

Abstract
Activated protein C functions directly as an anticoagulant and indirectly as a profibrinolytic enzyme. To determine whether the fibrin deposition previously observed in PC−/− murine embryos and neonates was mediated through the FXI pathway, PC+/−/FXI−/− mice were generated and crossbred to produce double-deficient progeny (PC−/−/FXI−/−). PC−/−/FXI−/− mice survived the early lethality observed in the PC−/−/FXI+/+ neonates, with the oldest PC−/−/FXI−/− animal living to 3 months of age. However, the majority of these animals was sedentary and significantly growth-retarded. On sacrifice or natural death, all of these PC−/−/FXI−/− mice demonstrated massive systemic fibrin deposition with concomitant hemorrhage and fibrosis, as confirmed through histological analyses. Several of these animals also presented with enlarged lymph nodes and extensive lymphatic fluid in the thoracic cavity. Thus, although a number of the PC−/−/FXI−/− mice survived the lethal perinatal coagulopathy seen in the PC−/− neonates, they nonetheless succumbed to overwhelming thrombotic disease in later life. This combined deficiency state provided the first clear indication that the course of a severe thrombotic disorder could be manipulated by blocking the intrinsic pathway and provided the first opportunity to study a total protein C deficiency in an adult animal.
Blood clotting at a site of vascular injury is initiated by formation of the Factor (F) VIIa/tissue factor complex, which catalyzes the activation of the zymogens, FIX and FX, to the serine proteases, FIXa and FXa, respectively. In subsequent steps, thrombin is generated, and this leads to fibrin deposition at the injured site. Although this mechanism rapidly generates FXa, it also allows for inhibition of the FVIIa/tissue factor complex by tissue factor pathway inhibitor in a FXa-dependent manner. 1 Thus, the continued ability of blood to clot requires another means of generation of FXa and thrombin. This is likely provided by the intrinsic system that functions through the FXIa-catalyzed formation of FIXa. 2 This latter enzyme serves as a FX activator, with collaboration of the cofactor, FVIIIa, along with Ca2+, on an acidic phospholipid membrane surface.
Other inhibitors regulate enzymes that are needed for coagulation. For example, the serpin, antithrombin III, is one agent that serves this role by direct inhibition of FXa and thrombin. Activated protein C (aPC) is an anticoagulant that functions in a different manner, namely by specific proteolytic inactivation of FV/FVa 3 and FVIII/FVIIIa. 4 aPC also displays profibrinolytic effects directly by inactivation of fibrinolytic inhibitors, such as plasminogen activator inhibitor-1 (PAI-1), 5 and indirectly through attenuation of thrombin formation, which is needed for activation of the zymogen, thrombin-activatable-fibrinolytic-inhibitor. 6,7 aPC has also been implicated in host-defense reactions that occur during intravascular inflammation. 8
Although a targeted deficiency of PC in mice led to early neonatal death, 9 a similar total deficiency of FXI did not result in severe consequences in the unchallenged animal. 10 Because of the nature of these phenotypes, an opportunity was present to examine the effect of an additional FXI deficiency on the phenotypes of PC-deficient animals. The issue to be addressed was the extent to which the FXI-dependent pathway contributed to the fibrin deposition observed in PC−/− mice. Thus, creation of PC/FXI double-deficient animals was seen as a means to address this question. This report is a summary of the phenotypic characteristics of these animals.
Materials and Methods
Animals
The generation of mice heterozygous for a null PC allele 9 and homozygous for the FXI null allele 10 has been described. Briefly, targeted replacement of the PC allele resulted in the deletion of all translatable exons, via substitution of exons 2 to 9 of the PC gene with a neomycin phosphotransferase gene (NEO). The FXI gene mutation resulted in truncation of the protein at exon 5, via disruption of the exon with a NEO cassette. Both transgenic lines were back-crossed to the F4 generation in C57BL/6J mice (ca., 94% C57BL/6J background). Mice heterozygous for PC and homozygous deficient for FXI (PC+/−/FXI−/−) were generated by cross-mating these F4 generation PC+/− and FXI−/− mice. The resulting mice were then subsequently mated to generate PC−/−/FXI−/− progeny. The animals were housed in microisolation cages on a constant 12-hour light/dark cycle with controlled temperature and humidity and given access to food and water ad libitum.
Genotypic Analyses
Ear punches, tails, or portions of embryonic tissue or yolk sac were isolated for the extraction of genomic DNA as previously described. 11 PC genotyping was performed by polymerase chain reaction in a 50-μl reaction using three different primers, 1) a common primer (CGT GAT GAG TTT CAG GCA GTG AGA G) for both the wild-type (WT) and null alleles; 2) a specific primer (GAG ATA AGC AGA TCC TGT GGA TTG C) for the WT allele situated in the 5′ flank of the PC gene; and 3) a primer (ATT CGC AGC GCA TCG CCT TCT ATC) included in the NEO cassette. Amplicons of 1,000 bp and 600 bp were obtained for the WT and null alleles, respectively. Genotyping for FXI was also performed by polymerase chain reaction using three different primers, 1) an oligonucleotide (TTG CAG CAA AGA TGA GTA CGT GAA C), located at the 5′ end of exon 5, served as a common primer for both the NEO and WT alleles; 2) a primer located in the NEO gene, which was the same as that used for PC genotyping; and 3) a primer (ATG GTC GAC ACT GGG AAA ATA CCC) located at the 3′ end of exon 5, which was used to amplify the WT allele. Amplicons of 160 bp and 400 bp were obtained for the WT and null alleles, respectively.
Timed Matings
Females were placed with nonsibling males in the late afternoon and the morning of vaginal plug detection was assigned E0.5dpc. Pregnant females were anesthetized with rodent cocktail (9 mg/ml ketamine, 1.8 mg/ml xylazine, 0.3 mg/ml acepromazine in saline), and the embryos harvested at E17.5dpc.
Necropsy
Embryos were dissected free of maternal tissue and their organs isolated and fixed in 10% neutral-buffered formalin overnight. Tails were excised for genotypic analysis. Animals that were sacrificed postnatally were first anesthetized with rodent cocktail. Blood was collected through vena cava puncture into 3.8% citrate at a ratio of 9:1 citrate. For histological analysis, animals were perfused through the left ventricle with saline followed by 10% neutral-buffered formalin. Lungs were additionally inflated with 10% neutral-buffered formalin through the trachea. Organs were then removed and fixed for an additional 3 hours in 10% neutral-buffered formalin.
