

Abstract
The tumor-suppressor protein Dpc4 (Smad4, Madh4) regulates gene expression. On binding of an extracellular ligand of the extensive transforming growth factor (TGF) superfamily to its cognate receptor complex, latent cytoplasmic Dpc4 is activated and translocated into the nucleus to function as part of various DNA-binding transcriptional activator complexes. The most relevant ligand/receptor pair to control the tumor suppressive function of Dpc4 remains uncertain, but is usually assumed to be TGF-β and its heteromeric receptor. We exploited a fortuitous experiment of nature to directly test this hypothesis: the TGF-β type II receptor gene is inactivated by mutation in nearly all colorectal carcinomas having microsatellite instability, as seen in hereditary nonpolyposis colorectal cancer (HNPCC) and in sporadic medullary colorectal cancers. Using a specific and sensitive immunohistochemical label for Dpc4, we examined nuclear localization of Dpc4 in 13 HNPCC, six medullary, and 41 sporadic nonmedullary colorectal carcinomas. In agreement with published rates, two (5%) of 41 sporadic tumors showed complete loss of Dpc4 protein, indicative of genetic inactivation. All 13 HNPCC and six medullary tumors had intact cytoplasmic and nuclear Dpc4 localization. The TGFBR2 gene was sequenced in three of the cancers from patients with HNPCC, and all of these harbored inactivating mutations. The specificity of the immunohistochemical assay was demonstrated in xenograft tumors of syngeneic cell lines that differed in DPC4 genetic status because of an engineered gene knockout. Thus, nuclear localization of Dpc4 can be maintained in cells with inactivated TGF-β type II receptors, suggesting the persistence of tumor-suppressive action of an upstream signaling input, most likely a ligand/receptor complex distinct from TGF-β. Identification of the relevant input would be expected to have implications for the understanding of tumorigenesis and the design of rational biological therapy.
Dpc4 is a phosphoprotein with sequence-specific DNA binding abilities and it is able to form complexes with a considerable variety of additional proteins. Somatic inactivation of this tumor-suppressor protein is common in pancreatic ductal adenocarcinomas and, albeit at lower frequencies, in multiple other tumor systems including colorectal cancer. 1,2 Germline DPC4 gene mutations cause juvenile polyposis. 3
The TGFβ genes form part of a large superfamily of extracellular ligands, and the corresponding receptors of each type of ligand also constitute distinct gene families. Signal transduction is initiated when a ligand binds and stabilizes the formation of the corresponding heterodimeric receptor pair. Transforming growth factor-β (TGF-β) has long been studied for its tumor-suppressive properties in a variety of cancer types and a lack of TGF-β-responsiveness has been demonstrated in many tumor lines. The importance of this signaling pathway in neoplasia is most strongly supported by the finding of biallelic inactivating mutations in the TGFBR2 gene in nearly all colorectal carcinomas having microsatellite instability. 4-6
A number of Smad genes mediate functions of the TGF-β superfamily (including those of bone morphogenic protein, activin, and TGF-β). 7-9 For example, Smad2 mediates TGF-β signals and Smad2 mutations have been identified in occasional colorectal cancers. Likewise, DPC4 is a common mediator of multiple TGF-β superfamily pathways. As depicted in Figure 1 ▶ , ligand binding to a heterodimeric receptor activates a serine-threonine kinase functional group. Certain Smad proteins interact with the receptors transiently, become phosphorylated, and then form a complex with Dpc4. These activated complexes are translocated into the nucleus where their binding to DNA stimulates the transcription of nearby genes. 10 There is now a considerable body of literature concerning experimental systems that confirms these biochemical relationships. 9 It was therefore attractive to assume that Dpc4 functioned as a tumor suppressor (ie, it was selectively inactivated in tumors) primarily because it mediated the TGF-β signals.
Figure 1.
