

Abstract
Chronic nephropathy is a recognized complication of sickle cell disease. Using a transgenic sickle mouse, we examined whether oxidative stress occurs in the sickle kidney, the origins and functional significance of such oxidant stress, and the expression of the oxidant-inducible, potentially protective gene, heme oxygenase-1 (HO-1); we also examined the expression of HO-1 in the kidney and in circulating endothelial cells in sickle patients. We demonstrate that this transgenic sickle mouse exhibits renal enlargement, medullary congestion, and a reduced plasma creatinine concentration. Oxidative stress is present in the kidney as indicated by increased amounts of lipid peroxidation; heme content is markedly increased in the kidney. Exacerbation of oxidative stress by inhibiting glutathione synthesis with buthionine-sulfoximine dramatically increased red blood cell sickling in the sickle kidney: in buthionine-sulfoximine-treated sickle mice, red blood cell sickling extended from the medulla into the cortical capillaries and glomeruli. HO activity is increased in the sickle mouse kidney, and is due to induction of HO-1. In the human sickle kidney, HO-1 is induced in renal tubules, interstitial cells, and in the vasculature. Expression of HO-1 is increased in circulating endothelial cells in patients with sickle cell disease. These results provide the novel demonstration that oxidative stress occurs in the sickle kidney, and that acute exacerbation of oxidative stress in the sickle mouse precipitates acute vaso-occlusive disease. Additionally, the oxidant-inducible, heme-degrading enzyme, HO-1, is induced regionally in the murine and human sickle kidney, and systemically, in circulating endothelial cells in sickle patients.
Sickle cell nephropathy is a cardinal complication of sickle cell disease, and up to 18% of patients so afflicted develop end-stage renal disease. 1-4 This latter outcome increases mortality by ∼50%, reduces patient survival to 4 years after its onset, and is associated with a median patient age of 27 years at the time of death. 4 Chronic renal insufficiency and end-stage renal failure are thus critical contributors to morbidity and mortality associated with sickle cell disease. 1-4
Renal involvement in sickle cell disease characteristically includes renal hypertrophy in the setting of prominent hemodynamic and vascular alterations. 1-3 Such hemodynamic and vascular processes impose a spectrum of effects on renal perfusion at the whole-organ and regional levels within the kidney: at one end of this spectrum, and in the cortical circulation, renal plasma flow rates and glomerular filtration rates (GFRs) are increased, whereas at the other, perfusion of the medulla is diminished because of vaso-occlusive disease. Such alterations are incriminated in the pathogenesis of specific phenotypic features of sickle cell nephropathy: increased glomerular perfusion and filtration impose hemodynamically-mediated damage to the glomerular compartment, whereas medullary ischemia impairs concentrating ability and other aspects of tubular function, induces histological tubulointerstitial disease, and may provoke frank papillary infarction. 1-3 Such vascular alterations—hyperemia in the case of the cortical circulation and ischemia in the case of the medullary circulation—may set in train inflammatory and fibrogenic processes that lead to progressive loss of renal function. Additionally, the kidney in sickle patients may be prone to intrinsic glomerulopathic processes manifested as focal segmental glomerulosclerosis and membranoproliferative glomerulonephritis. 1-3
Exploration of the mechanisms by which renal and other complications of sickle cell disease arise has been greatly facilitated by the recent availability of transgenic mice expressing sickle hemoglobin. 5-7 The use of these models has provided insights into mechanisms underlying sickling in vivo and the mechanisms contributing to end-organ damage. 5-7 These latter studies, for example, have revealed up-regulation of eNOS and iNOS in the kidneys of such mice, 8,9 and have raised the possibility that such alterations may contribute to the development of renal complications of sickle cell disease.
The present studies represent characterization of changes in the kidney in a more recently described transgenic sickle mouse. 10 This characterization includes, and focuses on, alterations in renal redox in this model. The rationale for examining renal redox resides in the recognition that the kidney in sickle cell disease is exposed to copious amounts of sickle hemoglobin. Because sickle hemoglobin is an unstable heme protein that may undergo scission to its heme and globin moieties, 11,12 we reasoned that the kidney would be exposed to large amounts of heme, the latter representing a lipophilic pro-oxidant. 11,12 In our examination of the involvement of oxidative stress in sickle cell nephropathy, we examined indices of oxidative stress along with heme content of the kidney.
We also examined expression of the redox sensitive enzyme, HO-1, as an index of oxidant stress. 13-15 Heme oxygenase (HO) is the rate limiting enzyme in the degradation of heme, converting heme to biliverdin in the course of which iron is released and carbon monoxide is emitted. HO consists of three isozymes, HO-1 representing the isozyme that is induced by oxidative stress, heme, and other stressors; HO-1 thus provides a relevant and sensitive index by which to assess alterations in cellular redox.
Materials and Methods
Transgenic Mouse Model of Sickle Cell Disease
This model is homozygous for deletion of mouse β-globin, and contains transgenes for human βS- and βS-antilles-globins linked to the transgene for human α-globin. 10 Studies were conducted in aged-matched control and sickle mice comprising similar numbers of male and female mice. Although the various studies of the kidney, in aggregate, involved mice that ranged in age from 0.5 to 1.5 years, each study was undertaken in similarly aged control and transgenic mice, each group comprising similar numbers of male and female mice.
Determination of Plasma Creatinine Concentration and Urinary Protein Concentration
Renal function was assessed by the concentration of plasma creatinine, the latter determined on plasma derived from tail-vein blood samples and using a Beckman Creatinine Analyzer II (Beckman Instruments, Inc., Fullerton, CA). 16 Urinary protein concentration was determined by the Coomassie method.