Histology, Histochemistry, and Immunohistochemistry
After fixation, samples were dehydrated, embedded in paraffin, and sectioned at 2 to 3 μm for hematoxylin and eosin (H&E) staining and 4 μm for all other analyses. Sections were stained with H&E to examine general tissue and cellular morphology, and with Masson’s trichrome for identification of collagen. 12 Iron was detected using a periodic acid-Schiff/Mallory method (PAS/Fe). 13,14 Blood and thoracic fluid smears were stained with Giemsa solution. 15
Fibrin was identified immunohistochemically with a polyclonal goat anti-mouse fibrin(ogen) antibody (Accurate Chemicals, Westbury, NY). Antigen retrieval was performed under high temperature and pressure with citrate buffer (Biogenex, San Ramon, CA), followed by endogenous peroxidase blocking with Peroxoblock (Zymed, South San Francisco, CA). Subsequent to incubation, first with rabbit serum and then with the primary antibody, slides were incubated with the secondary rabbit anti-goat IgG (DAKO, Carpinteria, CA), followed by goat peroxidase anti-peroxidase (DAKO). Peroxidase activity was detected with the substrate 3-amino, 9-ethylcarbazole (Biomeda, Foster City, CA) followed by a hematoxylin counterstain (Biomeda). Macrophages were identified immunohistochemically with a monoclonal rat anti-mouse Mac-3 antibody (Pharmingen, San Diego, CA), followed by a mouse anti-rat biotinylated IgG (Pharmingen) and a streptavidin horseradish peroxidase incubation (Biogenex). Activated platelets were identified via immunostaining for P-selectin using a rabbit anti-human polyclonal antibody (Pharmingen), followed by a swine anti-rabbit biotinylated IgG and streptavidin horseradish peroxidase (Biogenex). Similar antigen retrieval, peroxidase detection, and counterstain were used, as described previously for fibrin detection. Von Willebrand factor (vWF)-positive cell types were identified immunohistochemically using an EPOS anti-human vWF antibody conjugated to horseradish peroxidase (DAKO) and detected with 3-amino, 9-ethylcarbazole. In this case, antigen retrieval was performed with limited trypsin digestion.
Transmission Electron Microscopy
Ultrastructural analyses were performed on the heart of a 45-day FXI−/−/PC−/− mouse. The heart was perfused, fixed (12 hours) with Karnovsky solution, and postfixed (1.5 hours) in 1% osmium tetroxide. Dehydration was accomplished using a graded series of ethanol solutions, and embedded in epoxy resins (Polysciences, Warrington, PA). Ultrathin sections (90 nm) were stained in 2% uranyl acetate and Reynold’s lead stain. Sections were viewed and photographed using a Hitachi H600 transmission electron microscope (Hitachi, Tokyo, Japan) at 75 kV accelerating voltage.
Determination of Soluble Fibrin in Plasma
Plasma fibrin levels were detected using the COATEST soluble fibrin kit, as described by the manufacturer (Chromogenix, Molndal, Sweden). A volume of 2.5 μl of plasma was diluted in 50 μl of 0.1 mol/L Tris-HCl buffer, pH 7.9, and the solution added to a microtiter plate containing plasminogen, recombinant tissue-type plasminogen activator, plasmin inhibitor antibody, and the plasmin-selective chromogenic substrate, S-2403. After a 15-minute incubation, the reaction was quenched with 100 μl of 5% acetic acid. Absorbances, obtained from p-nitroaniline release from substrate S2403, are directly related to the amount of soluble fibrin in the plasma. The absorbance of the batch-specific control provided in the kit was then subtracted from the absorbance of the test samples, and values, expressed in soluble fibrin units, were Tris-buffered saline/Tween and then incubated with 100 μl of plasma sample (1:5 dilution in Tris-buffered saline) or standard (0 to 12.5 ng/ml murine PAI-1 (Molecular Innovations, Southfield, MI ) for 1 hour at room temperature. After a triple wash with Tris-buffered saline/Tween, the plates were incubated with 100 μl of 6 μg/ml anti-murine-PAI-1 polyclonal antibody (Molecular Innovations). The wells were then incubated with 1:1,000 in Tris-buffered saline goat anti-rabbit-IgG-AP (Biorad, Hercules, California) for 1 hour after three washes. Finally, the assay was developed with pNPP (Sigma Chemical Co., St. Louis, MO) for 30 minutes at room temperature and the plates were read in a plate reader at 405 nm.
Results
Generation of PC+/−/FXI−/− and PC−/−/FXI−/− Mice
Mice carrying the PC+/− genotype were mated with FXI−/− animals to generate PC+/−/FXI+/− progeny. These mice were then crossed with FXI−/− animals to produce PC+/−/FXI+/− mice, which were then bred to provide PC−/−/FXI−/− progeny. Animals possessing the combined PC+/−/FXI+/− and PC+/−/FXI−/− genotypes seemed healthy and bred normally, at least through the age of 6 months.
Postnatal genotypic analysis of litters sired from PC+/−/FXI−/− breedings led to identification of eight PC−/−/FXI−/− animals who survived the lethal coagulopathy observed in PC−/− neonates (Table 1) ▶ . The oldest animal (I107, 94 days) was sacrificed on presentation with severe inflammation, edema, and gangrene on its extremities such as its nose, tail (Figure 1a) ▶ , and paws (Figure 1b) ▶ . Although none of the other animals demonstrated this phenotype, the majority were sedentary and significantly growth retarded (Figure 1c) ▶ as compared to their littermates. They also demonstrated dyspnea, and either expired or were sacrificed between the ages of 13 and 94 days.
Table 1.
Characteristics of PC−/−/FXI−/− Animals
| Animal | Status | Age | Runt | Sex | Organs with obvious anomalies* | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Li | Lu | H | GB | LN | Bl | I | ST | Th | PG | K | Br | SO | T | V | P | SG | |||||
| I107 | sacrificed | 94 d | y | m | x | x | x | x | x | x | x | x | x | x | |||||||
| G506 | died | 49 d | y | m | x | x | x | x | x | x | x | x | x | x | |||||||
| I207 | sacrificed | 35 d | y | f | x | x | x | x | x | ||||||||||||
| G352-4 | sacrificed | 35 d | y | m | x | x | x | x | |||||||||||||
| G352-3 | died | 31 d | n | m | x | x | x | x | |||||||||||||
| G462-1 | sacrificed | 24 d | y | m | x | x | x | x | x | x | x | ||||||||||
| 314A | died | 15 d | y | f | x | x | x | x | x | ||||||||||||
| G352-2 | died | 13 d | y | m | x | x | x | x | x | ||||||||||||
*Li, liver; Lu, lung; H, heart; GB, gall bladder; I, intestines; ST, soft tissue; Th, thymus; PG, preputial gland; K, kidneys; Br, brain; SO, sex organs; T, thoracic fluid; V, vessels; P, pancreas; SG, salivary gland; LN, lymph nodes; Bl, bladder.
