

Abstract
Inogranic pyrophosphate (PPi) inhibits hydroxyapatite deposition, and mice deficient in the PPi-generating nucleoside triphosphate pyrophosphohydrolase (NTPPPH) Plasma cell membrane glycoprotein-1 (PC-1) develop peri-articular and arterial calcification in early life. In idiopathic infantile arterial calcification (IIAC), hydroxyapatite deposition and smooth muscle cell (SMC) proliferation occur, sometimes associated with peri-articular calcification. Thus, we assessed PC-1 expression and PPi metabolism in a 25-month-old boy with IIAC and peri-articular calcifications. Plasma PC-1 was <1 ng/ml by enzyme-linked immunosorbent assay in the proband, but 10 to 30 ng/ml in unaffected family members and controls. PC-1 functioned to raise extracellular PPi in cultured aortic SMCs. However, PC-1 was sparse in temporal artery lesion SMCs in the proband, unlike the case for SMCs in atherosclerotic carotid artery lesions of unrelated adults. Proband plasma and explant-cultured dermal fibroblast NTPPPH and PPi were markedly decreased. The proband was heterozygous at the PC-1 locus, and sizes of PC-1 mRNA and polypeptide, and the PC-1 mRNA-coding region sequence were normal in proband fibroblasts. However, immunoreactive PC-1 protein was relatively sparse in proband fibroblasts. In conclusion, deficient extracellular PPi and a deficiency of PC-1 NTPPPH activity can be associated with human infantile arterial and peri-articular calcification, and may help explain the sharing of certain phenotypic features between some IIAC patients and PC-1-deficient mice.
Physiological extracellular matrix (ECM) calcification is limited to bones, teeth, and nonarticular (growth) cartilages. 1,2 Moreover, ECM calcification must be tightly controlled, because normal calcium and phosphate concentrations in extracellular fluids are near the saturation point for the deposition of basic calcium phosphate crystals in the form of hydroxyapatite, and pathological calcification of the ECM can be observed in any tissue of the body. 1,2 The ECM of large arteries is the site most frequently affected by pathological calcification, which commonly occurs in longstanding atherosclerosis and diabetes mellitus. 1 In this regard, the majority of people older than 60 years of age have detectable calcifications from atherosclerotic plaques. 3 Furthermore, calcification of artery lesion plaques is associated with a higher morbidity and mortality in atherosclerosis. 4
Calcification of the arterial ECM has been observed in certain human diseases (and in mouse models) in the absence of atherosclerosis, diabetes, renal failure, or a primary endocrine disorder affecting serum calcium or phosphate levels. 1 In the human condition termed idiopathic infantile arterial calcification (IIAC), calcification (hydroxyapatite deposition) in the media of large muscular arteries is associated with a stenosing, fibroproliferative medial smooth muscle cell (SMC)-mediated process, maximal in the area of the internal elastic lamina. 5-9
In the more than 160 cases of IIAC that have been reported, the disease most often appeared by early infancy, and was often lethal by 6 months of age, generally because of ischemic cardiomyopathy, and other complications of obstructive arteriopathy including renal artery stenosis. 5-8 In more than a dozen reported cases of IIAC, peri-articular calcifications of large joints also developed in infancy. 5-8
One theory for the basis of pathological arterial ECM calcification has proposed an active mineralization process mediated by factors including de novo generation by cells within the arterial wall of oxidized lipids, promineralizing matrix vesicles, and ectopically expressed osteoblastic noncollagenous proteins such as osteopontin. 1,2 Alternatively, the physiological importance of genetically regulated inhibitors of calcification, ie, passive mineralization, has been illustrated in several human metabolic diseases (eg, hypoparathyroidism).
One of the major physiological inhibitors of calcification is PPi, which potently inhibits basic calcium phosphate (hydroxyapatite) crystal deposition in bone and cartilage. 10,11 Significantly, markedly decreased plasma PPi 9 and urinary PPi levels, 8 respectively, have each been reported in individual case studies of IIAC. Some IIAC patients also have responded to treatment with bisphosphonates, which are nonhydrolyzable analogues of PPi. 5,8
PPi is produced by a variety of biochemical reactions, and cells appear to channel intracellular PPi to the extracellular space. 10-12 Reactions that generate PPi include the nucleoside triphosphate pyrophosphohydrolase (NTPPPH) activities of a group of ecto-enzymes in the phosphodiesterase nucleotide pyrophosphatase (PDNP) family (EC 3.6.1.8, EC 3.1.4.1). 13,14 PDNP family NTPPPH isozymes produce free PPi by hydrolysis of the phosphodiester-I bond in both purine and pyrimidine nucleoside triphosphates, and have been recognized to contribute to the regulation of intracellular and extracellular PPi levels in several tissues. 10,12-14
PDNP family isozymes with NTPPPH activity are expressed as cell-bound class II (intracellular N-terminus) transmembrane glycoproteins of 120 to 130 kd that share a highly homologous extracellular domain containing two somatomedin B-like regions and a highly conserved catalytic site. 13,14 Furthermore, soluble enzymatically active NTPPPH species are liberated by proteolysis of the parent molecules. 13,14
The most widely distributed PDNP family NTPPPH isozyme is plasma cell membrane glycoprotein-1 (PC-1), 10,12-14 which is particularly abundant in fibroblasts, chondrocytes, osteoblasts, and hepatocytes, and also circulates in soluble form(s) in plasma. 15 The neural, enteric, and genitourinary tract PDNP family NTPPPH isozyme B10/PDNP3 also has been detected in a soluble form in plasma. 16 PC-1 and B10/PDNP3 have nonhomologous cytosolic domains but highly homologous extracellular domains, and are likely to have risen from antecedent gene duplication, as their respective genes lie in close proximity on human chromosome 6q21-23. 13,14
Another PDNP family NTPPPH isozyme, the alternatively spliced autotaxin/PD1α, has been identified and demonstrated to be particularly abundant in neural, intestinal, skin, and joint tissues. 13,14,17-19 Unlike PC-1 and B10/PDNP3, autotaxin is predominantly released from cells via proteolysis, 17,18 and autotaxin/PD-1α has both threonine type ATPase and NTPPPH activities. 19 Furthermore, unlike PC-1, forced expression by transfection of autotaxin did not elevate extracellular PPi in meniscal cells in vitro. 20
Several cell types express more than one NTPPPH. 10,12-14 However, the subcellular distributions and functions of each NTPPPH isozyme may differ in a given cell type, which is believed to reflect in large part substantial differences in the cytosolic tails of each isozyme. 12-14
PC-1 potently inhibits hydroxypatite crystal deposition by osteoblasts in vitro. 10 Moreover, PC-1 has been demonstrated to serve as a physiological mineralization inhibitor in vivo. 21 Specifically, in tiptoe-walking ttw/ttw mice, which have autosomal recessive inheritance of a naturally occurring nonsense truncation mutation in PC-1, hydroxyapatite-containing peri-articular calcifications develop in early life (by 3 weeks of age). 21,22 The ttw/ttw mice also develop synovial and articular cartilage, and arterial calcification in early life, and progressive fusion of spinal ligaments and of small and large peripheral joints in early adulthood. 21 A nearly identical phenotype to that of ttw/ttw mice was recently observed in PC-1 knockout mice. 22 Furthermore, deficient function of a nonenzymatic plasma-membrane PPi channel that supports extracellular PPi levels (termed ANK) has been associated with similar, progressive hyperostosis by early adulthood in ank/ank mice. 11
In this study, we characterized a patient with IIAC (and infantile peri-articular calcifications) in which extensive study revealed a PC-1/NTPPPH and extracellular PPi deficiency state. The results presented below indicate that PC-1 deficiency in humans and mice can be associated with similar phenotypic features.