Lipid Peroxidation/Heme Content
Lipid peroxidation was assessed by the thiobarbituric acid reactive substance assay, as previously described. 17 Heme content of whole kidney homogenate, and of various cellular fractions, was determined by the pyridine hemochromogen method, as previously described. 17
Measurement of HO and Other Antioxidant Enzyme Activities
As described in detail in our previous study, 18 HO activity was measured by the method of Pimstone and colleagues, 19 catalase by the method of Aebi and colleagues, 20 and glutathione peroxidase by the method of Lawrence and Burk. 21
Northern Analysis
RNA from kidneys was extracted using the Trizol method (Life Technologies, Inc., Gaithersburg, MD). Twenty micrograms of total RNA from each sample were separated on an agarose gel and transferred to a nylon membrane. 16 Membranes were hybridized overnight with 32P-labeled mouse HO-1 and mouse HO-2 cDNA probes. 22 Autoradiograms were standardized, as previously described, 22 by factoring the optical density of the message for HO-1 with the optical density of the 18S rRNA, the latter obtained on a negative of the ethidium bromide-stained nylon membrane.
Detection of HO-1 by Immunoperoxidase
Kidney sections, derived at autopsy, were studied for the expression of HO-1 by immunoperoxidase. The patient was female, 47 years old, had a history of modest severity (average of three pain crises per year), and died of a massive thromboembolism during an acute painful episode. Normal kidney tissue from a nephrectomized specimen served as a control. Kidney sections were stained for HO-1 using a monoclonal HO-1 antibody (OSA-111; Stressgen, Victoria, BC, Canada) as the primary antibody, a polyclonal goat anti-mouse IgG as the secondary antibody (SAB-100, Stressgen), and diaminobenzidine as substrate for localization, as previously described in detail. 16,22
Detection of HO-1 in Circulating Endothelial Cells (CECs) in Patients with Sickle Cell Disease
CECs were evaluated for expression of HO-1 using the methods we have previously described in detail. 23,24 Briefly, using fresh whole blood anticoagulated with ethylenediaminetetraacetic acid, we used immunomagnetic beads (Dynal, Oslo, Norway) coated with the anti-endothelial cell monoclonal antibody, P1H12, to prepare a CEC-enriched population of cells from the buffy coat. 23,24 After transfer to slides, cells were fixed with 4% paraformaldehyde and permeabilized with 0.4% Triton X-100. Cells were stained for HO-1 using a primary polyclonal antibody (SPA-895, Stressgen), followed by a rhodamide-labeled goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, Pa). A negative control was provided by use of an irrelevant primary antibody. Cells were scored as being “negative” if they showed no increase in staining higher than that of the negative control sample done in parallel. Cells were scored as being “low positive” if their staining intensity exceeded that of the negative control but was still less than that of the red autofluorescence of the immunomagnetic beads in the preparation (Figure 9 ▶ , top left). Cells were scored as being “high positive” if their staining was brighter than the bead autofluorescence (Figure 9 ▶ , top right). Results are expressed as percentage of CECs being negative, low positive, or high positive. Sufficient sample was evaluated for each donor so that at least 10 CECs were screened per normal donor and at least 20 CECs were screened per sickle donor; this difference is accounted for by the much smaller number of CECs per ml of blood in normal donors. 23 These studies were conducted in four healthy patients and in five patients with sickle cell disease.
Figure 9.

Immunoperoxidase staining for HO-1 in the kidney of a patient with sickle cell disease. Immunoperoxidase studies were undertaken in the absence (left) and presence (right) of HO-1 antibody. Right: Positive staining for renal distal tubules and interstitial cells in the sickle kidney. Original magnification, ×400.
The Effect of Acute Reduction in Renal Thiol Content in Control and Sickle Mice
Buthionine-sulfoximine (BSO), an inhibitor of γ-glutamylcysteine synthetase (the first step in glutathione synthesis), was administered every 12 hours for 3 days to control and sickle mice (5 mmol/kg i.p.). 25 The kidneys were then harvested for the assessment of thiol content and histological studies. Thiol content was determined by the method based on the reduction of the Ellman reagent, 5,5′-dithiobis (2-nitrobenzoic acid), as previously described. 26
Statistics
Results are presented as means ± SEM. For statistical analyses, the Student’s t-test or the Mann-Whitney test was used as appropriate. Results are considered significant for P < 0.05.
Results
Studies in Mice
The transgenic sickle mouse demonstrates increased kidney weight and increased body weight as compared to control mice: both the absolute kidney weight as well as the kidney weight factored for body weight were increased in sickle mice (Table 1) ▶ . The concentration of plasma creatinine was significantly lower in sickle mice, and in light of the greater body weight in sickle mice, this likely reflects a higher GFR in the transgenic sickle mouse. Neither the urinary protein concentration, nor the ratio of urinary concentration of protein/urinary concentration of creatinine, was altered in sickle mice (Table 1) ▶ .
Table 1.
Characteristics of Control and Sickle Mice at 6 to 8 Months of Age
| Control | Sickle | P value | |
|---|---|---|---|
| Body weight (g) | 23.7 ± 0.9 | 28.1 ± 2.0 | <0.05 |
| Hematocrit (%) | 49 ± 1 | 48 ± 2 | NS |
| Kidney weight (g) | 0.16 ± 0.01 | 0.23 ± 0.02 | <0.01 |
| Kidney weight/body weight ×10−3 | 6.8 ± 0.3 | 7.8 ± 0.4 | <0.05 |
| Plasma creatinine (mg/dl) | 0.34 ± 0.02 | 0.20 ± 0.01 | <0.0001 |
| Urinary protein concentration (mg/dl) | 73.7 ± 10.3 | 68.1 ± 9.7 | NS |
| Urinary protein concentration/urinary creatinine concentration | 2.4 ± 0.3 | 1.7 ± 0.3 | NS |
The control group comprised 12 to 14 mice; the sickle group comprised 9 to 10 mice.
NS, not significant.
Histological assessment of the kidney in the sickle mouse demonstrated congestion of the medullary vessels with spontaneous sickling of RBCs (Figure 1) ▶ . Sickle mice studied for up to 1.5 years of age did not develop progressive renal disease; in particular, there was no significant segmental or global glomerulosclerosis, interstitial fibrosis, atrophy of cortical or medullary tubules other than that associated with aging in control, aged-matched, mice.
Figure 1.