Figure 1.
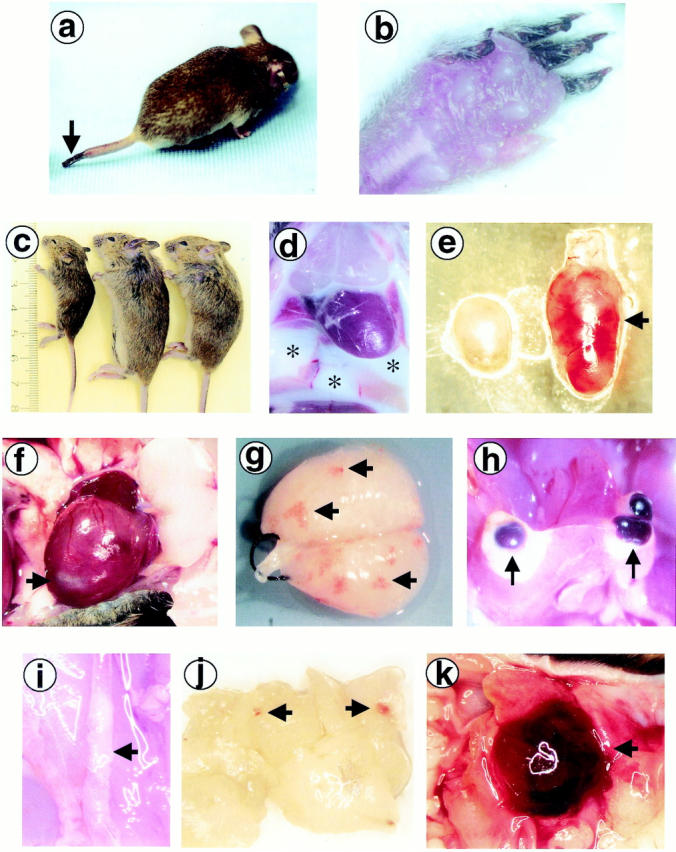
Gross appearance of PC−/−/FXI−/− animals. a and b: I107, the oldest surviving PC−/−/FXI−/− mouse (94 days), presented with gangrene on the tail tip (a; arrow) and toes (b), and severe inflammation and edema on the paws (b). c: The majority of the PC−/−/FXI−/− animals (left) were significantly growth retarded compared to their PC+/−/FXI−/− (middle) and PC+/+/FXI−/− (right) littermates. d: G352-2 and G506, two of the four PC−/−/FXI−/− animals discovered dead, presented with a significant volume of milky-white lymphatic fluid in the thoracic cavity (*). The population of cells was primarily lymphocytic. e: A common attribute observed in these animals was enlargement and apparent hemorrhage of lymph nodes (arrow). f: Hearts were also affected with various anomalies including infarcted tissue (arrow). g: After perfusion, lungs generally demonstrated hemorrhagic lesions (arrows). h: Hemorrhagic cysts were commonly found adjacent to fibrotic areas in the livers (arrows). i: Atherosclerotic areas were seen in some of the larger vessels such as the abdominal and thoracic aorta (arrow). j and k: Soft tissue hemorrhage (arrows) was also commonly observed, as seen in the pancreas (j) and the preputial gland (k).
At necropsy, PC−/−/FXI−/− animals displayed a wide array of pathologies (Table 1) ▶ . Perfusions were often difficult to perform, probably because of the presence of systemic microvascular clots. A total of two of the four animals that were discovered dead presented with a significant volume (∼1 ml) of lymphatic fluid in the thoracic cavity (Figure 1d) ▶ . The cellular population was primarily lymphocytic, with occasional erythrocytes, epithelial cells, monocytes, and neutrophils. Lymph nodes were significantly enlarged and showed areas of hemorrhage, particularly on the left side of the body near the salivary glands and kidneys, adjacent to the carotid bifurcation, and close to the femoral artery graft (Figure 1e) ▶ . Hearts from sacrificed PC−/−/FXI−/− animals were markedly enlarged as compared to littermate controls. Other anomalies included ventricular infarction (Figure 1f) ▶ , atrial clotting and fibrosis, and thrombosis near the truncus pulmonalis. After perfusion and inflation of the lungs, hemorrhagic lesions were readily apparent (Figure 1g) ▶ . In several animals, yellow patches of edemic tissue were also evident. Areas of fibrotic-appearing tissue and the presence of hemorrhagic cysts were commonly observed on the edges of the liver (Figure 1h) ▶ . Several animals also demonstrated significant thrombi in larger vessels such as the thoracic and abdominal aortas (Figure 1i) ▶ . Varying degrees of hemorrhage were also evident between the brain and the skull plate, and in other tissues such as the pancreas (Figure 1j) ▶ , preputial gland (Figure 1k) ▶ , stomach, intestines, bladder (along with mineralization), adrenal gland, prostate, testicles, epididymus, ovaries, uterus, thymus, salivary gland, fatty patches, and leg muscles. Analysis of blood smears indicated varying degrees of leukopenia, neutropenia, and neutrophil hypersegmentation. In addition, smears demonstrated reticulocytosis, anisocytosis, and a number of Howell-Jolly bodies. Because the spleens of these animals appeared normal, the presence of Howell-Jolly bodies may not be a significant pathological condition in these mice.
Histology
Histological analysis of paraffin-embedded tissues confirmed a wide array of pathologies in the PC−/−/FXI−/− animals. Concurrent with observations made at necropsy, analysis of H&E-stained tissue sections verified the systemic soft tissue hemorrhage. However, the most commonly affected organs were the liver, heart, lungs, and lymph nodes.