Materials and Methods
Skin Biopsy, Fibroblast Isolation, Fibroblast Cultures, Plasma Samples; and PPi, NTPPPH, and Alkaline Phosphatase Assays
For study of explant-cultured dermal fibroblasts, 1-cm dermal excision biopsies were taken under informed consent by institutionally approved protocol from the medial aspect of the upper forearm of the proband at age 25 months, each unaffected family member, and unrelated normal children (ages 2 to 6 years old) as controls.
Dermal samples were finely minced and then initially cultured for three passages in Chang in situ medium (Irvine Scientific, Irvine, CA), supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. For studies of NTPPPH and PPi metabolism cells were subcultured, from passages 4 to 10, in Medium 106 (Cascade Biologics, Portland, OR) supplemented with 100 U/ml penicillin and 50 μg/ml streptomycin (Omega Scientific, Tarzana, CA), and 1% v/v low serum growth supplement (Cascade Biologics).
PPi was determined and equalized for cell DNA, in samples immediately treated after isolation for 10 minutes at 65°C, conditions verified to heat-inactivate all PPi-synthesizing and PPi-degrading activities. 12 For measurements of intracellular PPi, fibroblasts were treated as previously described before the PPi assay, 12 which used differential adsorption on activated charcoal of UDP-d-[6-3H]-glucose (Amersham, Chicago, IL) from the PPi-stimulated reaction product 6-phospho-[6-3H]-gluconate.
Heparinized plasmas were obtained from the IIAC proband and family members, and unrelated normal children (ages 18 to 27 months) on several different days, and buffered 1:1 (for storage at −70°C) in 0.2 mol/L Tris, 1.6 mmol/L MgCl2, pH 8.1. 12 NTPPPH and alkaline phosphatase-specific activities (per μg plasma or cell protein) were determined by colorimetric assays at alkaline pH using synthetic substrates. 12 Protein assays (used to calculate specific activity) were performed using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL) using albumin as a standard. For PPi analyses, separate aliquots of plasma samples were heat-treated, as done above for the fibroblast samples.
All assays of constitutive NTPPPH and PPi metabolism, Western blots, and PC-1 mRNA expression, and confocal microscopy studies were done on fibroblasts at passages 4 to 6. Cells from subsequent passages only were used to provide adequate genomic DNA for sequencing purposes.
SMC Culture, Proliferation Assay, and Cell Transfection
Human second passage aortic SMCs from normal donors were obtained from Cascade Biologics and were grown in Medium 231 supplemented with 100 U/ml penicillin and 50 μg/ml streptomycin (Omega Scientific), and proprietary smooth muscle growth supplement (at 5% v/v) from Cascade Biologics. Cells were subcultured every 5 days, and media were resupplemented every 48 hours. For transfection studies, aliquots of 3 × 10 5 SMCs (fourth passage) were plated in a 35-mm culture dish and allowed to adhere overnight and then transfected with 1 μg of plasmid DNA by a previously described lifofectamine plus protocol. 12 Where fibroblasts were transfected, we used only fifth passage cells. We achieved a transfection efficiency of >55% in SMCs and >45% in fibroblasts in these studies.
To measure SMC proliferation, we plated aliquots of 5,000 cells in a 96-well plate and allowed them to adhere for 18 hours. Then the cells were transfected, as described above, incubated for 8 hours, and then carried in fresh medium for 24 hours, at which time the plate was treated with Hoescht H33258 (fluorogenic DNA binding dye) to determine cell numbers (excitation of 355 nm and emission 460 nm). A standard curve was used for each assay to extrapolate the values from fluorescence units to cell numbers.
Western Blotting and Immunoprecipitation Studies
Western blotting for immunodetection of PC-1 and B10/PDNP3 was performed as previously described, using the rabbit anti-PC-1 antibody R1769 specific for the 150 amino acid C-terminal domain of PC-1, 23 and a previously described rabbit antiserum to B10/PDNP3 amino acids 193 to 616. 12 For detection of autotaxin, we used a rabbit polyclonal antiserum collected after immunization with the KLH-conjugated peptide NH2-TEFLSNYLTNVDDITLVPGTLGR-COOH corresponding to amino acids 370 to 392 of autotaxin and common to full-length autotaxin and the PD-I alpha splice variant. 20
For immunoprecipitation of NTPPPH isozymes, 50 μg of plasma protein, or, where indicated, 50 μg (in protein) of fibroblast lysates (from ∼5 × 10 5 cells lysed in 0.1 mol/L Tris, 1.6 mmol/L MgCl2, 1% Triton X-100, pH 8.1) was precleared with 1:10 volume of protein G-Sepharose beads (Sigma, St. Louis, MO). Specific antibodies that recognized native human PC-1 (the monoclonal antibody 3E8, a gift of Dr. J. Goding, Monash Medical School, Prahran, Australia), 12 or the above-described antisera to autotaxin or B10/PDNP3 (1 μl) were added to each sample, and then the total volume was brought up to 0.25 ml in 0.1 mol/L Tris, 1.6 mmol/L MgCl2, pH 8.1. After constant rotation at 4°C overnight, protein G-Sepharose beads (25 μl) were added, and samples were maintained under constant rotation for an additional 24 hours at 4°C. The beads were then centrifuged, and washed three times with 0.1 mol/L Tris, 1.6 mmol/L MgCl2, pH 8.1 buffer (without 1% Triton X-100). To allow analysis of bead-associated proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting, beads were treated at 95°C in 0.1 ml of sodium dodecyl sulfate-containing buffer for 5 minutes.
