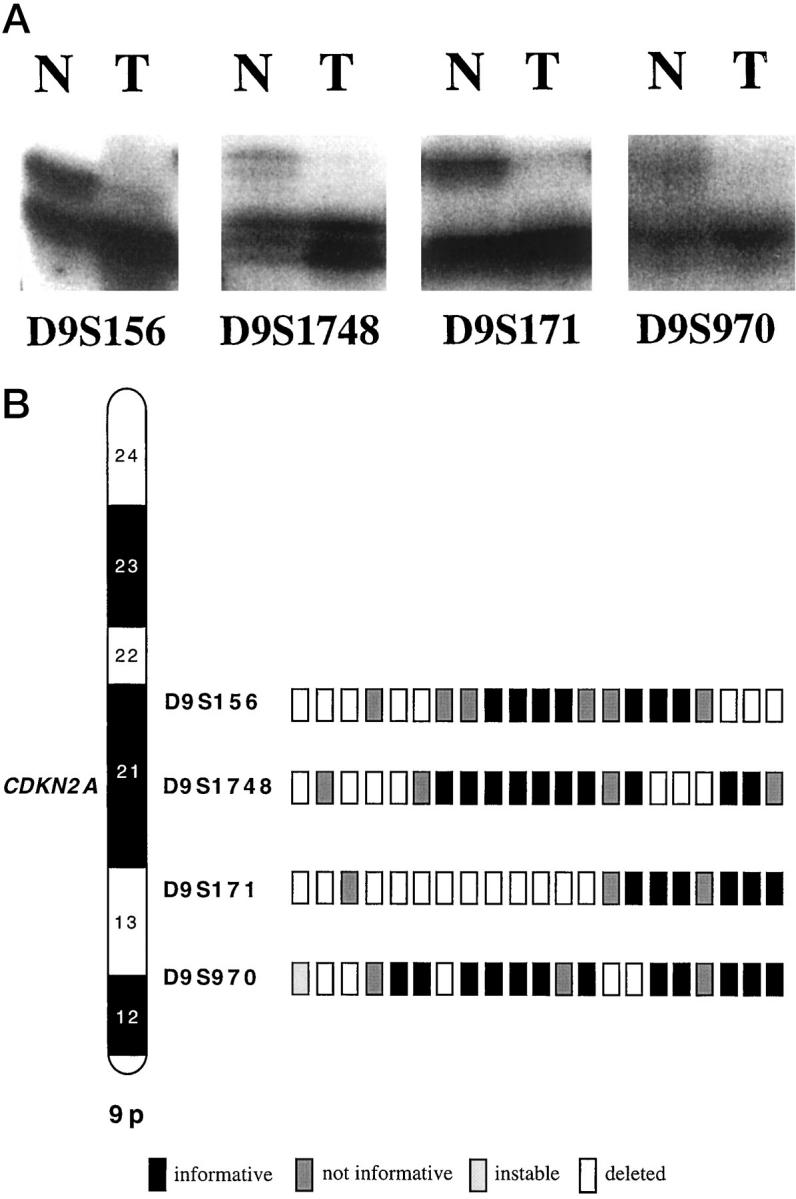
A: Four examples of renal carcinoma with LOH at microsatellites D9S156, D9S1748, D9S171, and D9S970. B: Allelic losses in a conventional (clear-cell) renal cell carcinoma at four 9p-specific microsatellite loci.
LOH at 9p was not associated with tumor stage, histological grade, and metastasis (Table 2) ▶ . Also, the separate analysis of the individual microsatellites did not reveal a relationship between LOH and morphological parameters. Tumor-specific survival data were available for 62 patients being informative for at least one 9p microsatellite. Fifteen of these patients showed LOH. Tumor-specific survival rates were only analyzed in stage pT3 RCCs, because there were too few tumor-related deaths in pT1/2 RCCs. In univariate survival analysis patient prognosis was associated with 9p deletion in stage pT3 RCCs. Tumor-specific 5-year survival was 58% for patients without 9p deletion (n = 30), whereas all patients with 9p deletions (n = 7) died of disease (P = 0.01, Figure 2 ▶ ). The analysis of pT3 CRCCs without metastasis at the time of nephrectomy also revealed a significant association between 9p deletions and poor prognosis (P = 0.02), although the number of tumors with deletions was small (n = 4) within this analysis. Cox proportional hazard analysis including the variables histological grade, presence of metastasis, and 9p deletion showed that LOH at 9p was not an independent predictor of poor prognosis in pT3 CRCCs (Table 3) ▶ .
Table 2.