The hearts of PC−/−/FXI−/− mice were often characterized with multiple small and large intravascular, epicardial, and lymphatic thrombus formation, as observed by positive fibrin (Figure 2, A and B) ▶ and vWF immunostaining (not shown), activated platelets trapped within the thrombi (Figure 2C) ▶ , arterial neointimal formation, acute hemorrhage, atrial fibrosis and mineralization, and ventricular fibrosis (Figure 2D) ▶ . Multifocal vacuolization, necrosis, and myocardial degeneration were also commonly observed. Inflammatory cell infiltrates (neutrophils, macrophages, mast cells) were seen surrounding affected vessels and within areas of degeneration and repair. In a single case, degeneration of the atrioventricular valves was observed. A transmission electron micrograph of a clot in the right auricle of a 45-day postnatal animal (Figure 3) ▶ offers confirmation of the histological and immunohistochemical findings and clearly shows the presence of activated platelets in a fibrin matrix.
Figure 2.
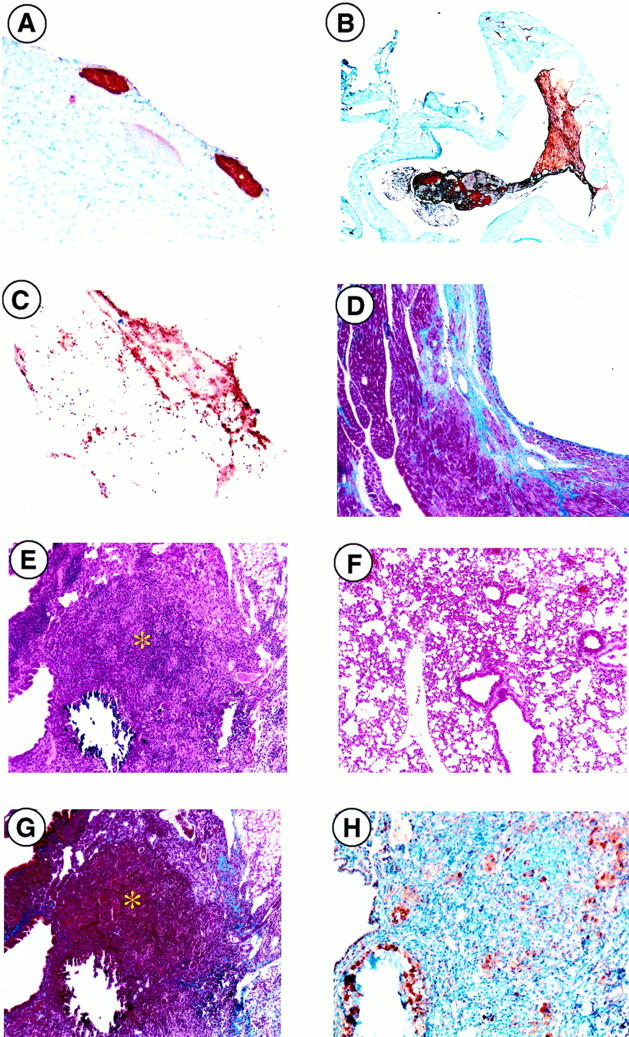
Microscopic analysis of PC−/−/FXI−/− hearts and lungs. Thrombus formation was readily observed (brown areas) via antifibrin(ogen) immunostaining in various regions of the heart, such as the epicardium (A) and atria (B). C: Anti-P-selectin immunostaining identified the presence of activated platelets within the thrombus. D: Ventricular fibrosis, seen macroscopically in Figure 1F ▶ , was verified through Masson’s Trichrome staining in blue. E: Lung tissue demonstrated focal areas of consolidation and mineralization (*) (H&E) as compared to a PC+/+/FXI−/− (F) control. G: Collagen deposition (blue) observed by Masson’s trichrome staining. H: Macrophage infiltration (brown) visualized by anti-Mac3 immunostaining was also observed. Original magnifications: ×40 (B), ×100 (A, D–F, G), ×200 (H), and ×400 (C).
Figure 3.
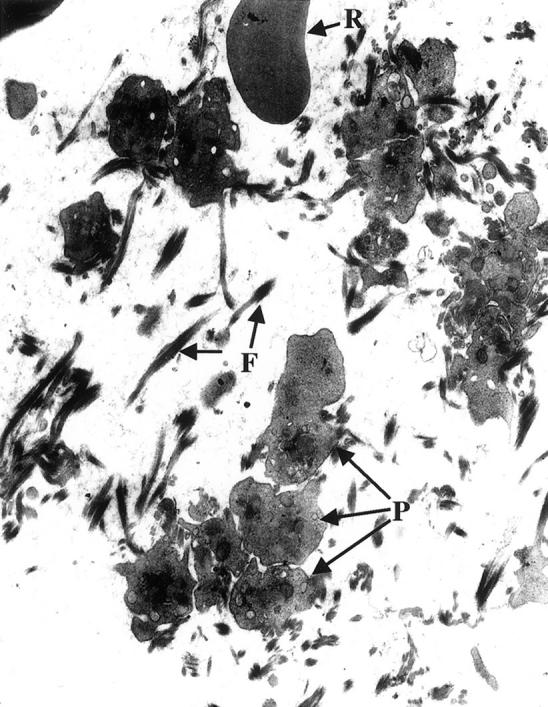
Transmission electron micrograph (original magnification, ×7,000) of the clots found in the right auricle of the heart of a FX−/−/PC−/− 45-day postnatal mouse. Note the platelets (P) aggregated among the bundles of fibrin fibers (F). An erythrocyte (R) can also be seen in this view of the clot.
Lungs (Figure 2E) ▶ demonstrated varying degrees of septal thickening, consolidation, alveolar hemorrhage, and fibroblast infiltration into the alveolar spaces, as compared to PC+/+/FXI−/− controls (Figure 2F) ▶ . Collagen deposition and focal mineralization (Figure 2G) ▶ , and more rarely, edema, were observed around these fibrotic lesions. In addition, acute and chronic inflammation (neutrophils, leukocytes, macrophages, giant cells) (Figure 2H) ▶ , erythrocytic phagocytosis, and pleuritis were evident. Perivascular lymphatic vessels were often distended and filled with lymphocytes. Degeneration of peritracheal muscle and fat were also observed. Small to moderately sized granulomas were commonly evident.
Livers (Figure 4 ▶ ; A, B, and C) consistently demonstrated intravascular thrombosis with multifocal areas of hyperplasia, mineralization, fibrosis, and hemorrhage. These areas were often separated from normal tissue with a sharp line of demarcation. The large blood-filled cysts observed in necropsy were not surrounded by normal vessel endothelium (via anti-vWF immunostaining, not shown), but instead appeared to be pockets of blood surrounded by capsular tissue found adjacent to large mineralized fibrotic areas (Figure 4D) ▶ . Infiltration of neutrophils, macrophages, and multinucleated giant cells were also common, particularly within and surrounding mineralized foci.