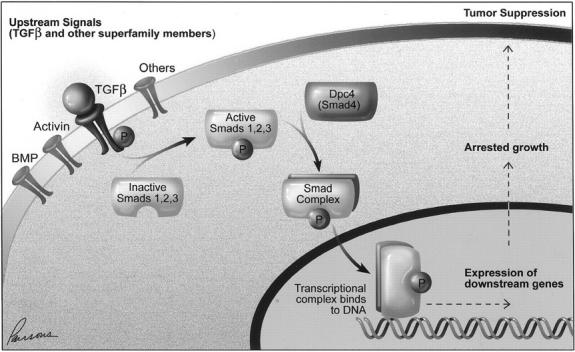
Diagram showing interrelationships between TGF-β superfamily members. Although TGF-β itself is known to lead to activation of Dpc4 in some systems, the data presented here support the presence of other input as well. The figure is schematic; the receptor is heterodimeric and requires additional proteins for efficient signaling, and the Smad complex also comprises multiple proteins to accomplish specific functions.
The identification of a sensitive and specific means of Dpc4 immunolocalization provided a new method to investigate these relationships directly in human tumors. A monoclonal antibody was recently validated as providing an in situ assay that closely mirrors the genetic status of the DPC4 gene. 11 Tissues with wild-type DPC4 status harbor nuclear and cytoplasmic labeling of Dpc4 consistent with our understanding of Dpc4 function. We used this immunohistochemical system to study Dpc4 localization as an assay for its functional inactivation in colorectal cancers, particularly in those expected to exhibit genetic inactivation of TGF-β signaling.
Materials and Methods
Xenografted Syngeneic Tumors
To generate xenograft tumors, actively growing cell lines were harvested and resuspended in serum-free McCoy’s 5A medium (Life Technologies, Inc., Grand Island, NY) at 5 × 10 7 cells per ml. The athymic nude mice (strain nu/nu; Harlan, Indianapolis, IN) were then injected subcutaneously with 0.1 ml HCT116 cells on one flank and with cells from cell line 5-60 or cells from cell line 5-18 on the other. HCT116 cells are known to have a truncating mutation of TGFBR2 but are DPC4+/+. 4 In contrast, the cell line 5-60 was prepared from HCT116 cells that have been genetically modified so they not only have a truncating mutation of TGFBR2 but are also DPC4+/−. The cell line 5-18 was prepared from HCT116 cells and they were genetically modified so they lack TGFBR2 and are DPC4−/−. The preparation of these cell lines has been previously described. 9 Fourteen days after injection, the animals were sacrificed. The tumors were removed and immediately preserved in 10% buffered formalin. Immunohistochemistry using the anti-Dpc4 antibody was then performed as below on each of the harvested tumors.
Specimen Selection
Slides and blocks of colorectal cancers resected from 13 patients with hereditary nonpolyposis colorectal cancer (HNPCC) 12 and from six patients with a medullary tumor phenotype (an appearance that has been associated with mismatch repair defects) were retrieved from the surgical pathology files of The Johns Hopkins Hospital. The 13 patients with HNPCC were previously shown to have DNA mismatch repair defects. 12 Medullary tumor phenotype was defined using criteria of Jass and colleagues 13 and Kim and colleagues. 14 These tumors appeared poorly differentiated with solid sheets of tumor cells, prominent intratumoral lymphocytic infiltration, and an intense peritumoral lymphoid response. Also retrieved and sectioned were materials from 41 sporadic colorectal carcinomas and their associated adenomas (n = 14) and lymph node metastases (n = 9).
Immunohistochemistry
Unstained 5-μm sections were cut from the paraffin blocks and deparaffinized using standard methods. Slides were treated with sodium citrate buffer (HIER buffer; Ventana-Bio Tek Solutions, Tucson, AZ) and steamed at 80°C. After cooling for 5 minutes, slides were labeled with monoclonal antibody to Dpc4 (clone B8; Santa Cruz Biotechnology, Santa Cruz, CA) using the Bio Tek-Mate 1000 automated stainer (Ventana-Bio Tek Solutions). Each slide was labeled with a 1:100 dilution of the antibody. The anti-Dpc4 antibody was detected using a biotinylated secondary antibody and 3,3′-diaminobenzidine as the chromagen with hematoxylin counterstaining.