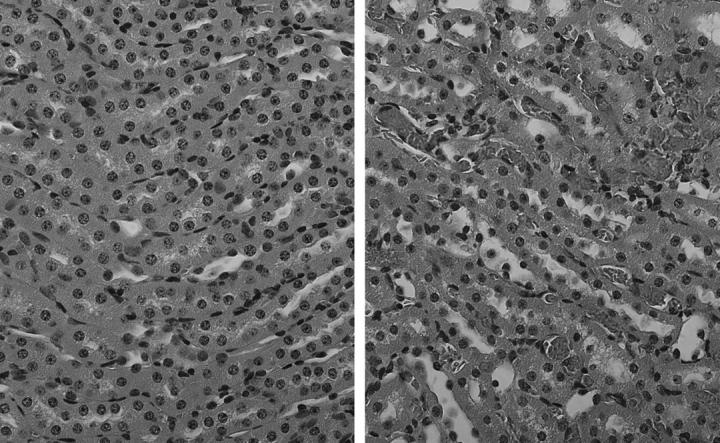
Histological sections of the kidney in control mice (left) and sickle mice (right) stained with H&E and demonstrating vascular congestion in the medulla in sickle mice. Original magnification, ×400.
We determined lipid peroxidation by the thiobarbituric acid-reactive substance assay and demonstrate a significant increase in the kidney in the sickle mouse (Figure 2) ▶ . The content of heme—the latter representing a strong pro-oxidant species—in the sickle kidney was uniformly increased in all cellular compartments including whole-kidney homogenates, and in cytosolic, mitochondrial, and microsomal fractions of the kidney (Figure 3) ▶ .
Figure 2.

Lipid peroxidation as measured by the thiobarbituric acid reactive substance assay in control mice (open column, n = 8) and sickle mice (striped column, n = 10). *, P < 0.05, control versus sickle mice.
Figure 3.

Heme content in whole kidney homogenate and subcellular fractions in control mice (open column) and sickle mice (striped column). In all studies, n = 8 in control mice and n = 10 in sickle mice. *, P < 0.05, control versus sickle mice.
Because heme content was increased in the sickle kidney in conjunction with increased oxidative stress, we assessed, by measuring HO activity, whether heme-degrading activity was altered in the sickle kidney. Such enzyme activity was increased in kidney microsomes prepared from sickle mice (181 ± 9 versus 236 ± 10 pmol/hour/mg protein; P < 0.05; n = 8 in control and sickle mice).
As HO is an antioxidant enzyme, we measured other antioxidant enzyme activity to determine whether such increase in HO activity was part of a more generalized antioxidant response. In this regard, catalase activity was unchanged in the sickle kidney (0.30 ± 0.02 versus 0.33 ± 0.02, k/mg protein; P = ns; n = 8 in control group and n = 10 in sickle mice) whereas glutathione peroxidase was reduced in the sickle kidney (589 ± 24 versus 509 ± 26, nmol/min/mg protein; P < 0.05; n = 8 in control group and n = 10 in sickle mice). Thus, the increase in HO activity does not seem to be part of a more widespread induction of antioxidant systems.
Increased HO activity may reflect induction of one of the HO isoforms. We thus assessed the expression of HO-1 and HO-2 by Northern analysis. HO-1 mRNA was increased some fivefold in the sickle kidney (Figure 4) ▶ , whereas mRNA expression for HO-2, the constitutively expressed isoform, was unaltered in the sickle kidney (mean standardized densitometric values: 7.91 ± 1.38 versus 8.40 ± 1.22; P = ns; n = 4 in control and sickle mice). Thus, induction of HO-1 accompanies the presence of oxidative stress in the sickle kidney.
Figure 4.

Northern analysis for HO-1 expression in control and sickle kidneys. Each lane represents RNA extracted from a single kidney of an individual mouse. Provided below the Northern analysis are the standardized, individual, densitometric readings and the mean standardized, densitometric readings for each group.
To determine the functional significance of oxidative stress in the sickle kidney, we depleted thiol content of the kidney in transgenic sickle mice by administering BSO, an agent that inhibits glutathione synthesis and promotes a pro-oxidant state. That BSO, so administered, did achieve reduction in kidney thiol content is shown in Figure 5 ▶ : kidney thiol content was significantly reduced in both groups, ∼50% reduction in control mice and 38% reduction in sickle mice. Such administration of BSO did not have any apparent histological effect in the kidney in control mice (data not shown); however, such administration of BSO markedly exacerbated RBC sickling in the kidney of the sickle mouse (Figures 6 and 7) ▶ ▶ . Medullary congestion was worsened in sickle mice treated with BSO as compared with sickle mice treated with vehicle (Figure 6) ▶ . Additionally, and strikingly, BSO-treated sickle mice—but not vehicle-treated sickle mice—exhibited extension of vascular congestion and RBC sickling from the medulla (where they are normally exhibited, and restricted to, in this unstressed transgenic sickle kidney) into cortical capillaries, cortical arterioles, and glomeruli (Figure 7) ▶ . Thus, acute oxidative stress in the kidney, as induced by reduction in kidney thiol content by BSO, exacerbates the sickling process that occurs in the kidney.
Figure 5.

Effect of BSO or vehicle on kidney thiol content in control and sickle mice. In all groups n = 8 except the BSO-treated sickle mice where n = 7. *, P < 0.05, vehicle-treated versus BSO-treated mice in the control and sickle groups.
Figure 6.
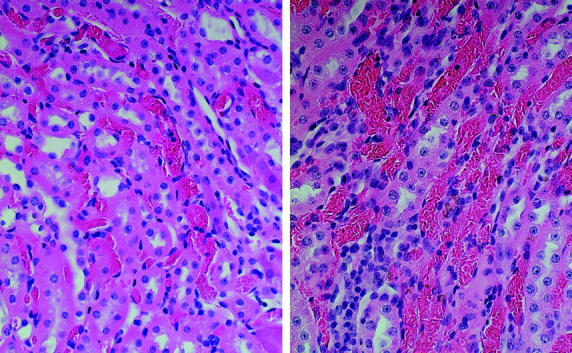
Vascular congestion and RBC sickling in the renal medulla in sickle mice treated with vehicle (left) or BSO (right). Original magnification, ×400.
Figure 7.