Figure 4.
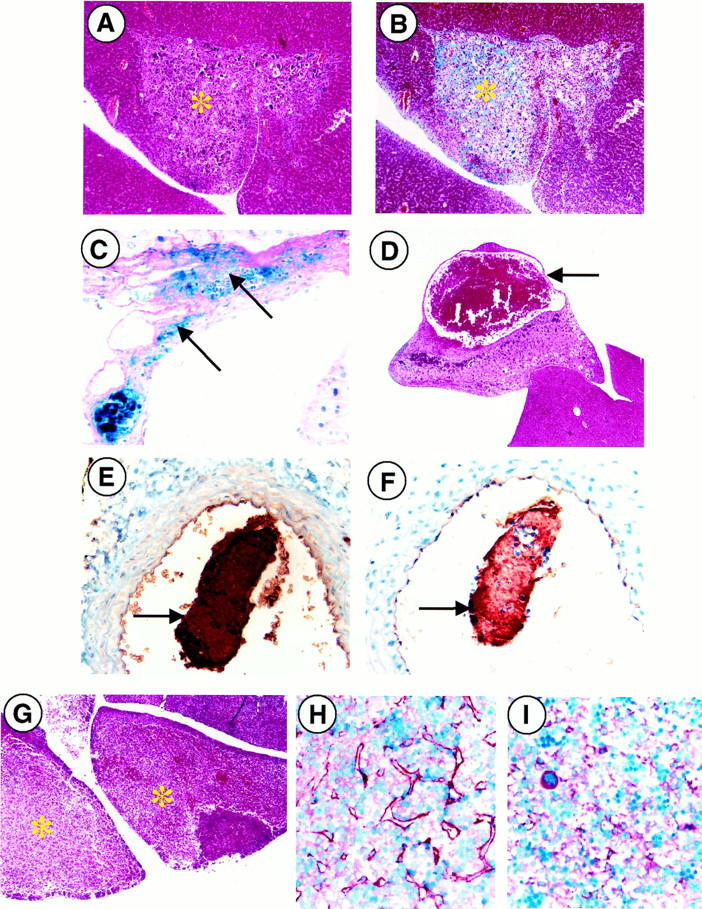
Microscopic analysis of PC−/−/FXI−/− livers and vessels. A and B: Fibrotic and mineralized areas of livers (*) were observed with a sharp line of demarcation from normal liver tissue (H&E and Masson’s Trichrome; original magnification, ×100). C: In addition, interstitial hemorrhage (arrows) was also a common feature of these animals, as observed by PAS/Fe histochemical staining (original magnification, ×400). D: Pockets of blood seen in necropsy were surrounded by capsular hepatic tissue (arrow) and were always found adjacent to large, mineralized fibrotic areas (H&E; original magnification, ×40). E and F: Intravascular thrombi (arrows) were visualized by antifibrin(ogen) and anti-vonWillebrand factor immunostaining (original magnification, ×400). G: H&E staining verified the presence of focal hepatic necrosis (*) in PC−/−/FXI−/− E17.5 embryonic livers (original magnification, ×40). H: Antifibrin(ogen) immunostaining demonstrates organized interstitial fibrin deposition in E17.5 embryonic livers (brown threads) versus the presence of fibrinogen alone in aged-matched PC+/+/FXI−/− controls (I).
Multifocal hemorrhage and fibrosis of lymph nodes were verified through H&E and Masson’s trichrome staining. Areas of organized intravascular thrombi were also commonly observed. Some nodes demonstrated mild cortical atrophy and edema. Sinuses were also heavily populated with macrophages, neutrophils, plasma cells, and mast cells. Germinal centers contained significant proliferating B cells. Lymphatic fluid also showed positive staining for fibrin. Intravascular thrombi were also observed in various large arteries, specifically the abdominal and thoracic aorta, as revealed by anti-fibrin(ogen) and anti-vWF immunostaining (Figure 4, E and F) ▶ .
Analysis of E17.5 Embryos
To determine the effect of FXI deficiency on PC−/− embryonic coagulopathy, timed matings between PC+/−/FXI−/− crosses were established and embryos harvested at E17.5dpc. Of 35 embryos harvested, eight were identified as PC−/−/FXI−/−. A total of two of the eight embryos seemed normal. The remaining six embryos presented with slightly opaque livers and two of these six embryos also showed some hemorrhage in their bladders and beneath the skull plate. H&E and anti-fibrin(ogen) immunostaining demonstrated varying degrees of hepatic necrosis (Figure 4G) ▶ and fibrin deposition (Figure 4H) ▶ in all of the embryos, as compared to PC+/+/FXI−/− controls (Figure 4I) ▶ . However, the systemic brain, kidney, atrial, and soft tissue fibrin deposition and hemorrhage observed in PC−/− embryos at this age were not seen in the PC−/−/FXI−/− embryos.
Soluble Fibrin
Clinically, soluble fibrin is regarded as a molecular marker for intravascular fibrin formation and, potentially, an elevated level may be a marker for a hypercoagulative state. Thus, the levels of soluble fibrin in plasma samples collected from PC−/−/FXI−/− animals were examined (Table 2) ▶ . The assay is based on the ability of fibrin to potentiate tissue-type plasminogen activator-catalyzed plasmin generation from plasminogen. Under normal nonhypercoagulative conditions, very low levels of soluble fibrin should be obtained. Indeed, pooled WT plasma did not indicate the presence of soluble fibrin. To determine whether the COATEST assay kit would recognize murine soluble fibrin, WT mice were injected with a sublethal dose of lipopolysaccharide, euthanized at different time points (time = 0.5, 1, 2, 5, 10, and 24 hours), and their plasma collected. Only at the last time point (24 hours) was there detectable levels of soluble fibrin (145 soluble fibrin units). All of the PC−/−/FXI−/− plasma samples that were tested (n = 5) demonstrated measurable levels of soluble fibrin greater than the WT control supplied by the manufacturers of the assay kit, and in two cases the soluble fibrin units were greater than that which was measured in plasma from the 24-hour LPS-injected WT animals (Table 2) ▶ . Not surprisingly, plasma samples from mice I208 (PC+/−/FXI−/−) and I209 (PC+/+/FXI−/−) did not demonstrate significant levels of soluble fibrin greater than WT levels.