Confocal Immunofluorescence Microscopy
Poly-d-lysine-coated coverslips were plated with either proband or control explant cultured dermal fibroblasts so as to achieve a cell density of 50 to 75% at 24 hours. In brief, the staining protocol involved fixing cells with 4% paraformaldehyde solution and blocking with 5% bovine serum albumin containing phosphate-buffered saline. PC-1 was detected using the monoclonal antibody 3E8, and B10/PDNP3 was detected using the rabbit polyclonal serum to human B10/PDNP3 cited above, as described. 12 Secondary antibodies used were Alexa 488 goat anti-mouse IgG conjugate or Alexa 568 goat anti-rabbit IgG conjugate, both from Molecular Probes (Eugene, OR). Fluorescent images of the cells were visualized by laser confocal microscopy, using a Zeiss Axiovert 100 mol/L laser scanning microscope, as previously described. 12
Enzyme-Linked Immunosorbent Assay (ELISA) for Soluble Plasma PC-1
To quantify soluble PC-1 in heparinized plasmas, ELISA plates were coated overnight at 4°C with an affinity-purified polyclonal antibody to PC-1, as previously described. 24 All samples were tested in a blinded manner. In brief, after washing with TBST buffer (20 mmol/L Tris, 150 mmol/L NaCl, and 0.05% Tween-20) to remove unbound antibody, wells were blocked with SuperBlock (Pierce Chemical, Rockford, IL). Next, human plasma (10 to 15 μl diluted to a total volume of 100 μl with 50 mmol/L HEPES, pH 7.6, 150 mmol/L NaCl, 0.1% Triton X-100, 1 mmol/L phenylmethyl sulfonyl fluoride, 1 mg/ml bovine serum albumin) was added to wells, and PC-1 in the plasma was allowed to bind overnight at 4°C. After extensive washing, biotinylated anti-PC-1 monoclonal antibody was added to wells for 2 hours. Wells were again washed, and then streptavidin horseradish peroxidase was added for 30 minutes. Next, the signal was enhanced using ELAST (Dupont NEN, Boston, MA), according to the manufacturer’s instructions. The signal was developed using 100 μl of TMB reagent (Kirkegaard-Perry, Gaithersburg, MD), and the reaction was stopped using 100 μl of 1 mol/L H3PO4, with absorbance measured at 451 nm.
Sequencing of PC-1, B10/PDNP3, and Autotaxin
Genomic DNAs from proband, appropriate age-related controls, and family members were PCR-amplified using intron-exon-specific primers for PC-1. 25 The entire coding region cDNA sequence of B10/PDNP3, and partial cDNA sequence of autotaxin (limited to domains encompassing the catalytic site and EF hand domain) also were determined using primers that spanned the cDNA regions, generating amplicons of 200 to 350 bp. All polymerase chain reaction products were electrophoretically separated, extracted, and then purified using Qiagen (Valencia, CA) gel extraction columns. Sequencing of all products was performed using dye primer chemistry on an ABI377 automated sequencer. The derived sequences were compared to the published sequences for each NTPPPH isozyme.
Northern Blot Analysis
Explant-cultured dermal fibroblasts were homogenized and total RNA prepared according to the manufacturer’s instructions using TRIzol reagent (Life Technologies, Inc., Gaithersburg, MD). The RNA samples (20 μg) were resolved on a 1% formaldehyde-agarose gel run at 5 V/cm and then transferred to MagnaGraph nylon membranes (Fisher Scientific, Tustin, CA) by capillary transfer. After transfer, the membrane was UV cross-linked at 254 nm according to the manufacturer’s instructions. Hybridization was performed at 42°C using a PC-1 cDNA probe with specific activity >10 8 cpm/μg, prepared by using a random primer labeling kit (Life Technologies, Inc.). The PC-1 cDNA probe encompassed the PC-1-specific region encoding the cytosolic tail, and was generated by digesting a pcDNA3 PC-1 construct 12 with BamHI and ScaI. Densitometric analysis of autoradiographs was performed as previously described. 10
Immunohistochemistry
For immunohistochemical analysis of artery lesion PC-1 and B10/PDNP3, formalin-fixed and paraffin-embedded tissues from the proband’s temporal artery biopsy and from unrelated adult atherosclerotic human carotid endarterectomy samples were sectioned at 3-μm thickness. After deparaffinization and rehydration, appropriate blocking buffers were applied to the sections. The sections were incubated with primary antibodies (1:200 of the rabbit antibody to B10/PDNP3 described above, and 1:100 of the monoclonal anti-human PC-1 antibody 3E8 cited above) for 14 hours at 4°C. After an extensive wash, the sections were blocked for endogenous peroxidase activity with Peroxo-Block (Zymed, South San Francisco, CA) and incubated with secondary species-specific antibody at 5 μg/ml for 1 hour. The sections were incubated with Vectastain ABC Elite solution (Vector Laboratories, Burlingame, CA) for 30 minutes and developed with 9-amino-3-ethylene-carbazole (Vector Laboratories) and counterstained with hematoxylin. We also used anti-α-actin antibody conjugated with horseradish peroxidase (DAKO, Carpinteria, CA), and the sections to be stained for SMC actin were incubated with the antibody for 1 hour, blocked with Peroxo-Block, and developed with 9-amino-3-ethylene-carbazole. Negative control sections were treated by using appropriate dilutions of the normal sera of the species in which the primary antibodies were made.
Gene Linkage Analyses for Matrix GLA Protein (MGP) and PC-1
To test if the disorder was caused by mutation(s) in the matrix GLA protein gene (MGP), the MGP gene was amplified using four sets of primers as described 27 and the products were analyzed for single-strand conformation polymorphism. No variant patterns were observed. Subsequently, possible genetic linkage of the disorder to the PC-1 locus on chromosome 6q was tested by analysis of polymorphic variable-number tandem repeats around the region of interest. The analyzed markers were D6S287, D6S1009, and D6S311. The markers were amplified using fluorescence-labeled markers and their length determined using a Perkin-Elmer ABI 310 automated DNA sequence apparatus (Perkin-Elmer, Emeryville, CA.
Results
Brief Case Summary
The case history of the proband was recently described in detail. 8 In brief, the proband is the male child of a consanguineous marriage (the fathers of each parent were brothers). Because of fetal distress, the proband was delivered preterm by caesarian section in the 36th week of gestation, in January 1997. The mother’s obstetrical history was G9, P4, A2 (spontaneous), with three additional stillborns. None of the surviving family members has developed IIAC.
At 5 days of age, the proband was diagnosed with congestive heart failure because of dilated cardiomyopathy. By 8 months of age, renal artery stenoses, aortic calcifications, and wrist and ankle peri-articular calcifications (Figure 1) ▶ had developed. The proband also started to experience episodes of acute peri-articular inflammation of the wrists and ankles.
Figure 1.

Radiological evidence for peri-articular calcifications in the IIAC proband. Lateral (left) and PA radiographic (right) views of the right wrist of the proband at 8 months of age are shown, and revealed dense globular calcifications. Similar findings also were seen in both wrists and ankles.
Temporal artery biopsy, done at 10 months of age, confirmed the clinical diagnosis of IIAC, via the findings of marked fibrointimal proliferation and luminal narrowing, and amorphous deposition of hydroxyapatite crystals in the arterial wall noted by electron microscopy. 8 A prolonged subsequent treatment course with etidronate, after our studies were completed, was associated with substantial resolution of the peri-articular calcification.
Blood karyotyping of the proband revealed a normal male karyotype in 50 cells studied. An extensive work-up in the proband ruled out a primary endocrine or metabolic disorder. Initially, we focused on the MGP gene as candidate locus for this disorder, because of the association of arterial calcification with MGP deficiency in mice. 28 A screen for mutations using a sensitive single-strand conformation polymorphism assay did not indicate presence of sequence changes in the IIAC proband or in his parents as compared to control individuals (data not shown).