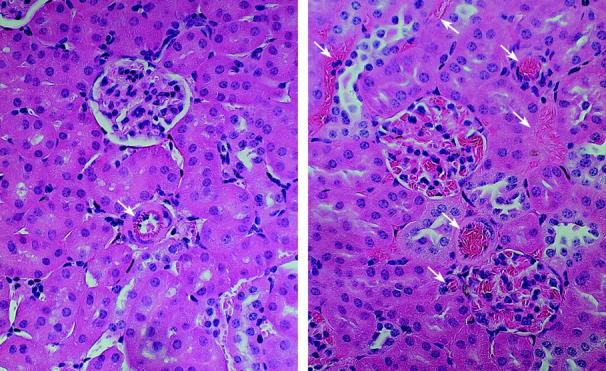
Vascular congestion and RBC sickling in the renal cortex in sickle mice treated with vehicle (left) or BSO (right). The white arrows point out arterioles and capillaries. BSO-treated sickle mice demonstrate vascular congestion and RBC sickling in glomeruli, arterioles, and capillaries in the renal cortex (right), findings not observed in glomeruli, arterioles, and capillaries in vehicle-treated sickle mice (left). Original magnification, ×400.
Studies in Patients with Sickle Cell Disease
To determine whether HO-1 is induced in the kidney in patients with sickle cell disease, we examined the expression of HO-1 in the kidney obtained from a sickle patient who died after a massive pulmonary embolus. The healthy human kidney does not express HO-1 as shown in Figure 8A ▶ . In contrast, the sickle kidney exhibits diffuse and widespread brown staining, that was HO-1 antibody-specific, in proximal and distal renal tubules (Figures 8B and 9) ▶ ▶ , in interstitial cells (Figures 8B and 9) ▶ ▶ , and in endothelial cells and in the smooth muscle cells in the media of small arteries (Figure 10) ▶ . Focal glomerular staining within isolated cells was also observed (data not shown); however, the nature of these cells—infiltrating mononuclear cells or mesangial cells—demonstrating such staining awaits further investigation.
Figure 8.

Immunoperoxidase staining for HO-1 in normal human kidney (A) and in the kidney of a patient with sickle cell disease (B). In A and B, immunoperoxidase studies were undertaken in the absence (left) and presence (right) of HO-1 antibody. B: Positive staining with the HO-1 antibody in renal proximal tubules in the kidney of the patient with sickle cell disease (right). Original magnification, ×400.
Figure 10.
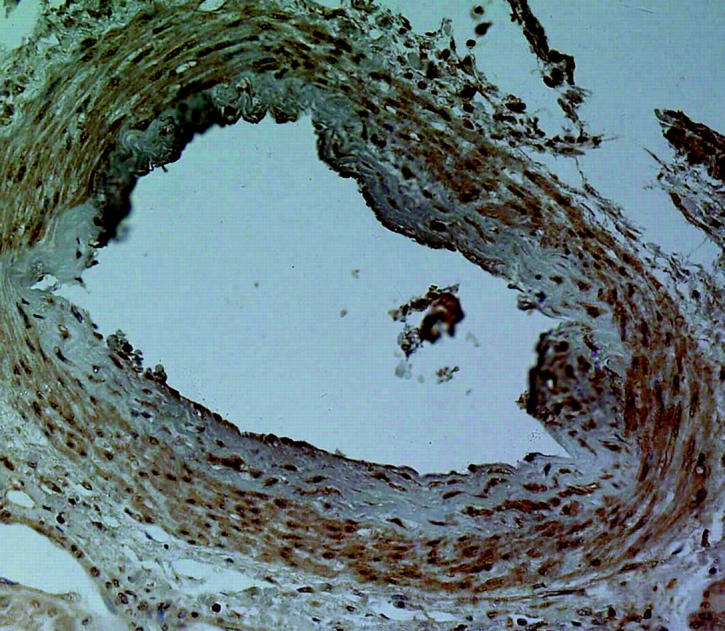
Immunoperoxidase staining for HO-1 in the vasculature of the kidney of a patient with sickle cell disease. This figure shows positive staining for HO-1 in the endothelium and the smooth muscle cells of the media of a small artery. Original magnification, ×400.
We also examined the expression of HO-1 in CECs in five patients with sickle cell disease and four healthy patients, the latter serving as controls. As demonstrated in Figures 11 and 12 ▶ ▶ , high positive staining for HO-1 in CECs was markedly greater in sickle patients as compared to healthy controls.
Figure 11.
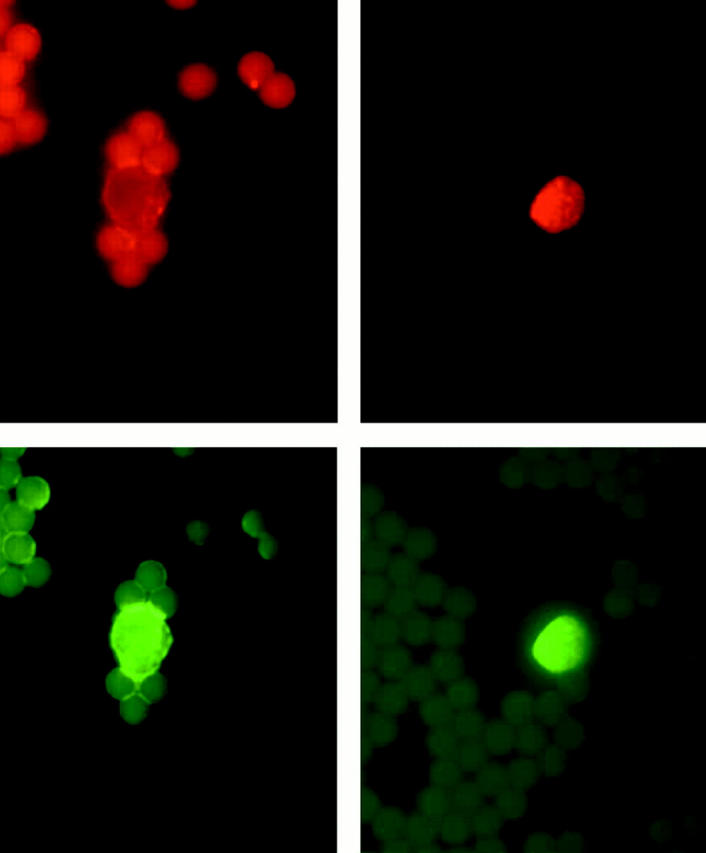
Representative dual-labeling studies of CECs stained with the antibody for HO-1 (red staining, top left and right) and with the endothelium-specific antibody, mAb-P1H12, (green staining, bottom left and right). Top left: Faint red staining with HO-1 antibody in an endothelial cell from a control subject, and designated “low positive” in scoring summarized in Figure 12 ▶ . Top right: Strong red staining with HO-1 antibody in an endothelial cell from a sickle patient, and designated “high positive” in scoring summarized in Figure 12 ▶ . Bottom left: Green staining with mAb-P1H12 of the cell shown in top left demonstrating that this is an endothelial cell. Bottom right: Green staining with mAb-P1H12 of the cell shown in top right demonstrating that this is an endothelial cell.