LOH Analysis: 9p Deletion and Tumor Phenotype in Conventional (Clear-Cell) RCC
| Number of tumors (n) | 9p LOH n (%) | P value | |
|---|---|---|---|
| Grade | |||
| G1 | 9 | 3 (33) | |
| G2 | 54 | 10 (19) | 0.87 |
| G3 | 25 | 8 (32) | |
| pT Stage | |||
| pT1/2 | 41 | 12 (29) | |
| pT3/4 | 47 | 9 (19) | 0.27 |
| Metastasis | |||
| pN0, cM0 | 76 | 19 (25) | |
| pN1 or cM1 | 12 | 2 (17) | 0.53 |
Figure 2.

Tumor-specific survival rates for patients with and without 9p deletion.
Table 3.
LOH Analysis: Proportional Hazard Analysis for pT3 Conventional (Clear-Cell) Renal Carcinoma
| Variable | Relative risk | P value |
|---|---|---|
| 9p-deletion | 1.6 | 0.5 |
| Histologic grade | 2.7 | 0.06 |
| Metastasis | 4.4 | 0.01 |
CDKN2A Sequence Alterations
There was a 24-bp deletion within exon 1 in 13 of 113 tumors (12%) (Figure 3A) ▶ . One patient had this deletion also in the normal DNA. This in-frame mutation results in a truncation of 8 amino acids of CDKN2A. Because of the special feature of the sequence in the start coding region of CDKN2A there were 25 possible variants of deletions leading to the same sequence alteration (Figure 3B) ▶ . The 24-bp deletion was not associated with tumor stage or presence of metastases. Interestingly, the deletion was not detected in grade 1 RCCs, but in 9% of grade 2 and 22% of grade 3 tumors (Table 4) ▶ . This trend did not reach significance (P = 0.07). There was no relationship between the 24-bp deletions and tumor-specific survival (P = 0.15).
Figure 3.

A: A 24-bp deletion in the start coding region of CDKN2A. The sequence alteration is indicated by an arrow. B: Twenty-five possible variants leading to the same altered sequence are shown. Repeat 1 and repeat 2 are indicated by arrows.
Table 4.
Sequence Analysis: 24-bp Deletion of Exon 1 and Tumor Phenotype
| Number of tumors (n) | 24-bp Deletion n (%) | P value | |
|---|---|---|---|
| pT Stage | |||
| pT1/2 | 48 | 6 (12%) | |
| pT3/4 | 65 | 7 (11%) | 0.7 |
| Histologic grade | |||
| G1 | 11 | 0 | |
| G2 | 70 | 6 (9%) | 0.07 |
| G3 | 32 | 7 (22%) | |
| Metastasis | |||
| pN0, cM0 | 90 | 10 (11%) | |
| pN1 or cM1 | 23 | 3 (13%) | 0.8 |
A G to A transition was detected in codon 140 of exon 2 in one patient. The mutation causes an amino acid exchange from alanine to threonine. This mutation was also detected in the normal DNA of the patient and possibly represents a polymorphism.
One hundred eight of 113 CRCCs (96%) showed a G to C transversion in the noncoding sequence of exon 3 that was also found in the matched normal DNA of each tumor. Eighty-nine of these tumors were homozygous and 19 were heterozygous for this transversion. There were only five tumors showing the wild-type sequence of the gene data base. 22 Interestingly, the homozygous C/C genotype was associated with a low grade of differentiation (P < 0.05) (Table 5) ▶ . To further evaluate the frequency of the C haplotype in the normal population, we analyzed 48 blood samples of healthy donors. The analysis revealed a C-allele frequency of 90% of the individuals tested.
Table 5.
Sequence Analysis: Homozygous Transversion in Exon 3 of p16 and Tumor Phenotype
| Number of tumors* (n) | Homozygous transversion n (%) | P value | |
|---|---|---|---|
| pT-Stage | |||
| pT1/2 | 46 | 37 (77%) | |
| pT3/4 | 62 | 52 (81%) | 0.6 |
| Histologic grade | |||
| G1 | 11 | 6 (55%) | |
| G2 | 67 | 56 (84%) | 0.03 |
| G3 | 30 | 27 (90%) | |
| Metastasis | |||
| pN0, cM0 | 85 | 68 (80%) | |
| pN1 or cM1 | 23 | 17 (91%) | 0.2 |
*Five tumors were excluded from analysis: 1 tumor was not interpretable, 4 tumors showed the wild type of exon 3.
CDKN2A Deletion by FISH Hybridization
High-quality hybridization signals for both centromeric and gene-specific probes were obtained in 54 tumors. Four of 54 CRCCs (7%) showed physical 9p21 deletions, but none of the tumors had a homozygous CDKN2A deletion according to our definition. There were two additional tumors with chromosome 9 monosomy.