Markedly low urine PPi levels were present in the proband relative to unrelated normals. 8 Thus, we pursued investigation of plasma- and explant-cultured fibroblast PPi, and of PPi-generating NTPPPH isozymes.
Deficiencies of PC-1, NTPPPH Activity, and Extracellular PPi Levels in the Proband
We first assessed levels of soluble plasma PC-1 by ELISA (in a blinded manner for the laboratory performing the ELISA). Because there is no normative data for PC-1 levels in plasma, sampling was generally done at more than one time point. Normal control children and unaffected family members had PC-1 plasma levels in the range of 10 to 30 ng/ml (Figure 2A) ▶ . In contrast, the proband consistently demonstrated trace PC-1 levels (<1 ng/ml) in plasma.
Figure 2.

ELISA for soluble plasma PC-1 (A), plasma PPi (B) and plasma NTPPPH (C) in the IIAC proband, unrelated control children, and unaffected family members. A: PC-1 ELISA was performed in triplicate, as described in Materials and Methods, in a blinded manner on heparinized plasma samples obtained on multiple dates from the IIAC proband (ME) and unrelated normal control children (left) and the unaffected family members (right). Studies were done on plasmas collected between 1998 to 1999. Each data point for each donor represents a different sampling date, and the dates of sampling for the proband in the left and right panels were completely different. Dates of birth for the IIAC proband and unaffected family members were: proband, January 1997; father, September 1963; mother, February 1959; sister no. 1, October 1987; sister no. 2, March 1990; and brother, December 1994. B: Plasma PPi values are indicated for the proband and unrelated control children (left) and the unaffected family members (right). For the PPi measurements, separate aliquots of heparinized plasma were prepared as described in Materials and Methods and then radiometrically measured for PPi, which was performed as described in Materials and Methods. Dates of collection were as in A. C: Plasma NTPPPH activities are indicated for the proband and unrelated control children (left) and the unaffected family members (right). For assessment of plasma NTPPPH, we studied aliquots of plasmas (collected on multiple dates, as above) that were buffered 1:1 in 0.2 mol/L Tris and 1.6 mmol/L MgCl2, pH 8.1. The figure shows the results of measurements of the specific activity of NTPPPH per μg of protein. Relative differences for results of NTPPPH activity/volume (not shown) were essentially the same.
To understand the implications of our findings of deficient PC-1 levels in plasma, we first assessed NTPPPH activity and PPi levels in proband plasma. Plasma PPi levels in the proband were consistently depressed. Specifically, proband plasma PPi was consistently 50% or less of the levels observed in unrelated children controls and in the unaffected family members (Figure 2B) ▶ . We also observed a consistent, relative depression total plasma NTPPPH-specific activity levels in the proband (a mean of <5 U/μg protein) (Figure 2C) ▶ , similar overall to the results for plasma PPi in the proband relative to unrelated controls. Plasma NTPPPH-specific activity levels also remained consistently lower (again a mean of <5 U/μg protein in three separate determinations) in the proband than in the unaffected family members (Figure 2C) ▶ . Results in all of these experiments were the same whether PPi or NTPPPH were measured as levels per μg protein, as shown, or per μl volume (not shown).
Some variability in plasma-specific activity of total NTPPPH was apparent in the proband’s mother (FE) and the brother (OE), whose NTPPPH-specific activity levels (∼6 and 8 U/μg protein each on separate occasions) approached to the range of plasma NTPPPH activity (∼4 to 5 U/μg protein) observed in the proband (Figure 2C) ▶ . Thus, we further studied NTPPPH-specific activity (and intracellular and extracellular PPi) in explant-cultured dermal fibroblasts (Figure 3) ▶ . We observed markedly lower NTPPPH-specific activity, and intracellular and extracellular PPi levels, in cells from the proband relative to all of the unaffected family members, and to two additional unrelated children controls.
Figure 3.
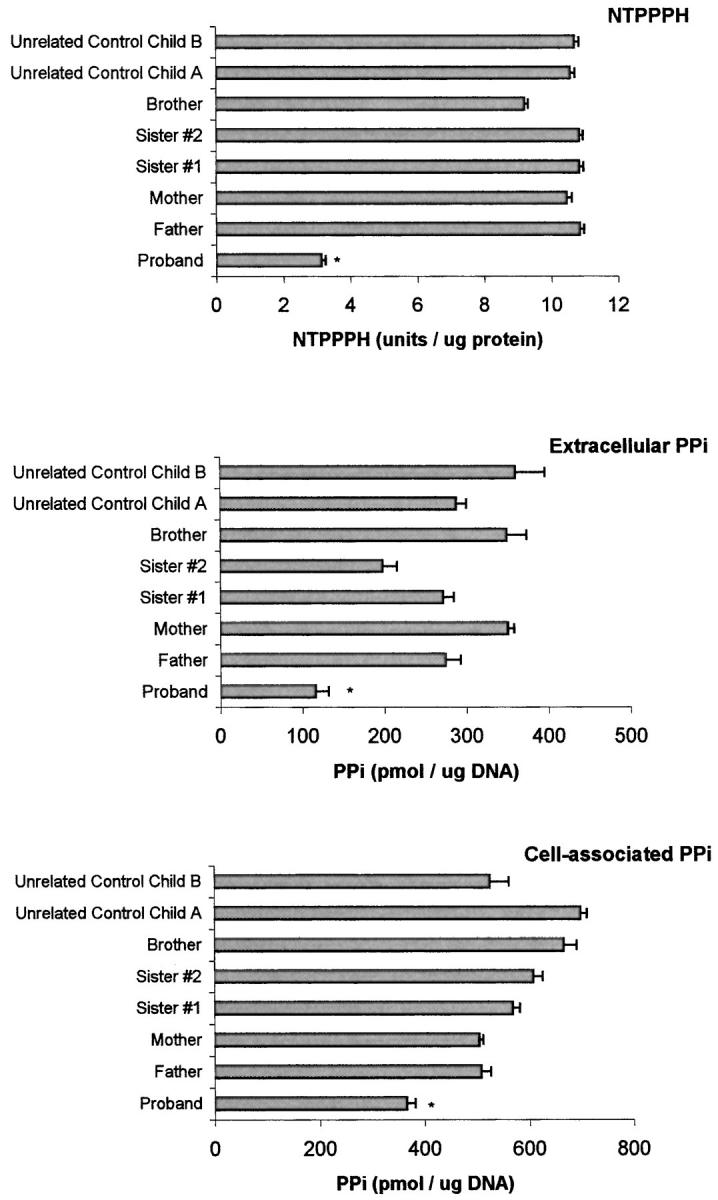
Explant-cultured dermal fibroblast NTPPPH-specific activity, and intracellular and extracellular PPi levels in the IIAC proband, unaffected family members, and additional unrelated control children. Explant-cultured dermal fibroblasts were obtained and cultured from excisional dermal forearm biopsies of all patients under informed consent, as described in Materials and Methods. NTPPPH-specific activity levels and intracellular and extracellular PPi for cultured fibroblasts from each donor were measured as described in Materials and Methods. The results shown are pooled from three determinations in triplicate from each donor. *, P < 0.05 for IIAC proband compared to each unaffected family member and each control.