Slides were reviewed by three of the authors (EM, REW, RHH) and recorded as positive or negative for both nuclear and cytoplasmic labeling as has been described. 11 For a slide to be interpreted as negative, no expression could be visible. Focal labeling was interpreted as positive. Normal colonic epithelium served as a positive control, and the primary antibody was omitted in negative controls. Pancreatic carcinomas with known Dpc4 genetic status were also included as positive and negative controls. 11
Epstein-Barr Virus (EBV) Testing
In situ hybridization was performed to detect EBV using a fluorescein-conjugated oligonucleotide probe to the EBV early RNA transcripts (Novocastra, Newcastle, UK). 15 After overnight incubation, probe binding was detected using an in situ hybridization detection kit (Novocastra) that uses an alkaline-phosphatase-conjugated rabbit F(ab′) anti-fluorescein isothiocyanate fragment followed by 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium substrate chromogen. Positive control tissues yielded blue/black nuclear staining.
Assessment of TGF-β Receptor Mutations
Sufficient archival material was available from three of the HNPCC colorectal cancers with nuclear Dpc4 expression for DNA sequencing. DNA was isolated from formalin-fixed paraffin-embedded tissues as previously described. 16 Evidence for mutations of the polyA tract of the TGFBR2 gene was determined by polymerase chain reaction amplification and end-labeling of the 5′ primer. The radiolabeled 73-bp polymerase chain reaction products were resolved on a 6% polyacrylamide gel. Constitutional DNA from the duodenum of two patients with pancreatic cancer served as negative controls and two pancreatic cancers with known polyA tract mutations 17 served as positive controls.
Results
Xenografted Specimens Prepared from Cell Lines with Known Genotype
The tumors prepared from HCT116 cells (which are known to have a truncating mutation of TGFBR2 but are DPC4+/+) had both nuclear and cytoplasmic labeling for Dpc4 (Figure 2A) ▶ . Those prepared from cell line 5-60 cells (lacking intact TGFBR2 but DPC4+/−) also had both nuclear and cytoplasmic labeling with the Dpc4 antibody (Figure 2B) ▶ . Xenografted tumors prepared from cell line 5-18 cells (lacking intact TGFBR2, DPC4−/−) showed neither cytoplasmic nor nuclear labeling with the Dpc4 antibody (Figure 2C) ▶ . This result confirmed unambiguously that the nuclear labeling was because of Dpc4 protein rather than cross-reacting antigen.
Figure 2.
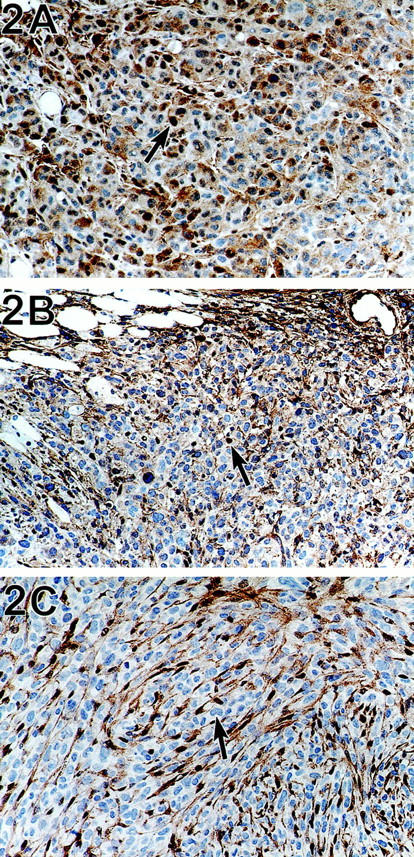
A: The tumors prepared from HCT116 cells (which are known to have a truncating mutation of TGFBR2 but are DPC4+/+) have both nuclear (arrow) and cytoplasmic labeling for Dpc4. Immunohistochemistry using anti-Dpc4 antibody, counterstained with hematoxylin. B: A xenograft prepared from cell line 5-60 cells (lacking intact TGFBR2 but DPC4+/−) has both nuclear (arrow) and cytoplasmic labeling with the Dpc4 antibody. Immunohistochemistry using anti-Dpc4 antibody, counterstained with hematoxylin. C: Xenografted tumor prepared from cell line 5-18 cells (lacking intact TGFBR2 and DPC4−/−) showed neither cytoplasmic nor nuclear labeling with the anti-Dpc4 antibody. The labeled cells in this photograph are ingrown stromal cells from the host animal; the large tumor cells (arrow) are unlabeled. Immunohistochemistry using anti-Dpc4 antibody, counterstained with hematoxylin.
