

Abstract
Inflammation and oxidative stress contribute to the pathogenesis of many human diseases including atherosclerosis. Advanced human atheroma contains high levels of the enzyme myeloperoxidase that produces the pro-oxidant species, hypochlorous acid (HOCl). This study documents increased numbers of myeloperoxidase-expressing macrophages in eroded or ruptured plaques causing acute coronary syndromes. In contrast, macrophages in human fatty streaks contain little or no myeloperoxidase. Granulocyte macrophage colony-stimulating factor, but not macrophage colony-stimulating factor, selectively regulates the ability of macrophages to express myeloperoxidase and produce HOCl in vitro. Moreover, myeloperoxidase-positive macrophages in plaques co-localized with granulocyte macrophage colony-stimulating factor. Pro-inflammatory stimuli known to be present in human atherosclerotic plaque, including CD40 ligand, lysophosphatidylcholine, or cholesterol crystals, could induce release of myeloperoxidase from HOCl production by macrophages in vitro. HOCl-modified proteins accumulated at ruptured or eroded sites of human coronary atheroma. These results identify granulocyte macrophage colony-stimulating factor as an endogenous regulator of macrophage myeloperoxidase expression in human atherosclerosis and support a particular role for the myeloperoxidase-expressing macrophages in atheroma complication and the acute coronary syndromes.
Inflammation and oxidative stress contribute to the pathogenesis of many human diseases including atherosclerosis. 1 Recent studies have demonstrated the presence of the active pro-oxidant enzyme myeloperoxidase (MPO) 2 and products of MPO-mediated reactions in human atherosclerosis. 3-6 Stimulated phagocytes secrete this enzyme at inflammatory sites, 7,8 where it generates a powerful reactive oxygen species, hypochlorous acid (HOCl), at physiological chloride concentrations. 9,10 HOCl can in turn serve as a metal-independent oxidizing agent in vivo. 4,5,10-12 Biasucci and colleagues 13 found significantly higher leukocyte intracellular MPO content in patients with coronary heart disease and determined that circulating phagocytes release MPO by degranulation in acute coronary syndromes. Thus, the MPO-HOCl system may contribute to oxidative stress in human atherosclerosis. Many lines of evidence implicate oxidation of low density lipoprotein in atherogenesis, 14,15 and recent studies establish MPO as one pathway for modifying low density lipoprotein in the artery wall. 2-5,11,12,15,16
Macrophages play critical roles in atherogenesis 1,17,18 and participate in the pathogenesis of the acute coronary syndromes. 19-21 Their pro-inflammatory properties likely contribute to destabilization of atheroma by promoting extracellular matrix (ECM) degradation and hence superficial erosion or rupture of plaques, causes of episodic progression of atherosclerosis and, ultimately, of occlusive thrombus formation. 21-23 During atherogenesis, circulating monocytes migrate into the arterial wall andmature into macrophages whose functional properties probably depend on the local microenvironment. 17,18,20,24 Daugherty and colleagues 2 found that macrophages in human atheroma contain MPO. However, traditional sources of MPO include blood neutrophils but not tissue macrophages, 7,25 thus raising the possibility that lesional macrophages in human atheroma differ in this respect in vivo and that MPO-containing macrophages might play a special role in pathogenesis of atherosclerosis by producing the MPO-derived reactive oxygen species, HOCl.
We hypothesized that MPO-containing macrophages and HOCl derived from these cells might influence atherosclerotic plaque stability. To test this hypothesis, we investigated the expression of MPO in various stagesof human atherosclerosis and the factor(s) that mayregulate macrophage MPO expression in human atherosclerosis. Our results indicate that MPO-containing macrophages might contribute to atherogenesis and particularly the acute coronary syndromes by augmentingoxidative stress.