Further Characterization of the Basis and Extent of PC-1 Deficiency in the Proband
Confocal immunofluorescence microscopy studies demonstrated at most trace levels of PC-1 in proband explant-cultured fibroblasts in comparison to several controls (Figure 4) ▶ . Furthermore, in proband fibroblasts, PC-1 was sparse in comparison to B10/PDNP3, as opposed to the concomitant abundance of both isozymes in control fibroblasts (Figure 4) ▶ .
Figure 4.
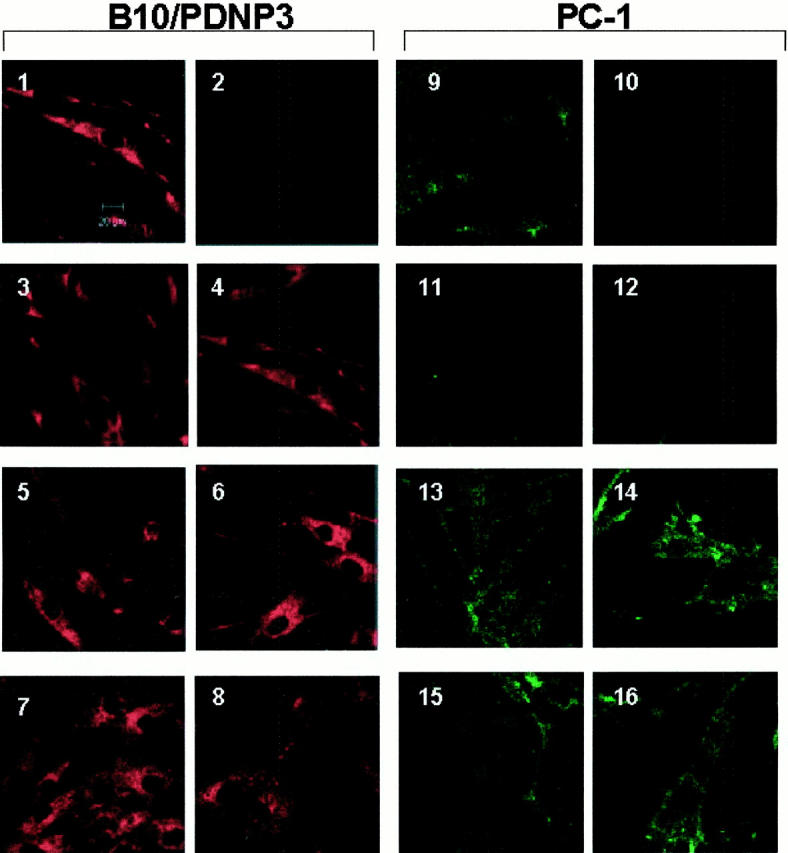
Comparison of B10/PDNP3 and PC-1 expression via laser scanning confocal microscopic images of proliferating dermal fibroblasts in culture. Explant cultured dermal fibroblasts (at 50 to 70% confluence) were analyzed at the same passage number via immunofluorescence for expression of B10/PDNP3 (panels 1–8) and PC-1 (panels 9–16), as described in Materials and Methods. As controls, cells were stained only with the secondary antibodies goat anti-mouse Alexa 488 (panel 2) and goat anti-rabbit Alexa 568 (panel 10) in the absence of the primary antibodies. Micrographs represent fibroblasts of: normal unrelated donor child (1 and 9), proband (3, 4, 11, and 12), father (5 and 13), mother (6 and 14), brother (7 and 15), sister no. 2 (8 and 16). Original magnifications, ×800 for each panel. Representative of four different studies for each individual for both B10/PDNP3 and PC-1.
To further assess the nature and extent of the concomitant PC-1 and NTPPPH deficiency in the proband, we used NTPPPH isozyme-specific antibodies 29 in crude immunoprecipitation assays. To accurately visualize the size of PC-1 in the proband, we needed to use abundant cell lysate protein to detect PC-1. These assays were not quantitative in nature, but allowed qualitative analysis of PC-1 immunoprecipitated from 50 μg of proband fibroblast lysate, which indicated that proband PC-1 was of normal size (∼130 kd under reducing conditions) (Figure 5) ▶ . We similarly recovered autotaxin/PD-1α and B10/PDNP3 of normal size from proband plasma, and fibroblasts, respectively (not shown). We also used immunoprecipitation to screen for the presence of NTPPPH enzymatic activity associated with individual NTPPPH isozymes. Using polyclonal antibodies specific for autotaxin/PD1α, as described in Materials and Methods, we consistently immunoprecipitated approximately half of the NTPPPH in normal plasma and >80% of plasma NTPPPH activity in the proband (not shown). Polyclonal antibodies specific for B10/PDNP3, and to a lesser degree monoclonal antibody specific for PC-1 each were able to immunoprecipitate small but significant levels of functional NTPPPH activity in fibroblast lysates from the proband (not shown).
Figure 5.

Qualitative Western blot analysis of immunoprecipitated explant-cultured dermal fibroblast PC-1. We immunoprecipitated PC-1 from 50 μg of protein derived from donor fibroblast lysates (each from aliquots of ∼5 × 10 5 cells), as described in Materials and Methods. We then performed sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting for PC-1 from each immunoprecipitate, as described in Materials and Methods.
Transfection of wild-type PC-1 in the proband explant-cultured fibroblasts induced a substantial increase in cellular NTPPPH and extracellular PPi levels (not shown). Furthermore, PC-1 mRNA was normal in size (3.5 kb) in the proband explant-cultured fibroblasts, although reduced in amount by 30 to 60% (assessed by densitometry) relative to each control (Figure 6) ▶ . The entire coding regions of PC-1 and B10/PDNP3 were sequenced, as were fragments of the autotaxin/PD1α coding region encompassing the catalytic site and EF hand domain, and we did not find significant coding region sequence mutations in the proband (not shown). Thus, the proband cells had deficient PC-1 expression but were capable of expressing normal PC-1 and of generating an increase in extracellular PPi in response to forced expression of PC-1.
Figure 6.

Northern blot analysis of PC-1 expression in explant-cultured dermal fibroblasts. We isolated total RNA from explant-cultured dermal fibroblasts from patients in the IIAC kindred as indicated and we transferred RNA samples (20 μg) from formaldehyde-agarose gels to nylon membranes and UV cross-linked the membranes as described in Materials and Methods. Hybridization was performed at 42°C using human PC-1 probe (top) with specific activity >10 8 cpm/μg, prepared by random primer labeling, as described in Materials and Methods. Loading of RNA is shown at the bottom. Estimated size of PC-1 mRNA was 3.5 kb.