HNPCC Colorectal Carcinomas and Carcinomas with Medullary Phenotype
All 13 (100%) carcinomas obtained from patients with HNPCC (Figure 3A) ▶ and all six carcinomas having medullary morphology showed both nuclear and cytoplasmic labeling for Dpc4. To assure that the six carcinomas with medullary morphology were not Epstein-Barr virus driven (a finding sometimes associated with medullary phenotype), we also performed in situ hybridization for Epstein-Barr virus, which was negative in all cases.
Figure 3.
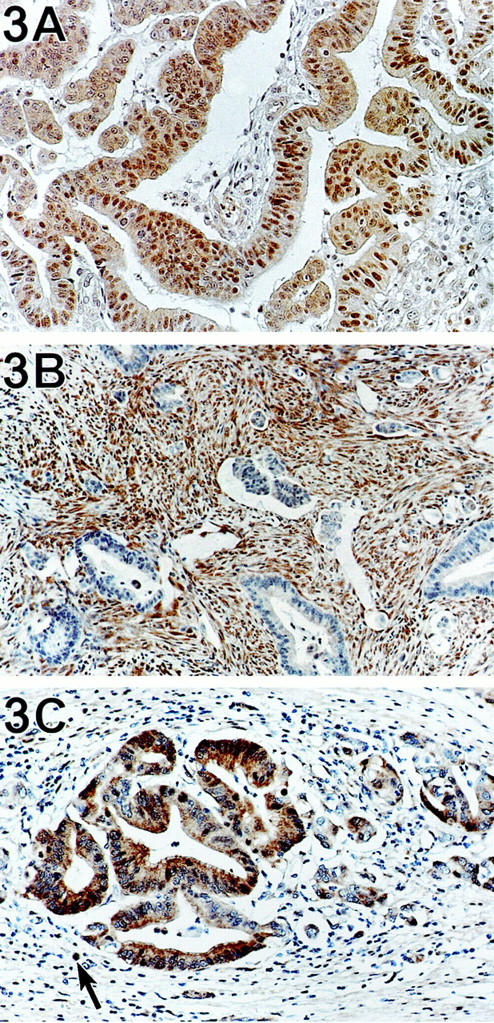
A: Colon carcinoma from a patient with HNPCC. This patient was shown to have a TGFBR2 mutation. Tumor cells contain both nuclear and cytoplasmic Dpc4. Immunohistochemistry using anti-Dpc4 antibody, counterstained with hematoxylin. B: A sporadic colorectal carcinoma completely lacking expression of Dpc4. The stromal and inflammatory cells within the desmoplastic stroma serve as internal controls that express Dpc4. Immunohistochemistry using anti-Dpc4 antibody, counterstained with hematoxylin. C: Sporadic colorectal adenocarcinoma with strong expression of Dpc4 in tumor cell nuclei and cytoplasm. The lymphoid cells (arrow) in the lamina propria also label for Dpc4.
Sporadic Carcinomas
Of the 41 sporadic cancers, 39 labeled with the Dpc4 antibody, and two (two of 41; 5%) completely lacked expression of the gene product by immunohistochemistry (Figure 3B) ▶ . Internal controls (normal colon epithelium and stroma) labeled in all cases. Fourteen associated sporadic adenomas all expressed the gene product. Two lymph node metastases, associated with the carcinomas that did not express Dpc4, were also negative for gene product, whereas the lymph node metastases from Dpc4-positive carcinomas (Figure 3C) ▶ were also positive.
TGFBR2 Gene Sequencing
Sufficient material was available from three of the 13 carcinomas from patients with HNPCC for TGFBR2 gene sequencing. All three cases tested had bi-allelic mutations of the polyA tract of TGFBR2. In all three cases, immunolabeling revealed intact nuclear localization of Dpc4 (Figure 3A) ▶ .