Figure 12.

Quantitation of HO-1 staining in CECs derived from healthy controls (open column, n = 4) and patients with sickle cell disease (striped column, n = 5). As described in the text and shown in Figure 11 ▶ , staining for HO-1 is categorized as “negative,” “low positive,” and “high positive.” *, P < 0.05, healthy control patient versus patients with sickle cell disease for that category of HO-1 staining.
Discussion
The transgenic sickle mouse characterized in the present study reproduces several of the salient features of sickle cell nephropathy: the kidney is enlarged, and spontaneous sickling of RBCs occur in the medulla; the serum creatinine is significantly lower in sickle mice, and in light of the greater body weight of sickle mice, this likely reflects a higher GFR in these animals. This latter finding, along with renal hypertrophy, are recognized features in patients with sickle cell disease. 1-3
In our characterization of this model, we demonstrate the presence of oxidative stress in the kidney. Our hypothesis that oxidative stress would occur in sickle cell disease drew on the notion that sickle hemoglobin, being an unstable heme protein, 11,12 would undergo scission and thus provide increased amounts of heme in organs such as the kidney; heme is a recognized lipophilic pro-oxidant. 11,12 Indeed, we demonstrate that heme content was markedly increased in kidney tissue, and is accompanied by increased amounts of lipid peroxidation. We suggest that such increased amounts of heme, especially in the setting of increased GFR, may further contribute to oxidative stress in this model. Increased GFR obligates increased sodium transport by the renal tubular epithelium, and as oxygen consumption is linked to the transport of sodium, increased sodium transport entails increased oxygen utilization; 27 increased oxygen consumption, in turn, may lead to increased generation of reactive species. 28
Oxidative stress in sickle cell disease may originate from other sources including sickle erythrocytes and leukocytes: sickle RBCs may themselves generate oxidants 29 or stimulate endothelial cells to generate oxidants 30 ; sickle RBCs rather than normal RBCs adhere to polymorphonuclear leukocytes and activate the respiratory burst. 31 Systemic levels of cytokines are elevated in sickle cell disease, even in the quiescent, crisis-free, phase, 32-36 and several of these cytokines, (for example, tumor necrosis factor-α and interleukin-6) are pro-oxidant. It is also possible that, insofar as repetitive episodes of vaso-occlusion occur in the sickle vasculature, and accordingly, expose tissues to ischemia/reperfusion injury, attendant generation of oxidants may occur through this mechanism.
To examine further the contribution of alterations in cellular redox to the complications of sickle cell disease, we decreased renal thiol content by administering BSO, an inhibitor of the synthesis of glutathione 26 ; glutathione represents the dominant thiol in tissues. Using this manipulation, we demonstrate marked effects in sickle but not control mice: the kidney in the sickle mouse, exposed to the glutathione-depleting agent, BSO, exhibited extension of vascular congestion from the inner medulla (where RBC congestion is normally exhibited in this unstressed transgenic sickle kidney) into the outer medulla, cortical capillaries, and glomerular capillaries. It would be of interest to determine whether worsening of renal function accompanies this exacerbation of vaso-occlusive disease in sickle mice after the administration of BSO. It should also be pointed out that BSO likely induces oxidative stress in other tissues besides the kidney, including, and in particular, the erythrocyte. The content of GSH in erythrocytes is likely decreased after the administration of BSO. Thus, the precipitation of vaso-occlusive disease in the kidney in sickle mice after the administration of BSO, in all likelihood, reflects the integrated effects of oxidative stress induced in the kidney as well as oxidative stress induced in erythrocytes. We thus provide the novel observation that acute oxidative stress, as imposed by acute depletion of kidney thiol content, and in all likelihood, in conjunction with depletion of thiol content of erythrocytes, markedly predisposes to vaso-occlusive disease in the sickle kidney. Based on these findings, we suggest that oxidative stress may be a mechanism by which recognized inducers of sickle crisis—sepsis, in particular, which alters cellular redox—provoke acute vascular congestion and vaso-occlusive disease.
Vaso-occlusive processes occupy a pivotal position in the evolution of acute and chronic complications of sickle cell disease. 37,38 Acute vaso-occlusive disease imposes ischemic/hypoxic, metabolic, and other insults, whereas recurrent cycles of occlusion and resolution of occlusion, subject tissues to ischemia-reperfusion pathobiology and other mechanisms that underlie chronic organ injury. 37,38 A number of pathophysiological processes contribute to vaso-occlusive disease including sickling of erythrocytes, abnormal erythrocyte rheology, proinflammatory and procoagulant processes, activation of the endothelium, enhanced adhesion of erythrocytes to the endothelium, increased production of vasoactive substances, and aberrant vascular and neurohumoral responses. 37,38 We provide the novel demonstration that alteration in cellular redox can markedly influence vaso-occlusive processes in sickle cell disease.