The proband was the offspring of a consanguineous marriage. Thus, we suspected that the proband would be homozygous by descent at and around the PC-1 and neighboring B10/PDNP3 loci. However, linkage analysis showed this was not the case (Figure 7) ▶ . Specifically, three haplotypes segregated in the nuclear family. Neither the proband nor his unaffected siblings were homozygous by descent. The proband had the same haplotype as his mother.
Figure 7.

Schematized results of polymorphic DNA analysis for PC-1 and B10/PDNP3 linkage in the IIAC proband and surviving unaffected members of the same kindred. Possible genetic linkage of the IIAC disorder to the PC-1 locus and the neighboring B10/PDNP3 on chromosome 6q was tested by analysis of polymorphic variable-number tandem repeats around the region of interest. The markers analyzed were D6S287, D6S1009, and D6S311. The markers were amplified using fluorescence-labeled markers and their length determined using a Perkin-Elmer ABI 310 automated DNA sequence apparatus. In the diagram, the numbers 1, 2, and 3 are used to designate specific differences in lengths when using each marker (A = spontaneous abortion).
PC-1 Expression in Proband Arterial Lesion SMCs: PC-1 and Regulation of Extracellular PPi in Cultured Arterial Vascular SMCs
Studies to this point demonstrated deficient PC-1/NTPPPH expression in the proband. Because IIAC was the dominant clinical feature in the proband, we immunohistochemically analyzed the temporal artery biopsy from the proband. We observed marked expansion of SMCs bordering the internal elastic lamina, abundant expression of the NTPPPH isozyme B10/PDNP3 by the expanded population of SMCs, but sparse PC-1/NTPPPH expression by the same cells (Figure 8) ▶ . In contrast, SMCs from human carotid atherosclerotic lesions of two adults unrelated to the proband demonstrated readily detectable PC-1 expression (and little B10/PDNP3 expression) in the media in association with SMCs (Figure 8) ▶ .
Figure 8.

Immunohistochemical analysis of PC-1, B10/PDNP3, and SMC α-actin expression in the temporal artery of the IIAC proband (A), and in atherosclerotic carotid artery lesions from unrelated adults (B). For immunohistochemical analysis of artery lesion PC-1 and B10/PDNP3, formalin-fixed and paraffin-embedded tissues from the proband’s temporal artery biopsy (A) and from unrelated human adult atherosclerotic carotid endarterectomy samples (B) were sectioned and stained as described in Materials and Methods, using specific antibodies for PC-1, B10/PDNP3, and α-actin antibody for SMC staining. Negative control sections shown in A (C, F, and I) were treated by using appropriate dilutions of the normal sera of the species in which the primary antibodies were made. A: Original magnifications, ×40 (A, C, D, F, G, and I), ×100 (B, E, and H). B: Serial sections from one donor. Original magnifications, ×100 (A, D, G, and J). Serial sections from another donor. Original magnifications, ×100 (B, E, H, and K). C, F, I, and L: Higher magnifications (×250) of the bottom right portion of the corresponding sections of B, E, H, and K. For comparative purposes, trichrome-stained sections are shown in J–L of B.
To identify the potential functional significance of deficient PC-1 expression in arterial SMCs, we studied normal human aortic SMCs in culture, which we observed to constitutively express PC-1 (not shown). We transfected the SMCs with human PC-1 and achieved increased NTPPPH activity. Under these conditions, increased PC-1 expression did not alter SMC proliferation (not shown), but increased PC-1 expression functioned in part to raise extracellular PPi in SMCs (Figure 9) ▶ .
Figure 9.

Direct effects of PC-1 expression on extracellular PPi in cultured human aortic SMCs. Human aortic SMCs were transfected with wild-type PC-1 cDNA or empty vector, and then cell-associated NTPPPH and extracellular PPi determined at 72 hours, as described in Materials and Methods. Results are pooled from three separate experiments done in triplicate. Control SMC extracellular PPi was 284.1 ± 17.0 pmol/μg DNA. *, P < 0.05.
Discussion
In this study, we identified a marked deficiency of PC-1 expression, NTPPPH activity and deficient intracellular and extracellular PPi in a human patient with IIAC and wrist and ankle peri-articular calcifications and episodic peri-arthritis. PC-1 is one of three PDNP family isozymes with NTPPPH activity. 13,14 However, the other NTPPPH isozymes, autotaxin/PD-1α and B10/PDNP3, have been observed to have distinct subcellular distributions and functions when compared to PC-1 in hepatocytes, chondrocytes, and osteoblasts. 12-14 For example, the autotaxin splice variant of the autotaxin/PD1α gene, is predominantly secreted. 18,19 Furthermore, B10/PDNP3, unlike PC-1, is not enriched in the chondrocyte plasma membrane and although B10/PDNP3 regulates intracellular PPi concentrations it does not seem to significantly regulate extracellular PPi in chondrocytes. 12 In addition, PC-1 but not B10/PDNP3 suppresses osteoblastic matrix vesicle-mediated mineralization in vitro. 29
The association of deficient PC-1 expression with human IIAC in the case studies occurred despite the findings that autotaxin-associated NTPPPH activity appeared to be abundant in proband plasma. Furthermore, B10/PDNP3, which had a normal cDNA sequence and was of normal polypeptide size in the proband, was abundantly expressed relative to PC-1 in both proband fibroblasts and the expanded population of SMCs in the arterial lesions of the proband. Thus, the physiological function of PC-1 as a mineralization inhibitor seems to be unique and not readily compensated for by NTPPPH activities of B10/PDNP3 or autotaxin/PD-1α.
We observed that PC-1, which was expressed by normal cultured aortic SMCs and SMCs in human atherosclerotic lesions, functioned at least in part to increase the extracellular levels of the mineralization inhibitor PPi in vitro. In this study, PC-1 expression was barely detectable in a markedly expanded population of SMCs in the proband temporal artery lesions. Taken together, the results suggested that a loss of PC-1-related homeostatic functions in the wall of the temporal artery might be central to IIAC pathogenesis at a local level in the artery wall. However, interpretation of these data are inherently limited because we did not carry out comparisons (to the proband) of relative PC-1 and B10/PDNP3 expression in temporal arteries of other family members and of unrelated children and adults.
Because transfection of wild-type PC-1 in proband cells induced a significant increase in both NTPPPH activity and extracellular PPi (data not shown), a primary PC-1 translational or posttranslational processing defect seemed unlikely in the proband. Defective function of the membrane PPi channel ANK is associated with low extracellular PPi and elevated intracellular PPi in fibroblasts. 11 However, both intracellular PPi and extracellular PPi were low in proband fibroblasts, which was consistent with a primary defect in PC-1/NTPPPH activity. 12 Therefore, ANK expression and function were not specifically explored in the proband.