CDKN2A Expression Tested on a Renal Tumor Tissue Microarray
To determine the frequency and potential implications of CDKN2A expression in RCC, we analyzed a cohort of 532 renal tumors on a tumor microarray. We observed six CRCCs with nuclear CDKN2A expression. These tumors showed neither CDKN2A mutations nor D9S1748 deletions. Papillary (n = 57), chromophobe (n = 23), and collecting duct carcinomas (n = 3) as well as oncocytoma (n = 17) did not show CDKN2A expression. Prostate and bladder carcinomas served as external positive controls. In these controls, a strong nuclear staining was detectable. Representative tumors are shown in Figure 4, A and B ▶ .
Figure 4.
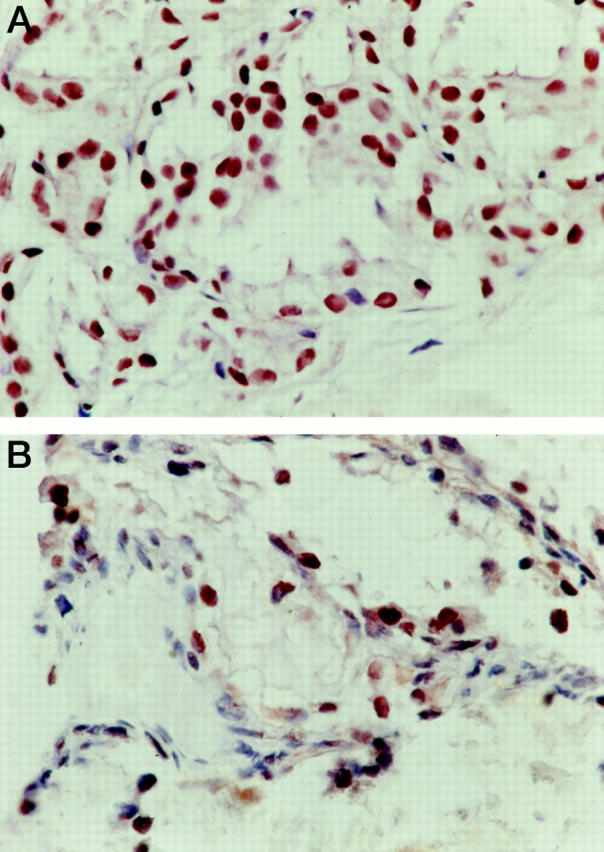
A: Immunohistochemical detection of CDKN2A expression in a case of prostate carcinoma (control case). B: Expression of CDKN2A in a conventional (clear-cell) renal carcinoma on the tumor array.
Discussion
We demonstrated an association between LOH on chromosome 9p with short patient survival. Chromosome 9p deletions were detected in 24% of CRCCs using four highly polymorphic microsatellite markers. A slightly higher rate of chromosome 9p deletions was reported by Cairns and colleagues, 1 who detected deletions in 14 of 42 primary CRCCs (33%). Other groups reported 9p LOH in 16 to 33% of CRCCs. 2,23-25 The significant difference in tumor-specific survival between tumors with and without 9p deletion suggests that a tumor suppressor gene on 9p is involved in tumor progression. In lung, bladder, head and neck cancer, and melanoma, 12,26 allele losses and homozygous deletions were most frequently observed at 9p21.
Allelic imbalance has generally been considered to represent loss of genetic material, but the use of PCR-based techniques makes it difficult to differentiate allele loss from allele gain. Although it cannot be excluded that some of our tumors had gains we think that the vast majority of our tumors analyzed showed LOH. In approximately half of the cases signal reduction of one allele was near absolute in the tumors. Only four of 24 tumors (17%) with LOH displayed a 50% reduction of one allele. In addition, our FISH results coincide with previous cytogenetic data in that regional or entire gains of chromosome 9 are rare events in kidney tumors. 27 Hence, signal reduction of one allele very likely reflects loss rather than gain of genetic material on chromosome 9.
CDKN2A has been implicated as a potential target for the deletion on 9p in a variety of neoplasms including RCCs, because CDKN2A was found inactivated by homozygous deletion and hypermethylation of the promoter region. 9,10 In our analysis, only seven of 73 CRCCs (10%) demonstrated allelic deletion at D9S1748 locus that lies within CDKN2A. A low frequency of CDKN2A deletions was also corroborated by our FISH analysis showing CDKN2A deletion in four of 54 CRCCs (7%). The absence of homozygous deletions by FISH is consistent with previous findings of Cairns and colleagues, 1 who detected inactivation of CDKN2A by homozygous deletion only in one of 42 primary RCCs. Although our FISH probe spans the minimal homozygously deleted region at CDKN2A described by Cairns and colleagues, 1 we cannot exclude the presence of homozygous deletions <180 kb. However, the large proportion of homozygous CDKN2A deletions in RCC cell lines described by Kinoshita and colleagues 4 may rather occur as result of a selective overgrowth of tumor cells with CDKN2A homozygous losses in tissue cultures.