Discussion
The findings in this study challenge the current concept that Dpc4 functions primarily to transduce TGF-β signals and highlight the need to re-explore the rationale behind accepted views on the relationship between Dpc4 and the TGF-β superfamily. Developmental systems studied in Drosophila and Xenopus have supported the concept that Dpc4 is a required mediator of TGF-β superfamily signals. In human tumors, breast carcinoma cell line 468 and colorectal cancer cell line SW480 cells have a defect in Dpc4 expression that results in their TGF-β unresponsiveness, 18-20 and somatic knockout of the DPC4 gene in a colorectal cancer cell line resulted in TGF-β unresponsiveness. 9
It is also known that Dpc4 can be influenced by other upstream signaling inputs. For example, developmental systems have principally concerned BMP-related ligands, not TGF-β itself. Similarly, the somatic knockout model of the DPC4 gene was not only TGF-β-unresponsive but also activin-unresponsive. 9
Indeed, there is considerable emerging evidence that raises doubts regarding, or at least complicates, the TGF-β-Dpc4 relationship. For example, DPC4-null pancreatic cancer and colorectal cancer cell lines do not, as a rule, lose TGF-β responsiveness, irrespective of whether one looks at transcriptional responses or growth suppression. 20,21 The ras genes, the MAP kinase pathway, and the MKK4-mediated stress-activated protein kinase pathway are implicated in these responses and would not involve the Dpc4 protein. 20-22 DPC4-null transgenic mouse cells have classes of TGF-β responses that remain intact. 23 Both pancreatic and colorectal tumors show instances in which there is co-existence of genetic inactivation of both a TGF-β receptor gene and the DPC4 gene. 24 This would not be expected if inactivation of one were sufficient to obviate the function of the other. These data together suggest considerable branching of the pathways uniting TGF-β receptors and Dpc4 (Figure 1) ▶ . Furthermore, preliminary immunohistochemical studies of pancreatic cancer also demonstrate nuclear localization of Dpc4 in the rare tumors having genetic inactivation of the TGF-β receptors (RE Wilentz, unpublished data).
The immunohistochemical labeling results in the current study raise the question of whether TGF-β responsiveness is necessary for Dpc4 function. We found intact nuclear localization of Dpc4 in human carcinomas with inactivating mutations in a TGF-β receptor gene and confirmed these observations using xenografted tumors from appropriate human cell lines. Although the mere presence of Dpc4 in the nucleus does not necessarily imply the existence of conditions sufficient for its tumor-suppressive function, there is evidence from studies of engineered cell lines that the direct manipulation of Dpc4 to relocalize to the nucleus, in the absence of exogenous administration of ligand, is sufficient to cause apoptosis and cell cycle inhibition. 25
The accumulated data therefore suggest that TGF-β signaling is not always required for the major tumor-suppressive functions of the DPC4 gene. It would now be important to determine whether there is a dominant signaling input to Dpc4 and what this input might be. On such an understanding would rest the better comprehension of some of the key driving forces in tumorigenesis. This, in turn, would lead to more precise identification of the molecular targets on which we might base hopes for the development of rational therapy for human cancer. Undoubtedly, additional direct observational studies of human tumors will be critical to the evaluation of competing hypotheses.
Footnotes
Address reprint requests to Scott E. Kern, M.D., The Johns Hopkins University, 451 Cancer Research Building, Baltimore, MD 21231. E-mail: sk@jhmi.edu.
Supported in part by CA62924 and CA68228 from the National Institutes of Health.