In the temporal artery of the IIAC proband, we observed marked expansion of the SMC population. Ingestion by SMCs of hydroxyapatite, deposited in the arterial wall in IIAC associated with deficient PPi production, could by itself stimulate SMC proliferation, analogous to mitogenic effects of ingested hydroxyapatite in fibroblasts. 30 However, we speculate that PC-1 might function to prevent arterial calcification by not only modulating PPi generation but also by other effects on SMC differentiation and/or regulation of SMC matrix proteoglycan synthesis via PC-1 nucleotide pyrophosphatase activity on the sulfation donor phosphoadenosine phosphosulfate. 13,14 Although transfection of PC-1 did not affect proliferation of normal cultured SMCs in vitro, it remains to be determined if marked PC-1 deficiency affects SMC proliferation in vivo.
The PC-1-deficient IIAC proband studied here in early childhood had peri-articular calcifications, which also develop in early childhood in PC-1-deficient mice. 21-23 Certain phenotypic features, including osteophytes, spinal ligament hyperostosis, and articular cartilage calcifications, develop at later points in life in PC-1-deficient mice. 21-23 More prolonged observation of the proband will be of interest, but the future phenotype of the proband will likely be affected by his treatment with bisphosphonates. It is possible that PC-1 may not play as broad a regulatory role for calcification in humans, as compared to mice, but it may not be possible to determine this by future study of the proband.
Immunoreactive soluble PC-1 was dramatically reduced in proband plasma and full-length PC-1, although detectable by immunoprecipitation, also was relatively sparse in cultured fibroblasts from the proband. Interestingly, the proband was heterozygous for PC-1. Given the heterozygosity of the IIAC proband for PC-1, it was not surprising to observe that the IIAC proband dermal fibroblasts expressed at least some PC-1 polypeptide (of normal size). In addition, proband PC-1 mRNA was normal in size.
The PC-1 expression defect of the proband seemed to be more profound for circulating PC-1 and for PC-1 expression in the diseased temporal artery wall than in the proband fibroblasts cultured under proliferative conditions in serum-containing media. Further studies to determine the molecular genetic basis for the relative but not absolute PC-1 deficiency in the proband will be of interest. Although the combination of the proband’s PC-1 haplotypes was shared with his mother, it remains possible that their PC-1 genotype will be found to demonstrate distinctions. Genetic linkage studies will be limited by the fact that only one surviving member of the kindred was affected. We were unable to identify abnormalities in the coding region sequence of PC-1 in the proband. However, it is recognized that a variety of types of mutations can be difficult to identify, particularly in heterozygous patients. 31 Conversion approaches 31 to identifying a possible refractory PC-1 mutation in the proband will be of interest to apply in the future. Included among anomalies that could be envisaged in the proband are polymorphism(s) or mutation(s) of the PC-1 promoter that might result in decreased transcription activation in response to PC-1 expression-inducing growth factors such as transforming growth factor-β, a-fibroblast growth factor and b-fibroblast growth factor in developing arteries and peri-articular tissues. 32 Another possibility would be mutation of a receptor or transcription factor that functions to drive (or suppress) PC-1 expression, particularly in vascular SMCs.
In mice, PC-1 is one of several genes for which homologous recombination-induced deficiency has been noted to be associated with spontaneous arterial calcification. 1,2 However, mice deficient in the osteoclast differentiation inhibitory factor osteoprotegerin develop arterial calcification but not peri-articular calcification in early life. 33 Although mice deficient in MGP develop spontaneous arterial calcification they also manifest pathological cartilage calcification. 28 Furthermore, deficient MGP expression in humans (Keutel’s syndrome) has thus far not been associated with arterial calcification. 34 Screening for MGP mutations was negative in the affected kindred. Because we did not sequence the entire MGP or osteoprotegerin genes in our family, or screen for their expression in arterial lesions, we did not exclude a primary or secondary contribution of mutations or abnormalities in expression of either of these genes to IIAC in the proband. However, given the phenotype of combined IIAC and peri-articular calcification in the proband and the marked abnormalities in NTPPPH activity and PPi metabolism, the possibility that either MGP deficiency or osteoprotegerin deficiency were the driving force for the calcification disorder seems unlikely.
In conclusion, the results of this study indicate the utility of screening for plasma and tissue PC-1, NTPPPH, and PPi levels in evaluating IIAC, particularly when associated with peri-articular calcification. PC-1 may ultimately serve as a therapeutic target in certain cases of IIAC.
Acknowledgments
We thank Dr. F. Gerresheim, Department of Molecular Human Genetics, Ruhr-University, Bochum, Germany for assistance in the isolation of fibroblasts; Dr. M. Albrecht, Department of Pediatric Surgery, Dortmund Municipal Hospital, Germany for performing the skin biopsies and the arterial biopsy; and Dr. C. August, Gerhard Domagk-Institute of Pathology, University of Muenster, Germany, for assistance in examination of the arterial biopsy by light and electron microscopy to confirm the diagnosis of IIAC in the proband.
Footnotes
Address reprint requests to R. Terkeltaub M.D., VA Medical Center, 3350 La Jolla Village Dr., San Diego, CA 92161. E-mail: rterkeltaub@ucsd.edu.
Supported by grants from the Department of Veterans Affairs, National Institutes of Health (P01AGO7996, HL61731, DK52999), the Arthritis Foundation, the Swiss National Science Foundation (grant 31-57272.99) and the Ministry of Education, Science, Sports, and Culture of Japan.
F.R. and S.V. contributed equally to this work.