The most common mechanism for CDKN2A inactivation in human cancer cell lines is through homozygous deletion, whereas intragenic mutations occur in a smaller proportion of tumors and cell lines. 9,11 Cairns and colleagues 11 were not able to detect point mutations of CDKN2A in 42 RCCs with chromosome 9p losses and argued that CDKN2A is not the primary target of the 9p21 loss. Importantly, we detected three different CDKN2A sequence alterations, one in exon 1, one in exon 2, and an additional alteration in the 3′-untranslated region of exon 3. Thirteen of 113 CRCCs (12%) showed a 24-bp deletion in exon 1, causing the loss of amino acid residues 4 to 11.
The same mutation was also detected at a lower rate in melanoma and prostate cancer. Kumar and colleagues 28 found a 24-bp deletion in two of 31 (6%) sporadic melanomas and Komiya and colleagues 29 reported a 24-bp deletion in one of 51 (2%) prostate cancers. Because of the special sequence structure of the two repeat units located at the 5′ end of exon 1, there are 25 possible variants leading to the same sequence alteration (Figure 3B) ▶ . The relevance of these mutations for gene expression or protein function of CDKN2A is unclear. This repeat region seems to be unstable because duplications of this 24-bp repeat were found in familial melanomas. 30-32 Based on the functional studies of CDKN2A, this mutant form was functionally normal with respect to CDK4 binding. 33 As both 24-bp insertion and deletion lie outside the ankyrin-repeat region of CDKN2A, these mutations may not have any negative effects on the interaction between CDKN2A and CDK4. Whether the 24-bp deletion in CRCC weakens CDKN2A expression in tumors remains to be determined. The association of these mutations with higher grades raises the possibility that this mutation is associated with progression or genetic instability of RCC.
The ratio of wild-type and mutated CDKN2A alleles was approximately equal in the majority of the tumors suggesting a mechanism that leads to a gain of heterozygosity. As some tumors showed less amounts of the mutant allele the existence of a small subpopulation of heterozygous or homozygous tumor cells cannot be excluded. Therefore, the observed mutation rate in our tumors might be a slight underestimation.
The G to A transition in codon 140 of exon 2 is likely to be a genetic polymorphism rather than a somatic mutation. This polymorphism has been described in many primary tumors. 34-36 The transition results in an amino acid exchange from alanine to threonine. This point mutation may not influence CDKN2A function, because the altered protein inhibits the CCND1-CDK4/6 complex in the late G1 phase of the cell cycle similarly to the wild-type protein. 37
The G to C transversion in the noncoding sequence (3′-UTR) of exon 3 (nucleotide 540) represents a known polymorphism, which was present in tumor and normal tissue of 108 patients. The same transversion was found by Ueki and colleagues 38 in eight of 30 primary astrocytomas and in 11 of 50 CEPH DNAs. The transversion was heterozygous in all these cases. This represents a G-allele frequency of 0.12 and a C-allele frequency of 0.88. Kumar and colleagues 28 described a G-allele frequency of 0.18 and a C-allele frequency of 0.82 in sporadic melanoma. In our study, 96% of CRCCs showed a heterozygous polymorphism, the C-allele frequency was 0.88 and the G-allele frequency was 0.12. The identical G and C allele frequencies in DNA extracted from blood of 48 healthy donors argues against a role of this polymorph-ism for a CRCC disposition.
Methylation of the promoter region is another mechanism for CDKN2A inactivation in cell lines and primary tumors. 10 Merlo and colleagues 39 demonstrated methylation in one RCC cell line, suggesting that inactivation because of hypermethylation may play a role in RCC. However, a previous analysis by Clifford and colleagues 40 detecting CDKN2A hypermethylation only in one of 44 primary RCCs (2%) suggested that this mechanism may not be relevant for CDKN2A inactivation in primary CRCC.