The presence of oxidative stress and increased amounts of heme in the sickle kidney led us to examine the expression of HO-1 in the sickle kidney. HO is the rate-limiting enzyme in the degradation of heme, and the isozyme, HO-1, is readily responsive to oxidative stress and heme. 13-15 We demonstrate up-regulation of HO-1 in the sickle kidney accompanied by increased HO activity. This response seemed relatively specific because expression of the constitutive form of HO, HO-2, was unaltered, whereas activities of other antioxidant enzymes such as catalase and glutathione peroxidase were not increased. In complementary studies undertaken in a patient with sickle cell disease, HO-1 is also up-regulated in the kidney as evidenced by increased expression of HO-1 in renal tubular epithelial cells, in interstitial cells, and in the vasculature; focal staining for HO-1 was observed in glomeruli.
We speculate that such induction of HO may retard pathways of injury in the sickle kidney. HO activity degrades heme and assists in the cellular handling of iron; 13-15 HO-1 is also an antioxidant and thereby protects against the injurious effects of oxidative stress. 13-15 HO-1 facilitates the synthesis of ferritin that is the main chelator of iron in tissues. 13,14 HO-1 generates carbon monoxide, and through this gaseous product, HO-1 promotes vasodilatation, 39,40 the latter serving to retard the evolution of vaso-occlusive disease. HO-1 is anti-inflammatory through the recognized effects of its products, carbon monoxide and bilirubin; 41,42 for example, carbon monoxide is a cytoprotectant, 42 that can suppress proinflammatory cytokines such as platelet-derived growth factor and endothelin-1; 43 bilirubin can attenuate adhesion of leukocytes to the venular endothelium. 44 The particular importance of HO-1 in defending the kidney against heme protein-induced, acute or chronic nephrotoxicity is underscored by our recent studies in HO-1 knockout mice: HO-1 knockout mice demonstrate markedly worse renal injury and increased mortality as compared to wild-type mice when subjected either to the acute nephrotoxic effects of hemoglobin, 16 or to the chronic inflammatory effects of repetitively administered hemoglobin. 45 Drawing on these findings, we speculate that HO-1, induced in the sickle kidney, confers cytoprotectant, anti-inflammatory actions in the sickle kidney, the latter exposed as it is to large amounts of heme proteins.
Up-regulation of HO-1 in patients with sickle cell disease was not restricted to the kidney in that CECs in sickle patients demonstrate increased expression of HO-1. Recent studies of CECs have added new insights into the pathobiology of sickle cell disease. 23,24,46 CECs are detected in sparse numbers in health, whereas in patients with sickle cell disease, the numbers of these cells increase fivefold in the steady state, and 10-fold in sickle crisis. 23 Moreover, these endothelial cells display an activated phenotype as reflected by their up-regulation of adhesion molecules and selectins. 23,24,46,47 Added evidence in support of an inflammatory systemic milieu in sickle cell disease is provided by elevated leukocytic counts which occur in these patients, and which correlate with their propensity to vaso-occlusive complications and overall mortality in sickle cell disease. 48 These findings, in conjunction with elevated circulating levels of cytokines and acute phase response proteins, 49,50 in aggregate, have led to the concept of sickle cell disease as a proinflammatory disease. Interestingly, many of the key participants in inflammation—chemokines, adhesion molecules, and selectins—may be induced by oxidative stress: 51 oxidative stress occurring in sickle cell disease may thus contribute to the inflammatory phenotype of this disease. In this regard, the induction of HO-1, an antioxidant gene with anti-inflammatory properties, 13-15 in CECs and in the kidney may offer a countervailing response in a proinflammatory milieu.
The lack of histological features of progressive renal injury in this transgenic sickle mouse model, despite the presence of vascular congestion in the medulla of the sickle kidney, merits comment. Firstly, it should be pointed out that only a subset of patients with sickle cell disease develop progressive nephropathy: this mouse model, in its basal unstressed state, may thus be more representative of those sickle patients who are not afflicted by progressive renal disease; as such, this transgenic sickle model may be particularly useful in examining mechanisms that provoke progressive renal injury in sickle cell disease. Perhaps, the severity of oxidative stress serves as a determinant of progressive renal disease; and in this regard, it is possible that exacerbation of oxidative stress in this transgenic sickle mouse by more sustained administration of BSO would eventuate in progressive renal injury. Secondly, the induction of HO-1 in the sickle kidney may mitigate chronic inflammatory processes that may otherwise occur. Relevant to this consideration is our recent demonstration that HO-1 knockout mice, but not wild-type mice, demonstrate marked tubulointerstitial inflammation when repetitively exposed to hemoglobin. 45 Thirdly, models of chronic renal histological injury that are established in rats, when applied to mice, are often surprisingly mild and unprepossessing: for example, the remnant kidney model in the mouse fails to evince significant proteinuria, glomerulosclerosis, tubulointerstitial disease, or vascular sclerosis when followed for up to 44 weeks 52 ; Mpv 17-deficient mice, which are characterized by heavy and sustained proteinuria, do not develop tubulointerstitial disease, when examined for up to 52 weeks of age. 53
That expression of HO-1 is increased in sickle cell disease may also be relevant to the long-recognized but never explained finding of increased amounts of carboxyhemoglobin in patients with sickle cell disease. 54 Carboxyhemoglobin levels reflect, in part, production of carbon monoxide, 55 and the main biochemical system in the body that produces carbon monoxide is HO. 13 We thus suggest that elevated levels of carboxyhemoglobin observed in sickle patients reflect induction of HO-1 in sickle cell disease.
CECs were used in examining expression of HO-1 for two main reasons: firstly, the use of these cells addresses the question whether cells other than the kidney in sickle cell disease express HO-1; secondly, the study of these cells has afforded insights in a number of other conditions attended by vascular injury including myocardial infarction, sepsis, traumatic vascular injury, and venous insufficiency. 23,24,56 Given the presence of vascular injury in renal diseases in general, and the contribution of vascular injury to the morbidity and mortality associated with renal diseases, the study of CECs merits consideration as an approach in the investigation of vascular injury in renal diseases.
In summary, alterations in cellular redox occur in the sickle kidney. Such redox alterations, we posit, contribute to vascular pathobiological processes and inflammatory pathways that conspire in the pathogenesis of sickle cell nephropathy. These redox alterations are countered by the induction of HO-1, a system that, quite remarkably, possesses attributes that may oppose many of the pathogenetic mechanisms underlying renal and other complications of sickle cell disease.