References
- 1.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA, Jr, Meltzer PS, Hahn SA, Kern SE: DPC4 gene in various tumor types. Cancer Res 1996, 56:2527-2530 [PubMed] [Google Scholar]
- 2.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1 [see comments]. Science 1996, 271:350-353 [DOI] [PubMed] [Google Scholar]
- 3.Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P, Tomlinson IP, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA: Mutations in the SMAD4/DPC4 gene in juvenile polyposis [see comments]. Science 1998, 280:1086-1088 [DOI] [PubMed] [Google Scholar]
- 4.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res 1995, 55:5548-5550 [PubMed] [Google Scholar]
- 5.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV: Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability [see comments]. Science 1995, 268:1336-1338 [DOI] [PubMed] [Google Scholar]
- 6.Myeroff LL, Parsons R, Kim SJ, Hedrick L, Cho KR, Orth K, Mathis M, Kinzler KW, Lutterbaugh J, Park K, Bang Y-J, Lee HY, Park J-G, Lynch HT, Roberts AB, Vogelstein B, Markowitz SD: A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res 1995, 55:5545-5547 [PubMed] [Google Scholar]
- 7.Postlethwaite AE, Raghow R, Stricklin G, Ballou L, Sampath TK: Osteogenic protein-1, a bone morphogenic protein member of the TGF-beta superfamily, shares chemotactic but not fibrogenic properties with TGF-beta. J Cell Physiol 1994, 161:562-570 [DOI] [PubMed] [Google Scholar]
- 8.Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH: A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400:687-693 [DOI] [PubMed] [Google Scholar]
- 9.Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, Kern SE, Kinzler KW, Vogelstein B: Targeted deletion of Smad4 shows it is required for transforming growth factor beta and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA 1998, 95:2412-2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heldin CH, Miyazono K, ten Dijke P: TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390:465-471 [DOI] [PubMed] [Google Scholar]
- 11.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000, 156:37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton SR, Vogelstein B, Kinzler KW: Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients [see comments]. Nat Med 1996, 2:169-174 [DOI] [PubMed] [Google Scholar]
- 13.Jass JR, Smyrk TC, Stewart SM, Lane MR, Lanspa SJ, Lynch HT: Pathology of hereditary non-polyposis colorectal cancer. Anticancer Res 1994, 14:1631-1634 [PubMed] [Google Scholar]
- 14.Kim H, Jen J, Vogelstein B, Hamilton SR: Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994, 145:148-156 [PMC free article] [PubMed] [Google Scholar]
- 15.Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, Kadkol SS, Yeo CJ, Choti M, Zahurak M, Johnson K, Tascilar M, Offerhaus GJ, Hruban RH, Kern SE: Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol 2000, 156:1641-1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goggins M, Hruban RH, Kern SE: BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications [In Process Citation]. Am J Pathol 2000, 156:1767-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE: Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998, 58:5329-5332 [PubMed] [Google Scholar]
- 18.Zhang Y, Feng X, We R, Derynck R: Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 1996, 383:168-172 [DOI] [PubMed] [Google Scholar]
- 19.de Winter JP, Roelen BA, ten Dijke P, van der Burg B, van den Eijnden-van Raaij AJ: DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-beta1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997, 14:1891-1899 [DOI] [PubMed] [Google Scholar]
- 20.Dai JL, Schutte M, Bansal RK, Wilentz RE, Sugar AY, Kern SE: Transforming growth factor-beta responsiveness in DPC4/SMAD4-null cancer cells. Mol Carcinog 1999, 26:37-43 [DOI] [PubMed] [Google Scholar]
- 21.Hocevar BA, Brown TL, Howe PH: TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J 1999, 18:1345-1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulder JW, Offerhaus GJ, de Feyter EP, Floyd JJ, Kern SE, Vogelstein B, Hamilton SR: The relationship of quantitative nuclear morphology to molecular genetic alterations in the adenoma-carcinoma sequence of the large bowel. Am J Pathol 1992, 141:797-804 [PMC free article] [PubMed] [Google Scholar]
- 23.Sirard C, Kim S, Mirtsos C, Tadich P, Hoodless PA, Itie A, Maxson R, Wrana JL, Mak TW: Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor beta-related signaling. J Biol Chem 2000, 275:2063-2070 [DOI] [PubMed] [Google Scholar]
- 24.Kern SE: Advances from genetic clues in pancreatic cancer. Curr Opin Oncol 1998, 10:74-80 [DOI] [PubMed] [Google Scholar]
- 25.Dai JL, Bansal RK, Kern SE: G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA 1999, 96:1427-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]