Mutations affecting CDKN2A, homozygous deletions, or methylation of the promoter region might result in a lack of CDKN2A expression. In this analysis, none of the normal kidney tissues and only six CRCCs in the renal tumor microarray displayed detectable levels of CDKN2A. It has been reported that CDKN2A expression is low or even undetectable in most normal human tissues that reflects the basal physiological level of CDKN2A. 41,42 Therefore, the negative phenotypes of CRCCs found in this study may rather reflect the normal physiological state than a reduced expression. Frequent expression of CDKN2A can be seen in prostate, 43 lung, 44 ovarian, 45 and breast carcinoma. 46 CDKN2A expression has been associated with tumor progression and poor prognosis in ovarian 47 and breast cancer 46 and a higher likelihood of tumor recurrence in prostate cancer. 43 Up-regulation of CDKN2A, resulting in expression of CDKN2A, may develop through different mechanisms. Cellular stress, 48 hyperthermia, 49 and UV irradiation 50 have been reported to trigger CDKN2A expression. Some data suggest an association between CDKN2A and retinoblastoma protein (RB1) expression, in which the absence of functional RB1 limits CDKN2A activity. Indirect mechanisms for CDKN2A expression might be because of E2F1 or CCND1 expression. 7
In summary, CDKN2A alterations (mutations and/or deletions) are present in a subgroup of primary CRCCs. Importantly, LOH was more frequent at D9S171, which was initially mapped to 9p21 (Genome Database; Gene Map ‘96). Subsequent mapping has placed this marker to 9p13 ∼10 cM centromeric to CDKN2A (GeneMap’99). The significantly lower rates of allelic losses found centromeric (D9S970) and telomeric (D9S1748 and D9S156) to D9S171 strongly support the existence of an unknown tumor suppressor gene proximal to the CDKN2A locus. Such a tumor suppressor gene might also be involved in other tumor types because preferential allelic loss at D9S171 has been observed in lung, 51 head and neck, 52 brain, 53 breast, 54 and esophagus carcinomas, 55 as well as in melanoma, 56 mesothelioma, 57 neuroblastoma, 58,59 pituitary adenoma, 60 and childhood acute lymphoblastic leukemia. 61
Acknowledgments
We thank C. Egenter, M. Mirlacher, H. Novotny, H. Oggier, A. Stalder, M. Storz, and the staff of the Institute for Pathology, University Basel, for their technical support.
Footnotes
Address reprint requests to Holger Moch, M.D., Institute of Pathology, University Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland. E-mail: hmoch@uhbs.ch.
Supported by Swiss National Science Foundation (31-50752.97).
References
- 1.Cairns P, Tokino K, Eby Y, Sidransky D: Localization of tumor suppressor loci on chromosome 9 in primary human renal cell carcinomas. Cancer Res 1995, 55:224-227 [PubMed] [Google Scholar]
- 2.Thrash-Bingham CA, Greenberg RE, Howard S, Bruzel A, Bremer M, Goll A, Salazar H, Freed JJ, Tartof KD: Comprehensive allelotyping of human renal cell carcinomas using microsatellite DNA probes. Proc Natl Acad Sci USA 1995, 92:2854-2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moch H, Presti JC, Jr, Sauter G, Buchholz N, Jordan P, Mihatsch MJ, Waldman FM: Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res 1996, 56:27-30 [PubMed] [Google Scholar]
- 4.Kinoshita H, Yamada H, Ogawa O, Kakehi Y, Osaka M, Nakamura E, Mishina M, Habuchi T, Takahashi R, Sugiyama T: Contribution of chromosome 9p21-22 deletion to the progression of human renal cell carcinoma. Jpn J Cancer Res 1995, 86:795-799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bissig H, Richter J, Desper R, Meier V, Schraml P, Schäffer A, Sauter G, Mihatsch M, Moch H: Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am J Pathol 1999, 155:267-274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foulkes WD, Flanders TY, Pollock PM, Hayward NK: The CDKN2A (p16) gene and human cancer. Mol Med 1997, 3:5-20 [PMC free article] [PubMed] [Google Scholar]
- 7.Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J: Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 1995, 375:503-506 [DOI] [PubMed] [Google Scholar]