Table 2.
Soluble Fibrin and PAI-1 Levels
| Sample | Genotype | Age (days) | Soluble fibrin units | PAI-1 (ng/ml) |
|---|---|---|---|---|
| I207 | PC−/−/FXI−/− | 35 | 180 | 7.4 |
| I208 | PC+/−/FXI−/− | 35 | 50 | 0.05 |
| I209 | PC+/+/FXI−/− | 35 | 50 | 0.9 |
| G352-4 | PC−/−/FXI−/− | 35 | 95 | 7.9 |
| G462-1 | PC−/−/FXI−/− | 24 | 175 | 92 |
| Mouse WT pool | PC+/+/FXI+/+ | NA | 47 ± 12* | ND |
| Mouse WT pool | PC+/+/FXI+/+ | 35 | ND | 0 |
| Mouse WT pool | PC+/+/FXI+/+ | 24 | ND | 15 |
| Mouse WT pool | PC+/+/FXI+/+ | 7 | ND | 84 |
*A batch-specific assigned value provided by the Chromogenix Quality Control Laboratory and is not obtainable from the standard curve. NA, not applicable; ND, not determined.
Cytokine Levels
Because PC has been implicated as a link between coagulation and inflammation, and secondly, because of the significant neutrophilia and recruitment of other inflammatory cell infiltrates into affected organs, enzyme-linked immunosorbent assay and ribonuclease protection assay were performed on plasma and serum samples, and lung and liver RNA, respectively, to assess possible alterations in systemic and tissue-specific cytokine expression levels.
The same five plasma samples tested for soluble fibrin levels were assayed for the presence of interleukin (IL)-6 (plasma) and tumor necrosis factor (TNF)-α (serum). Plasma samples from lipopolysaccharide-injected WT animals were again used as positive controls for elevated levels of these two cytokines. PC−/−/FXI−/− plasma and serum samples were tested in a dilute solution (1:1 in sample buffer) or tested at an undiluted concentration. In either case, neither IL-6 nor TNF-α was detectable, at least to the pg range. IL-6 and TNF-α levels were also undetectable in pools of WT mouse plasma and serum, respectively.
RPA analysis was performed using total lung and liver RNA collected from a single PC−/−/FXI−/− 15-day-old animal (314A), along with its PC+/− and PC+/+ littermates. Blood smears collected from this former animal demonstrated similar leukopenia and neutropenia, as observed in the other PC−/−/FXI−/− animals with significant inflammatory cell infiltration into the lungs and liver. Total RNA was hybridized with 32P-labeled RNA probes of various cytokines [TNF-β, lymphotoxin-β, TNF-α, interferon (IFN)-γ, IFN-β, transforming growth factor (TGF)-β1, and TGF-β2], digested with RNase, and the complexes resolved on a denaturing acrylamide gel. Exposure of the gel to film throughout an extended period of time (4 to 6 weeks) demonstrated that there were no changes in the expression levels of any of these cytokines in this PC−/−/FXI−/− animal as compared to littermate controls.
PAI-1 Levels
The effects of a hypercoagulative state on fibrinolysis is dominated by increased levels of PAI-1, leading to insufficient fibrinolysis. Thus, plasma samples collected from PC−/−/FXI−/− animals were analyzed for increased PAI-1 levels (Table 2) ▶ . In addition, pools of WT mouse plasma from various ages were also assessed, because PAI-1 levels are thought to be higher in neonates. 16 As is evident in Table 2 ▶ , PAI-1 concentrations (84 ng/ml) in 7-day WT mice are higher than in 24-day WT mice (15 ng/ml), as compared to 35-day WT mice (undetectable). Mice I207 and G352-4, two 35-day PC−/−/FXI−/− animals, demonstrated higher levels of PAI-1 (7.4 ng/ml and 7.9 ng/ml, respectively), as compared to an age-matched WT control (undetectable PAI-1). Control plasma samples from mice I208 (PC+/−/FXI−/−) and I209 (PC+/+/FXI−/−) did not exhibit elevated PAI-1 levels. In addition, G462-1 (24-day PC−/−/FXI−/−) also possessed a level of PAI-1 (92 ng/ml) greater than its age-matched WT control (15 ng/ml).
Discussion
The crucial role for PC as an anticoagulant is underscored by extensive thrombotic events observed in homozygotes or compound heterozygotes of PC deficiency. However, PC has also been regarded as a link between coagulation and inflammation, 17 because uncontrolled coagulation results in thrombosis, as well as promotion of cellular proliferation and an inflammatory response. 18 Although increased neutrophil infiltration was observed in PC−/− E18.5dpc embryos, an inflammatory-related cause of death was not thoroughly explored.
To further understand the consequences of total PC deficiency, our interest was to determine whether the additional loss of FXI, the initiator of the intrinsic maintenance pathway of coagulation, would result in increased survival for PC−/− mice. PC+/−/FXI−/− mice were generated and crossbred to create PC−/−/FXI−/− progeny. At necropsy, E17.5dpc PC−/−/FXI−/− embryos did not demonstrate any significant pathology as compared to PC−/− embryos at this age. 9 However, histological analysis did demonstrate varying degrees of focal necrosis and fibrin deposition in the livers of these PC−/−/FXI−/− mice. Thus, the results obtained suggest that levels of embryonic FXI can contribute to coagulopathy in utero, because loss of FXI results in a decrease in severity of the embryonic coagulopathy seen in PC−/− embryos. In addition, a significant number of PC−/−/FXI−/− neonates survived beyond the time period at which the majority of the PC−/− neonates expired (day 0 to 2). Nonetheless, all of these mice eventually demonstrated varying degrees of systemic coagulopathy and inflammatory cell infiltration, resulting in their deaths between the ages of 13 to 94 days. Thus, it is clear that removal of the intrinsic pathway initiator, FXI, can result in at least a partial rescue of the coagulopathy associated with a PC deficiency, but more importantly allowed more mature mice to be studied that possessed a total PC deficiency.