Acknowledgments
We thank Mrs. Sharon Heppelmann for secretarial expertise in the preparation of this work.
Footnotes
Address reprint requests to Dr. Karl A. Nath, Mayo Clinic, 200 First St., SW, 542 Guggenheim Bldg., Rochester, MN 55905. E-mail: nath.karl@mayo.edu.
Supported by National Institutes of Health grants HL-55552 (to K. A. N. and R. P. H.), DK-47060 (to K. A. N.), DK-16105 (to J. P. G.), and HL-53524 (to Z. S. K.).
References
- 1.Falk RJ, Jennette JC: Sickle cell nephropathy. Adv Nephrol 1994, 23:133-147 [PubMed] [Google Scholar]
- 2.Saborio P, Scheinman JI: Sickle cell nephropathy. J Am Soc Nephrol 1999, 10:187-192 [DOI] [PubMed] [Google Scholar]
- 3.Pham T-TT, Pham P-CT, Wilinson AH, Lew SQ: Renal abnormalities in sickle cell disease. Kidney Int 2000, 57:1-8 [DOI] [PubMed] [Google Scholar]
- 4.Powars DR, Elliot-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, Johnson C: Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med 1991, 115:614-620 [DOI] [PubMed] [Google Scholar]
- 5.Fabry ME, Nagel RL, Pachnis A, Suzuka SM, Costantini F: High expression of human β S- and α-globins in transgenic mice. I. Hemoglobin composition and hematological consequences. Proc Natl Acad Sci USA 1992, 89:12150-12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabry ME, Costantini FD, Pachnis A, Suzuka SM, Bank N, Aynedjian HS, Factor S, Nagel RL: High expression of human βs and α-genes in transgenic mice. II. Red cell abnormalities, organ damage, and the effect of hypoxia. Proc Natl Acad Sci USA 1992, 89:12155-12159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagel RL: A knockout of a transgenic mouse: animal models of sickle cell anemia. N Engl J Med 1998, 339:194-195 [DOI] [PubMed] [Google Scholar]
- 8.Bank N, Aynedjian HS, Qiu J-H, Osei SY, Ahima RS, Fabry ME, Nagel RL: Renal nitric oxide synthases in transgenic sickle cell mice. Kidney Int 1996, 50:184-189 [DOI] [PubMed] [Google Scholar]
- 9.Bank N, Kiroycheva M, Ahmed F, Anthony GM, Fabry ME, Nagel RL, Singhal PC: Peroxynitrite formation and apoptosis in transgenic sickle cell mouse kidneys. Kidney Int 1998, 54:1520-1528 [DOI] [PubMed] [Google Scholar]
- 10.Fabry ME, Sengupta A, Suzuka SM, Costantini F, Rubin EM, Hofrichter J, Christoph G, Manci E, Culberson D, Factor SM, Nagel RL: A second generation transgenic mouse model expressing both hemoglobin S (HbS) and HbS-Antilles results in increased phenotypic severity. Blood 1995, 86:2419-2428 [PubMed] [Google Scholar]
- 11.Hebbel RP: The sickle erythrocyte in double jeopardy: autooxidation and iron decompartmentalization. Semin Hematol 1990, 27:51-69 [PubMed] [Google Scholar]
- 12.Hebbel RP: Membrane-associated iron. Embury SH Hebbel RP Mohandas N Steinberg MH eds. Sickle Cell Disease: Basic Principles and Clinical Practice, ch 12. 1994, :pp 163-172 Raven Press, Ltd., New York [Google Scholar]
- 13.Platt JL, Nath KA: Heme oxygenase: protective gene or trojan horse. Nat Med 1998, 4:1364-1365 [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Lavrovsky Y, Venkatachalam MA, Roy AK: Heme oxygenase-1 in tissue pathology. Am J Pathol 2000, 156:1485-1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal A, Nick HS: Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J Am Soc Nephrol 2000, 11:965-973 [DOI] [PubMed] [Google Scholar]
- 16.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J: The indispensability of heme oxygenase-1 (HO-1) in protecting against heme protein-induced toxicity in vivo. Am J Pathol 2000, 156:1527-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath KA, Grande JP, Croatt AJ, Likely S, Hebbel RP, Enright H: Intracellular targets in heme protein induced renal injury. Kidney Int 1998, 53:100-111 [DOI] [PubMed] [Google Scholar]
- 18.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME: Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 1992, 90:267-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pimstone NR, Engel P, Tenhunen R, Seitz PT, Marver HS, Schmid R: Inducible heme oxygenase in the kidney: a model for the homeostatic control of hemoglobin catabolism. J Clin Invest 1971, 50:2042-2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aebi H: Catalase. Bergmeyer HU eds. Methods of Enzymatic Analysis, 1974, vol 2.:pp 673-684 Academic Press, Inc., New York [Google Scholar]
- 21.Lawrence RA, Burk RF: Glutathione peroxidase activity in selenium deficient rat liver. Biochem Biophys Res Commun 1976, 95:351-358 [DOI] [PubMed] [Google Scholar]
- 22.Haugen EN, Croatt AJ, Nath KA: Angiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int 2000, 58:144-152 [DOI] [PubMed] [Google Scholar]
- 23.Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP: Circulating activated endothelial cells in sickle cell anemia. N Engl J Med 1997, 337:1584-1590 [DOI] [PubMed] [Google Scholar]
- 24.Solovey A, Gui L, Key NS, Hebbel RP: Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest 1998, 101:1899-1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minchinton AI, Rojas A, Smith KA, Soranson JA, Shrieve DC, Jones NR, Bremner JC: Glutathione depletion in tissues after administration of buthionine sulphoximine. Int J Radiat Oncol Biol Phys 1984, 10:1261-1264 [DOI] [PubMed] [Google Scholar]
- 26.Nath KA, Enright H, Nutter L, Fischereder MF, Zou JN, Hebbel RP: Effect of pyruvate on oxidant injury to isolated and cellular DNA. Kidney Int 1994, 45:166-176 [DOI] [PubMed] [Google Scholar]