- 8.Cairns P, Tokino K, Eby Y, Sidransky D: Homozygous deletions of 9p21 in primary human bladder tumors detected by comparative multiplex polymerase chain reaction. Cancer Res 1994, 54:1422-1424 [PubMed] [Google Scholar]
- 9.Kamb A, Liu Q, Harshman K, Tavtigian S, Cordon-Cardo C, Skolnick M: Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science 1994, 264:416-417 [Google Scholar]
- 10.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB: Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 55:4525-4530 [PubMed] [Google Scholar]
- 11.Cairns P, Mao L, Merlo A, Lee DJ, Schwab D, Eby Y, Tokino K, van der Riet P, Blaugrund JE, Sidransky D: Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science 1994, 265:415-417 [DOI] [PubMed] [Google Scholar]
- 12.Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabrielson E, Tockman M, Cho KR, Hedrick L, Bova GS, Isaacs W, Koch W, Schwab D, Sidransky D: Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 1995, 11:210-212 [DOI] [PubMed] [Google Scholar]
- 13.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, III, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 14.Kovacs G, Akhtar M, Beckwith B, Bugert B, Cooper C, Delahunt B, Eble J, Fleming S, Ljungberg B, Medeiros L, Moch H, Reuter V, Ritz E, Roos G, Schmidt D, Srigley J, Störkel S, Van den Berg E, Zbar B: The Heidelberg classification of renal cell tumours. J Pathol 1997, 183:131-133 [DOI] [PubMed] [Google Scholar]
- 15.Thoenes W, Stoerkel S, Rumpelt H: Histopathology and classification of renal cell tumors (adenomas, oncocytomas, and carcinomas): the basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract 1986, 181:125-143 [DOI] [PubMed] [Google Scholar]
- 16.: UICC: TNM Classification of Malignant Tumours, ed 5 1997, C Wittekind. New York, Chichester, Weinheim, Brisbane, Singapore, Toronto, Wiley-Liss, Edited by L Sobin
- 17.Mao L, Merlo A, Bedi G, Shapiro GI, Edwards CD, Rollins BJ, Sidransky D: A novel p16INK4A transcript. Cancer Res 1995, 55:2995-2997 [PubMed] [Google Scholar]
- 18.Schraml P, Muller D, Bednar R, Gasser T, Sauter G, Mihatsch MJ, Moch H: Allelic loss at the D9S171 locus on chromosome 9p13 is associated with progression of papillary renal cell carcinoma. J Pathol 2000, 190:457-461 [DOI] [PubMed] [Google Scholar]
- 19.Sauter G, Moch H, Carroll P, Kerschmann R, Mihatsch M, Waldman F: Chromosome-9 loss detected by fluorescence in situ hybridization in bladder cancer. Int J Cancer 1995, 64:99-103 [DOI] [PubMed] [Google Scholar]
- 20.Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP: Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998, 4:844-847 [DOI] [PubMed] [Google Scholar]
- 21.Moch H, Schraml P, Bubendorf L, Mirlacher M, Kononen J, Gasser T, Mihatsch MJ, Kallioniemi OP, Sauter G: High-throughput tissue microarray analysis to evaluate genes uncovered by cDNA microarray screening in renal cell carcinoma. Am J Pathol 1999, 154:981-986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serrano M, Hannon GJ, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366:704-707 [DOI] [PubMed] [Google Scholar]
- 23.Bugert P, Kovacs G: Molecular differential diagnosis of renal cell carcinomas by microsatellite analysis. Am J Pathol 1996, 149:2081-2088 [PMC free article] [PubMed] [Google Scholar]
- 24.Thrash-Bingham CA, Salazar H, Freed JJ, Greenberg RE, Tartof KD: Genomic alterations and instabilities in renal cell carcinomas and their relationship to tumor pathology. Cancer Res 1995, 55:6189-6195 [PubMed] [Google Scholar]
- 25.Schullerus D, Herbers J, Chudek J, Kanamaru H, Kovacs G: Loss of heterozygosity at chromosomes 8p, 9p, and 14q is associated with stage and grade of non-papillary renal cell carcinomas. J Pathol 1997, 183:151-155 [DOI] [PubMed] [Google Scholar]
- 26.Fountain JW, Karayiorgou M, Ernstoff MS, Kirkwood JM, Vlock DR, Titus-Ernstoff L, Bouchard B, Vijayasaradhi S, Houghton AN, Lahti J, Kidd VJ, Housman DE, Dracapoli NC: Homozygous deletions within human chromosome band 9p21 in melanoma. Proc Natl Acad Sci USA 1992, 89:10557-10561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitelman F: Catalog of Chromosome Aberrations in Cancer, ed 5 1994, Chichester, Brisbane, Toronto, Singapore, Wiley-Liss, New York