Although it was difficult to assign a pattern of systemic insult correlated to age, the most commonly affected organs of PC−/−/FXI−/− mice were the liver, heart, lungs, and lymph nodes, which consistently demonstrated varying fibrin deposition, fibrosis, mineralization, and inflammatory cell infiltration, as observed through histological analyses. Several PC−/−/FXI−/− animals presented with atherosclerotic lesions and thrombosis in major vessels and in vitro analysis of plasma samples confirmed the presence of increased levels of soluble fibrin and enhanced PAI-1 levels. In addition, hemorrhage, secondary to coagulopathy, was occasionally observed in the stomach, intestines, preputial gland, sex organs, and soft tissues.
Not surprisingly, analysis of blood samples from PC−/−/FXI−/− animals demonstrated a paucity of inflammatory cells that were likely because of heavy recruitment into organs. In addition, the severe systemic hemorrhage resulted in anemia, as evidenced by the presence of higher than expected levels of reticulocytes, Howell-Jolly bodies, and the recruitment of immature erythrocytes in the blood.
Because the occurrence of coagulopathy in these animals is a likely indicator of up-regulated prothrombin activation, other aspects of thrombin-related functions were investigated, such as those that would address the apparent central role of thrombin in the progression of an inflammation-coagulation autoamplification loop. 19 In this model, it is suggested that in addition to catalyzing fibrin formation, thrombin also stimulates inflammatory processes. Inflammation, in turn, can then feedback and promote coagulation, in part through the resulting damaged endothelium, which becomes prothrombotic. In addition, the cytokines released during inflammation also influence coagulation through up-regulation of tissue factor, 20 as well as FVIII and fibrinogen, 21 and down-regulation of thrombomodulin, 22 which would attenuate PC activation. Further contributing to the thrombotic state, the thrombin present can inhibit fibrinolysis by catalyzing activation of thrombin-activatable-fibrinolytic-inhibitor. In a setting such as a PC−/−/FXI−/− deficiency, it might be expected that this inflammation-coagulation cross-talk would result in a self-propagating and deleterious feedback loop, resulting in thrombosis, ischemia, free radical damage, and in the most severe cases, death. In addition, PC has been suggested to play a role in the inflammatory response. Although it has been shown that the aPC/protein S complex up-regulated the proinflammatory cytokines, IL-6 and IL-8, in human endothelial cells, 23 another study 24 showed the prevention, by (A)PC/EPCR binding, of IL-6 and IL-8 up-regulation in Escherichia coli-challenged animals. It has been further demonstrated that APC, but not PC, prevented TNF-α up-regulation 25 and CD14 down-regulation in macrophages issued inflammatory challenges, 25 and, additionally, that PC functioned in the blocking of neutrophil activation. 26 From another perspective, the fact that TNF-α and IL-1 down-regulate PC production in mice further suggests that the proinflammatory markers can lead to an increase in the procoagulant potential. 27 From these studies, it seems that the relationships between PC/aPC and cytokine production are complex and involve the protease capabilities of aPC in some cases and/or perhaps different cellular receptors for PC/aPC.
Given the potentially strong relationships between (a)PC and coagulation/inflammation, it was of interest to assess possible alterations in systemic and tissue-specific cytokine expression levels in mice with a total PC deficiency that survived the neonatal coagulopathy and showed clear signs of inflammation. Interestingly, no evidence was found of alterations in plasma TNF-α and IL-6 levels of various-aged PC−/−/FXI−/− animals. In addition, no changes were observed in TNF-α, IL-6, TNF-β, LT-β, IFN-γ, IFN-β, TGF-β1, and TGF-β2 in lung and liver from a 15-day PC−/−/FXI−/− animal, as compared to littermate controls. These results were somewhat surprising, particularly because histological analysis demonstrated significant neutrophilia and other inflammatory cell infiltration. Nonetheless, there was no evidence of enhanced cytokine levels in the animals tested, even with sufficient levels of thrombin to induce coagulopathy. It is possible that the role of PC in inflammation does not exclusively rely on the cytokines examined in this report; that the cytokine responses after clot formation are transient 28 and were no longer elevated in the 15-day PC-deficient animal, which contained clots much earlier in life; and/or the additional absence of FXI in these animals has established a more complex relationship between PC and inflammation that does not depend on these particular cytokines. Unfortunately, it is not possible to test this latter possibility because adult animals with a total PC deficiency do not survive without the corresponding total FXI deficiency. However, other PC−/− rescue strategies are being probed in this laboratory that may in the future provide the necessary animals.
The analysis of combined gene deficiencies is a powerful approach to understanding the pathologies of the individual gene deletion phenotypes, thus, further delineating the functions of these proteins. In a similar type of study, an additional FVII deficiency did not compensate for the coagulopathy observed in PC−/− embryos. 29 In fact, the coagulopathy in these double-deficient animals was significantly enhanced as compared to the single PC−/− phenotype. This is most likely because of the loss of the anticoagulant function of the tissue factor pathway inhibitor inhibitory complex, which requires FVIIa to function. In contrast, a FVII deficiency has been shown to rescue the coagulopathy observed in embryos deficient in tissue factor pathway inhibitor. 30 In this latter case, the effects of lost anticoagulant function of tissue factor pathway inhibitor were not significant in the additional absence of a procoagulant, FVII. Although the significance of the FXI-initiated intrinsic pathway may not have been thoroughly appreciated in the past, 31 the results presented in this report clearly demonstrate that alterations in FXI levels, even during embryogenesis, can affect the degree of disseminated intravascular coagulation in PC-deficiency. The coagulopathy observed in the absence of FXI and PC likely results from a continuous activation of FV and FVIII through loss of PC function, and activation of FIX and FX from catalytic levels of thrombin generated from the FVIIa/tissue factor initiated extrinsic pathway. The loss of FXI seems to partially alleviate the neonatal mortality observed in the PC−/− animals; however, the resulting later stage coagulopathy still results in their demise in early adulthood.
Acknowledgments
The authors wish to thank Drs. Harm HogenEsch and James Foster for assistance with the histopathological analyses.
Footnotes
Address reprint requests to Dr. Francis J. Castellino, W. M. Keck Center for Transgene Research and the Department of Chemistry and Biochemistry, 229 Nieuwland Science Hall, University of Notre Dame, Notre Dame, Indiana, 46556. E-mail: castellino.1@nd.edu.
Supported by National Institutes of Health grants HL-19982 (to F. J. C.) and HL-63682 (to V. A. P.), a grant from the W. M. Keck Foundation (to F. J. C.), and by the Kleiderer/Pezold Family Endowed Professorship (to F. J. C.).