- 27.Gullans SR, Hebert SC: Metabolic basis of ion transport. Brenner BM eds. The Kidney, ch 5. 1996, :pp 211-246 WB Saunders Co., Philadelphia [Google Scholar]
- 28.Nath KA, Fischereder M, Hostetter TH: The role of oxidants in progressive renal injury. Kidney Int 1994, 45:S111-S115 [PubMed] [Google Scholar]
- 29.Hebbel RP, Eaton JW, Balasingam M, Steinberg MH: Spontaneous oxygen radical generation by sickle erythrocytes. J Clin Invest 1982, 70:1253-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK: Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood 1998, 92:3924-3935 [PubMed] [Google Scholar]
- 31.Hofstra TC, Kalra VK, Meiselman HJ, Coates TD: Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood 1996, 87:4440-4447 [PubMed] [Google Scholar]
- 32.Malave I, Perdomo Y, Escalona E, Rodriguez E, Anchustegui M, Malave H, Arends T: Levels of tumor necrosis factor α/cachectin (TNFα) in sera from patients with sickle cell disease. Acta Haematol 1993, 90:172-176 [DOI] [PubMed] [Google Scholar]
- 33.Taylor SC, Shacks SJ, Mitchell RA, Banks A: Serum interleukin-6 levels in the steady state of sickle cell disease. J Interferon Cytokine Res 1995, 15:1061-1064 [DOI] [PubMed] [Google Scholar]
- 34.Kuvibidila S, Gardner R, Ode D, Yu L, Lane G, Warrier RP: Tumor necrosis factor α in children with sickle cell disease in stable condition. J Natl Med Assoc 1997, 89:609-615 [PMC free article] [PubMed] [Google Scholar]
- 35.Graido-Gonzalez E, Doherty JC, Bergreen EW, Organ G, Telfer M, McMillen MA: Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood 1998, 92:2551-2555 [PubMed] [Google Scholar]
- 36.Croizat H, Nagel RL: Circulating cytokines response and the level of erythropoiesis in sickle cell anemia. Am J Hematol 1999, 60:105-115 [DOI] [PubMed] [Google Scholar]
- 37.Embury SH, Hebbel RP, Steinberg MH, Mohandas N: Pathogenesis of vasoocclusion. Embury SH Hebbel RP Mohandas N Steinberg MH eds. Sickle Cell Disease: Basic Principles and Clinical Practice, ch 21. 1994, :pp 311-326 Raven Press, Ltd., New York [Google Scholar]
- 38.Hebbel RP: Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest 1997, 100:S83-S86 [PubMed] [Google Scholar]
- 39.Motterlini R, Gonzales A, Foresti R, Clark JE, Green CJ, Winslow RM: Heme oxygenase-1-derived carbon monoxide contributes to the suppression of acute hypertensive responses in vivo. Circ Res 1998, 83:568-577 [DOI] [PubMed] [Google Scholar]
- 40.Kozma F, Johnson RA, Zhang F, Yu C, Trong X, Nasjletti A: Contribution of endogenous carbon monoxide to regulation of diameter in resistance vessels. Am J Phsyiol 1999, 276:R1087-R1094 [DOI] [PubMed] [Google Scholar]
- 41.Nath KA: Heme oxygenase-1: a redoubtable response that limits reperfusion injury in the transplanted adipose liver. J Clin Invest 1999, 104:1485-1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otterbein LE, Mantell LL, Choi AM: Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol 1999, 276:L688-L694 [DOI] [PubMed] [Google Scholar]
- 43.Morita T, Kourembanas S: Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest 1995, 96:2676-2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T, Kitajima M, Makino N, Ishimura Y, Suematsu M: Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress. Circ Res 1999, 85:663-671 [DOI] [PubMed] [Google Scholar]
- 45.Nath KA, Vercellotti G, Grande JP, Miyoshi H, Paya C, Manivel JC, Haggard JJ, Croatt AJ, Payne W, Alam J: Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int 2001, 59:106-117 [DOI] [PubMed] [Google Scholar]
- 46.Lubin BH: Sickle cell disease and the endothelium. N Engl J Med 1997, 337:1623-1625 [DOI] [PubMed] [Google Scholar]
- 47.Hebbel RP: Blockade of adhesion of sickle cells to the endothelium by monoclonal antibodies. N Engl J Med 2000, 342:1910-1912 [DOI] [PubMed] [Google Scholar]
- 48.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP: Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med 1994, 330:1639-1644 [DOI] [PubMed] [Google Scholar]
- 49.Singhal A, Doherty JF, Raynes JG, McAdam KP, Thomas PW, Serjeant BE, Serjeant GR: Is there an acute-phase response in steady-state sickle cell disease? Lancet 1993, 341:651-653 [DOI] [PubMed] [Google Scholar]
- 50.Bourantas KL, Dalekos GN, Makis A, Chaidos A, Tsiara S, Mavridis A: Acute phase proteins and interleukins in steady state sickle cell disease. Eur J Haematol 1998, 61:49-54 [DOI] [PubMed] [Google Scholar]
- 51.Haugen E, Nath KA: The involvement of oxidative stress in the progression of renal injury. Blood Purif 1999, 17:58-65 [DOI] [PubMed] [Google Scholar]
- 52.Kren S, Hostetter TH: The course of the remnant kidney model in mice. Kidney Int 1999, 56:333-337 [DOI] [PubMed] [Google Scholar]
- 53.O’Bryan T, Weiher H, Rennke HG, Kren S, Hostetter TH: Course of renal injury in the Mpv 17-deficient mouse. J Am Soc Nephrol 2000, 11:1067-1074 [DOI] [PubMed] [Google Scholar]
- 54.Craft JA, Alessandrini E, Kenney LB, Klein B, Bray G, Luban NLC, Meek R, Nadkarni VM: Comparison of oxygenation measurements in pediatric patients during sickle cell crisis. J Pediatr 1994, 124:93-95 [DOI] [PubMed] [Google Scholar]
- 55.Engel RR, Rodkey FL, Krill CE, Jr: Carboxyhemoglobin levels as an index of hemolysis. Pediatrics 1971, 47:723-730 [PubMed] [Google Scholar]
- 56.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP: Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest 2000, 105:71-77 [DOI] [PMC free article] [PubMed] [Google Scholar]