- 28.Kumar R, Lundh Rozell B, Louhelainen J, Hemminki K: Mutations in the CDKN2A (p16INK4a) gene in microdissected sporadic primary melanomas. Int J Cancer 1998, 75:193-198 [DOI] [PubMed] [Google Scholar]
- 29.Komiya A, Suzuki H, Aida S, Yatani R, Shimazaki J: Mutational analysis of CDKN2 (CDK4I/MTS1) gene in tissues and cell lines of human prostate cancer. Jpn J Cancer Res 1995, 86:622-625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pollock PM, Spurr N, Bishop T, Newton-Bishop J, Gruis N, van der Velden PA, Goldstein AM, Tucker MA, Foulkes WD, Barnhill R, Haber D, Fountain J, Hayward NK: Haplotype analysis of two recurrent CDKN2A mutations in 10 melanoma families: evidence for common founders and independent mutations. Hum Mutat 1998, 11:424-431 [DOI] [PubMed] [Google Scholar]
- 31.Flores JF, Pollock PM, Walker GJ, Glendening JM, Lin AH, Palmer JM, Walters MK, Hayward NK, Fountain JW: Analysis of the CDKN2A, CDKN2B and CDK4 genes in 48 Australian melanoma kindreds. Oncogene 1997, 15:2999-3005 [DOI] [PubMed] [Google Scholar]
- 32.Walker GJ, Gabrielli BG, Castellano M, Hayward NK: Functional reassessment of P16 variants using a transfection-based assay. Int J Cancer 1999, 82:305-312 [DOI] [PubMed] [Google Scholar]
- 33.Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, McLaughlin J, Hogg D, Lassam NJ: CDKN2A mutations in multiple primary melanomas. N Engl J Med 1998, 338:879-887 [DOI] [PubMed] [Google Scholar]
- 34.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Jr, Tucker MA, Dracopoli NC: Germline p16 mutations in familial melanoma. Nat Genet 1994, 8:15-21 [DOI] [PubMed] [Google Scholar]
- 35.Wagner SN, Wagner C, Briedigkeit L, Goos M: Homozygous deletion of the p16INK4a and the p15INK4b tumour suppressor genes in a subset of human sporadic cutaneous malignant melanoma. Br J Dermatol 1998, 138:13-21 [DOI] [PubMed] [Google Scholar]
- 36.Heidenreich A, Gaddipati JP, Moul JW, Srivastava S: Molecular analysis of P16(Ink4)/CDKN2 and P15(INK4B)/MTS2 genes in primary human testicular germ cell tumors. J Urol 1998, 159:1725-1730 [DOI] [PubMed] [Google Scholar]
- 37.Ranade K, Hussussian CJ, Sikorski RS, Varmus HE, Goldstein AM, Tucker MA, Serrano M, Hannon GJ, Beach D, Dracopoli NC: Mutations associated with familial melanoma impair p16INK4 function. Nat Genet 1995, 10:114-116 [DOI] [PubMed] [Google Scholar]
- 38.Ueki K, Rubio MP, Ramesh V, Correa KM, Rutter JL, von Deimling A, Buckler AJ, Gusella JF, Louis DN: MTS1/CDKN2 gene mutations are rare in primary human astrocytomas with allelic loss of chromosome 9p. Hum Mol Genet 1994, 3:1841-1845 [DOI] [PubMed] [Google Scholar]
- 39.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D: 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995, 1:686-692 [DOI] [PubMed] [Google Scholar]
- 40.Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER: Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosom Cancer 1998, 22:200-209 [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA: Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995, 55:4531-4535 [PubMed] [Google Scholar]
- 42.Yao J, Pollock RE, Lang A, Tan M, Pisters PW, Goodrich D, El-Naggar A, Yu D: Infrequent mutation of the p16/MTS1 gene and overexpression of cyclin-dependent kinase 4 in human primary soft-tissue sarcoma. Clin Cancer Res 1998, 4:1065-1070 [PubMed] [Google Scholar]
- 43.Lee CT, Capodieci P, Osman I, Fazzari M, Ferrara J, Scher HI, Cordon-Cardo C: Overexpression of the cyclin-dependent kinase inhibitor p16 is associated with tumor recurrence in human prostate cancer. Clin Cancer Res 1999, 5:977-983 [PubMed] [Google Scholar]
- 44.Betticher DC, White GR, Vonlanthen S, Liu X, Kappeler A, Altermatt HJ, Thatcher N, Heighway J: G1 control gene status is frequently altered in resectable non-small cell lung cancer. Int J Cancer 1997, 74:556-562 [DOI] [PubMed] [Google Scholar]