References
- 1.Broze GJ, Warren LA, Novotny WF, Higuchi DA, Girard JJ, Miletich JP: The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of action. Blood 1988, 71:335-343 [PubMed] [Google Scholar]
- 2.Broze GJ, Gailani D: The role of factor XI in coagulation. Thromb Haemost 1993, 70:72-74 [PubMed] [Google Scholar]
- 3.Kisiel W: Human plasma protein C. J Clin Invest 1979, 64:761-769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vehar GA, Davie EW: Preparation and properties of bovine factor VIII (antihemophilic factor). Biochemistry 1980, 19:401-410 [DOI] [PubMed] [Google Scholar]
- 5.Taylor FB, Lockhart MS: A new function for activated protein C: activated protein C prevents inhibition of plasminogen activators by releasate from mononuclear leukocytes-platelet suspensions stimulated by phorbol diester. Thromb Res 1985, 37:639-649 [DOI] [PubMed] [Google Scholar]
- 6.Bajzar L, Nesheim M: The effect of activated protein C on fibrinolysis in cell-free plasma can be attributed specifically to attenuation of prothrombin activation. J Biol Chem 1993, 268:8608-8616 [PubMed] [Google Scholar]
- 7.Bajzar L, Morser J, Nesheim M: TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 1996, 271:16603-16608 [DOI] [PubMed] [Google Scholar]
- 8.Taylor FB, Chang A, Esmon CT, D’Angelo A, Vigano-D’Angelo S, Blick KE: Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J Clin Invest 1987, 79:918-925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jalbert LR, Rosen ED, Moons L, Chan JCY, Carmeliet P, Collen D, Castellino FJ: Inactivation of the gene for anticoagulant protein C causes lethal perinatal consumptive coagulopathy in mice. J Clin Invest 1998, 102:1481-1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gailani D, Lasky NM, Broze GJ, Jr: A murine model of factor XI deficiency. Blood Coagul Fibrinolysis 1997, 8:134-144 [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC: Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994, 368:419-424 [DOI] [PubMed] [Google Scholar]
- 12.Masson P: Trichrome stainings and their preliminary technique. J Tech Methods 1929, 12:75-90 [Google Scholar]
- 13.Mallory FB, Wright JH: Pathological Techniques. 1924:p 207 W. B. Saunders, Philadelphia
- 14.McManus JFA: Histological and histochemical uses of periodic acid. Stain Tech 1948, 23:99. [DOI] [PubMed] [Google Scholar]
- 15.Sturmia MM: A rapid universal blood stain. J Lab Clin Med 1936, 21:930-936 [Google Scholar]
- 16.Koh SC, Cheong YC, Arulkumaran S, Ratnam SS: Coagulation activation, fibrinolysis and inhibitors in neonates. Ann Acad Med Singapore 1997, 26:767-771 [PubMed] [Google Scholar]
- 17.Esmon CT, Taylor FB, Snow TR: Inflammation and coagulation: linked processes potentially regulated through a common pathway mediated by protein C. Thromb Haemostas 1991, 66:160-165 [PubMed] [Google Scholar]
- 18.Hancock WW, Grey ST, Hau L, Akalin E, Orthner C, Sayegh MH, Salem HH: Binding of activated protein C to a specific receptor on human mononuclear phagocytes inhibits intracellular calcium signaling and monocyte-dependent proliferative responses. Transplantation 1995, 60:1525-1532 [DOI] [PubMed] [Google Scholar]
- 19.Esmon CT, Fukudome K, Mather T, Bode W, Regan LM, Stearns-Kurosawa DJ, Kurosawa S: Inflammation, sepsis, and coagulation. Haematologica 1999, 84:254-259 [PubMed] [Google Scholar]
- 20.Kirchhofer D, Tschopp TB, Hadvary P, Baumgartner HR: Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in homogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Invest 1994, 93:2073-2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirano T, Akira S, Taga T, Kishimoto T: Biological and clinical aspects of interleukin 6. Immunol Today 1990, 11:443-449 [DOI] [PubMed] [Google Scholar]
- 22.Lentz SR, Tsiang M, Sadler JE: Regulation of thrombomodulin by tumor necrosis factor-alpha: comparison of transcriptional and posttranscriptional mechanisms. Blood 1991, 77:542-550 [PubMed] [Google Scholar]
- 23.Hooper WC, Phillips DJ, Renshaw MA, Evatt BL, Benson JM: The up-regulation of IL-6 and IL-8 in human endothelial cells by activated protein C. J Immunol 1998, 161:2567-2573 [PubMed] [Google Scholar]
- 24.Taylor FB, Stearns-Kurosawa DJ, Kurosawa S, Ferrell G, Chang ACK, Laszik Z, Kosanke S, Peer G, Esmon CT: The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood 2000, 95:1680-1686 [PubMed] [Google Scholar]
- 25.Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW: Selective inhibitory effects of the anticoagulant activated protein C on the responses of human mononuclear phagocytes to LPS, IFN-γ, or phorbol ester. J Immunol 1994, 153:3664-3672 [PubMed] [Google Scholar]
- 26.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki KB: Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood 1996, 87:642-647 [PubMed] [Google Scholar]
- 27.Yamamoto K, Shimokawa T, Kojima T, Loskutoff DJ, Saito H: Regulation of murine protein C gene expression in vivo: effects of tumor necrosis factor-alpha, interleukin-1, and transforming growth factor-beta. Thromb Haemost 1999, 82:1297-1301 [PubMed] [Google Scholar]
- 28.Wakefield TW, Greenfield LJ, Rolfe MW, DeLucia A, Strieter RM, Abrams GD, Kunkel SL, Esmon CT, Wrobleski SK, Kadell AM: Inflammatory and procoagulant mediator interactions in an experimental baboon model of venous thrombosis. Thromb Haemost 1993, 69:164-172 [PubMed] [Google Scholar]
- 29.Chan JCY, Cornelissen I, Collen D, Ploplis VA, Castellino FJ: Combined factor VII/protein C deficiency results in intrauterine coagulopathy in mice. J Clin Invest 2000, 105:897-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan JCY, Carmeliet P, Moons L, Rosen ED, Huang Z-F, Broze GJ, Collen D, Castellino FJ: Factor VII deficiency rescues the intrauterine lethality in mice associated with a tissue factor pathway inhibitor deficit. J Clin Invest 1999, 103:475-482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, Taylor FB, Colman RW: The contact system contributes to hypotension but not to disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest 1993, 91:61-68 [DOI] [PMC free article] [PubMed] [Google Scholar]