References
- 1.Schinke T, McKee MD, Kiviranta R, Karsenty G: Molecular determinants of arterial calcification. Ann Med 1998, 30:538-541 [PubMed] [Google Scholar]
- 2.Schinke T, McKee MD, Karsenty G: Extracellular matrix calcification: where is the action? Nat Genet 1999, 21:150-151 [DOI] [PubMed] [Google Scholar]
- 3.Stary HC: Natural history of calcium deposits in atherosclerosis progression and regression. Zeitschrift fur Kardiologie 2000, 89(Suppl 2):S28-S35 [DOI] [PubMed] [Google Scholar]
- 4.Bini A, Mann KG, Kudryk BJ, Schoen FJ: Noncollagenous bone matrix proteins, calcification, and thrombosis in carotid artery atherosclerosis. Arterioscler Thromb Vasc Biol 1999, 19:1852-1861 [DOI] [PubMed] [Google Scholar]
- 5.Maayan C, Peleg O, Eyal F, Mogle P, Rosenmann E, Bar Ziv J: Idiopathic infantile arterial calcification: a case report and review of the literature. Eur J Pediatr 1984, 142:211-215 [DOI] [PubMed] [Google Scholar]
- 6.Van Dyck M, Proesmans W, Van Hollebeke E, Marchal G, Moerman P: Idiopathic infantile arterial calcification with cardiac, renal and central nervous system involvement. Eur J Pediatr 1989, 148:374-377 [DOI] [PubMed] [Google Scholar]
- 7.Vera J, Lucaya J, Garcia Conesa JA, Aso C, Balaguer A: Idiopathic infantile arterial calcification: unusual features. Pediatr Radiol 1990, 20:585-587 [DOI] [PubMed] [Google Scholar]
- 8.Rutsch F, Schauerte P, Kalhoff H, Petrarulo M, August C, Diekmann L: Low levels of urinary inorganic pyrophosphate indicating systemic pyrophosphate deficiency in a boy with idiopathic infantile arterial calcification. Acta Paediatrica 2000, 89:1265-1269 [DOI] [PubMed] [Google Scholar]
- 9.Stuart AG: Idiopathic arterial calcification of infancy and pyrophosphate deficiency. J Pediatr 1993, 123:170-171 [DOI] [PubMed] [Google Scholar]
- 10.Johnson K, Moffa A, Chen Y, Pritzker K, Goding J, Terkeltaub R: Matrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J Bone Miner Res 1999, 14:883-892 [DOI] [PubMed] [Google Scholar]
- 11.Ho AM, Johnson MD, Kingsley DM: Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289:265-270 [DOI] [PubMed] [Google Scholar]
- 12.Johnson K, Vaingankar S, Chen Y, Moffa A, Goldring MB, Sano K, Jin-Hua P, Sali A, Goding J, Terkeltaub R: Differential mechanisms of PPi production by plasma cell membrane glycoprotein-1 (PC-1) and B10 in chondrocytes. Arthritis Rheum 1999, 42:1986-1997 [DOI] [PubMed] [Google Scholar]
- 13.Goding J, Terkeltaub R, Maurice M, Deterre P, Sali A, Belli S: Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and nonlymphoid cells: structure and function of the PC-1 family. Immunol Rev 1998, 161:11-26 [DOI] [PubMed] [Google Scholar]
- 14.Goding JW: Ecto-enzymes: physiology meets pathology. J Leukoc Biol 2000, 67:285-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belli SI, van Driel IR, Goding JW: Identification and characterization of a soluble form of the plasma cell membrane glycoprotein PC-1 (5′-nucleotide phosphodiesterase). Eur J Biochem 1993, 217:421-428 [DOI] [PubMed] [Google Scholar]
- 16.Meerson NR, Delautier D, Durand-Schneider AM, Moreau A, Schilsky ML, Sternlieb I, Feldmann G, Maurice M: Identification of B10, an alkaline phosphodiesterase of the apical plasma membrane of hepatocytes and biliary cells, in rat serum: increased levels following bile duct ligation and during the development of cholangiocarcinoma. Hepatology 1998, 27:563-568 [DOI] [PubMed] [Google Scholar]
- 17.Kawagoe H, Stracke ML, Nakamura H, Sano K: Expression and transcriptional regulation of the PD-Ia/autotaxin gene in neuroblastoma. Cancer Res 1997, 57:2516-2521 [PubMed] [Google Scholar]
- 18.Stracke ML, Clair T, Liotta LA: Autotaxin, tumor motility-stimulating exophosphodiesterase. Adv Enzyme Regul 1997, 37:135-144 [DOI] [PubMed] [Google Scholar]
- 19.Clair T, Lee HY, Liotta LA, Stracke ML: Autotaxin is an exoenzyme possessing 5′-nucleotide phosphodiesterase/ATP pyrophosphatase and ATPase activities. J Biol Chem 1997, 272:996-1001 [DOI] [PubMed] [Google Scholar]
- 20.Johnson K, Hashimoto S, Pritzker K, Lotz M, Terkeltaub R: Central role of plasma cell membrane glycoprotein-1 in the pathogenesis of meniscal CPPD deposition disease of the knee. Arthritis Rheum 1999, 42(Suppl):S255 [Google Scholar]
- 21.Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S: Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet 1998, 19:271-273 [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto M, Hosoda Y, Kojimahara K, Yamazaki T, Yoshimura Y: Arthritis and ankylosis in twy mice with hereditary multiple osteochondral lesions: with special reference to calcium deposition. Pathol Int 1994, 44:420-427 [DOI] [PubMed] [Google Scholar]
- 23.Sali A, Favaloro JM, Terkeltaub R, Goding JW: Germline deletion of the nucleoside triphosphosphate (NTPPPH) plasma cell membrane glycoprotein (PC-1) produces abnormal calcification of periarticular tissues. Vanduffel L Lemmens R eds. Ecto-ATPases and Related Ectonucleotidases. 1999, :pp 267-282 Shaker Publishing BV, Maastricht [Google Scholar]
- 24.Frittitta L, Camastra S, Baratta R, Costanzo BV, D’Adamo M, Graci S, Spampinato D, Maddux BA, Vigneri R, Ferrannini E, Trischitta V: A soluble PC-1 circulates in human plasma: relationship with insulin resistance and associated abnormalities. J Clin Endocrinol Metab 1999, 84:3620-3625 [DOI] [PubMed] [Google Scholar]
- 25.Nakamura I, Ikegawa S, Okawa A, Okuda S, Koshizuka Y, Kawaguchi H, Nakamura K, Koyama T, Goto S, Toguchida J, Matsushita M, Ochi T, Takaoka K, Nakamura Y: Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum Genet 1999, 104:492-497 [DOI] [PubMed] [Google Scholar]
- 26.Munroe PB, Olgunturk RO, Fryns J-P, Van Maldergem L, Ziereisen F, Yuksel B, Gardiner RM, Chung E: Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat Genet 1999, 21:142-144 [DOI] [PubMed] [Google Scholar]
- 27.Knisely AS, Gannuch GM, Rossi A, Steinmann B, Superti-Furga A: The matrix GLA protein gene: two polymorphisms, but no pathogenic mutation, in a patient with idiopathic arterial calcification of infancy (abstract). Lab Invest 1999, 80:37A [Google Scholar]
- 28.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G: Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386:78-81 [DOI] [PubMed] [Google Scholar]
- 29.Johnson K, Wennberg C, Hessle L, Mauro S, Goding J, Millan JL, Terkeltaub R: Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Cell Physiol 2000, 269:R1365-R1377 [DOI] [PubMed] [Google Scholar]
- 30.Nair D, Misra RP, Sallis JD, Cheung HS: Phosphocitrate inhibits a basic calcium phosphate and calcium pyrophosphate dihydrate crystal-induced mitogen-activated protein kinase cascade signal transduction pathway. J Biol Chem 1997, 272:18920-18925 [DOI] [PubMed] [Google Scholar]
- 31.Yan H, Kinzler KW, Vogelstein B: Genetic testing—present and future. Science 2000, 289:1890-1892 [DOI] [PubMed] [Google Scholar]
- 32.Solan JL, Deftos LJ, Goding JW, Terkeltaub RA: Expression of the nucleoside triphosphate pyrophosphohydrolase PC-1 is induced by basic fibroblast growth factor (bFGF) and modulated by activation of the protein kinase A and C pathways in osteoblast-like osteosarcoma cells. J Bone Miner Res 1996, 11:183-192 [DOI] [PubMed] [Google Scholar]
- 33.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998, 12:1260-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teebi AS, Lambert DM, Kaye GM, Al-Fifi S, Tewfik TL, Azouz EM: Keutel syndrome: further characterization and review. Am J Med Genet 1998, 78:182-187 [PubMed] [Google Scholar]