- 45.Fujita M, Enomoto T, Haba T, Nakashima R, Sasaki M, Yoshino K, Wada H, Buzard GS, Matsuzaki N, Wakasa K, Murata Y: Alteration of p16 and p15 genes in common epithelial ovarian tumors. Int J Cancer 1997, 74:148-155 [DOI] [PubMed] [Google Scholar]
- 46.Dublin EA, Patel NK, Gillett CE, Smith P, Peters G, Barnes DM: Retinoblastoma and p16 proteins in mammary carcinoma: their relationship to cyclin D1 and histopathological parameters. Int J Cancer 1998, 79:71-75 [DOI] [PubMed] [Google Scholar]
- 47.Dong Y, Walsh MD, McGuckin MA, Gabrielli BG, Cummings MC, Wright RG, Hurst T, Khoo SK, Parsons PG: Increased expression of cyclin-dependent kinase inhibitor 2 (CDKN2A) gene product P16INK4A in ovarian cancer is associated with progression and unfavourable prognosis. Int J Cancer 1997, 74:57-63 [DOI] [PubMed] [Google Scholar]
- 48.Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G: Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 1996, 16:859-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valenzuela MT, Nunez MI, Villalobos M, Siles E, McMillan TJ, Pedraza V, Ruiz de Almodovar JM: A comparison of p53 and p16 expression in human tumor cells treated with hyperthermia or ionizing radiation. Int J Cancer 1997, 72:307-312 [DOI] [PubMed] [Google Scholar]
- 50.Wang XQ, Gabrielli BG, Milligan A, Dickinson JL, Antalis TM, Ellem KA: Accumulation of p16CDKN2A in response to ultraviolet irradiation correlates with late S-G(2)-phase cell cycle delay. Cancer Res 1996, 56:2510-2514 [PubMed] [Google Scholar]
- 51.Merlo A, Gabrielson E, Askin F, Sidransky D: Frequent loss of chromosome 9 in human primary non-small cell lung cancer. Cancer Res 1994, 54:640-642 [PubMed] [Google Scholar]
- 52.Lydiatt WM, Davidson BJ, Schantz SP, Caruana S, Chaganti RS: 9p21 deletion correlates with recurrence in head and neck cancer. Head Neck 1998, 20:113-118 [DOI] [PubMed] [Google Scholar]
- 53.Maruno M, Yoshimine T, Muhammad AK, Tokiyoshi K, Hayakawa T: Loss of heterozygosity of microsatellite loci on chromosome 9p in astrocytic tumors and its prognostic implications. J Neurooncol 1996, 30:19-24 [DOI] [PubMed] [Google Scholar]
- 54.An HX, Niederacher D, Picard F, van Roeyen C, Bender HG, Beckmann MW: Frequent allele loss on 9p21-22 defines a smallest common region in the vicinity of the CDKN2 gene in sporadic breast cancer. Genes Chromosom Cancer 1996, 17:14-20 [DOI] [PubMed] [Google Scholar]
- 55.Tarmin L, Yin J, Zhou X, Suzuki H, Jiang HY, Rhyu MG, Abraham JM, Krasna MJ, Cottrell J, Meltzer SJ: Frequent loss of heterozygosity on chromosome 9 in adenocarcinoma and squamous cell carcinoma of the esophagus. Cancer Res 1994, 54:6094-6096 [PubMed] [Google Scholar]
- 56.Ohta M, Berd D, Shimizu M, Nagai H, Cotticelli MG, Mastrangelo M, Shields JA, Shields CL, Croce CM, Huebner K: Deletion mapping of chromosome region 9p21–p22 surrounding the CDKN2 locus in melanoma. Int J Cancer 1996, 65:762-767 [DOI] [PubMed] [Google Scholar]
- 57.Cheng JQ, Jhanwar SC, Lu YY, Testa JR: Homozygous deletions within 9p21–p22 identify a small critical region of chromosomal loss in human malignant mesotheliomas. Cancer Res 1993, 53:4761-4763 [PubMed] [Google Scholar]
- 58.Marshall B, Isidro G, Martins AG, Boavida MG: Loss of heterozygosity at chromosome 9p21 in primary neuroblastomas: evidence for two deleted regions. Cancer Genet Cytogenet 1997, 96:134-139 [DOI] [PubMed] [Google Scholar]
- 59.Takita J, Hayashi Y, Kohno T, Yamaguchi N, Hanada R, Yamamoto K, Yokota J: Deletion map of chromosome 9 and p16 (CDKN2A) gene alterations in neuroblastoma. Cancer Res 1997, 57:907-912 [PubMed] [Google Scholar]
- 60.Farrell WE, Simpson DJ, Bicknell JE, Talbot AJ, Bates AS, Clayton RN: Chromosome 9p deletions in invasive and noninvasive nonfunctional pituitary adenomas: the deleted region involves markers outside of the MTS1 and MTS2 genes. Cancer Res 1997, 57:2703-2709 [PubMed] [Google Scholar]
- 61.Takeuchi S, Koike M, Seriu T, Bartram CR, Slater J, Park S, Miyoshi I, Koeffler HP: Homozygous deletions at 9p21 in childhood acute lymphoblastic leukemia detected by microsatellite analysis. Leukemia 1997, 11:1636-1640 [DOI] [PubMed] [Google Scholar]