Materials and Methods
Rabbit polyclonal antibody (pAb) MPO was purchased from Biodesign, Inc. (Kennebunk, ME). Monoclonal antibody (mAb) CD68 was obtained from DAKO Corp. (Carpinteria, CA). mAb GM-CSF was obtained from Genzyme (Cambridge, MA). mAb CD66b was acquired from Coulter (Hialeah, FL). mAb HOCl-modified protein (HOP-1, clone 2D10G9) 26 was provided by Dr. Ernest Malle and Dr. Roland Stocker (Graz, Austria). Recombinant CD40 ligand (CD40L) was a gift from Dr. Marilyn R. Kehry, Boehringer Ingelheim Pharmaceuticals, Inc., (Ridgefield, CT). The secondary antibodies for immunostaining, ABC-kits, and AB-blocking reagent were acquired from Vector Laboratories, Burlingame, CA. Purified MPO and other chemicals were obtained from Sigma Chemical Co., St. Louis, MO. Recombinant granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) were gifts from Genetics Institute (Cambridge, MA). Zymosan was opsonized by incubation with human serum at 37°C for 60 minutes.
Human Arterial Samples
Human nonatherosclerotic arteries and atherosclerotic arteries (coronary arteries, carotid arteries, and aortas) were obtained from patients who underwent transplantation and carotid endarterectomy or from autopsy. The arteries were immediately rinsed with phosphate-buffered saline (PBS), embedded in OCT compound, and stored at −80°C. Ruptured or eroded coronary arteries were obtained from sudden cardiac death patients and fixed in formaldehyde. We histologically classified these into nondiseased arteries (n = 7), diffuse intimal thickening (type I, n = 14), fatty streaks (type II, n = 14), atheromatous plaques (type Va, n = 25), fibromuscular plaques (type Vc, n = 17), eroded plaques (type VI, n = 7), and ruptured plaques (type VI, n = 8) by the American Heart Association histological criteria.
Immunohistochemistry
Frozen sections were cut in 6-μm-thick sections and fixed in 4% paraformaldehyde. Paraffin sections of ruptured or erosive plaques were cut in 6-μm-thick sections, dewaxed in xylene, and rehydrated in ethanol. The sections were incubated in 0.5% bovine serum albumin, 2.5% nonfat dried milk, 5% nonimmune serum/PBS for 30 minutes to block nonspecific binding of the primary antibody. Nonspecific avidin and biotin binding was diminished by AB-blocking reagent. Sections were incubated for 1 hour at room temperature with the primary antibodies and immunostaining was performed with an avidin-biotin-alkaline phosphatase system with fast red as a chromogen. In some experiments, we performed double immunostaining using pAb MPO as the first primary antibody and mAb CD68 or CD66b as the second primary antibody. At first, MPO immunoreactivity was detected by an avidin-biotin-alkaline phosphatase system (red), and then the sections were treated with AB-blocking reagent again and incubated with the second primary antibody at 4°C overnight. The next day, the second target immunoreactivity was detected by an avidin-biotin-glucose oxidase system (blue). (dilution of antibodies: 1:250 for MPO, CD68; 1:300 for GM-CSF; 1:100 for CD66b; 1:10 for HOP-1). Nuclei were counterstained with hematoxylin for single staining or with methyl green for double staining. By examining low-power fields (×100) in serial sections, MPO-positive cells were counted and GM-CSF-immunoreactive areas were selected and quantitated by computer-assisted image analysis (Image-Pro Plus, Media Cybernetics). Linear regression analysis was performed to examine the correlation between the number of MPO-positive cells (cells/low-power field) and GM-CSF-immunoreactive areas (mm2/low-power field).
Isolation of Human Monocytes
Human peripheral blood mononuclear cells were isolated from plateletpheresis residues of normal donors (provided by Dr. Steven Clinton at the Dana-Farber Cancer Institute) by Ficoll density gradient centrifugation (density = 1.077 g/ml, 400 × g for 45 minutes). The mid-layer cells were collected and suspended in Ca, Mg-free Hanks’ balanced salt solution. The mononuclear cells were again overlayered on separation media (density = 1.070 g/ml) and recentrifuged for 15 minutes at 400 × g to separate a lymphocyte-rich fraction and to limit neutrophil contamination in the monocyte-rich fraction. After centrifugation, the monocyte-rich fraction was collected from the mid-floating layer, and the cells were plated on 6-well plates (Falcon 3046, Falcon, Oxnard, CA) and incubated for 1 hour at 37°C to allow adhesion. Then, the preparation was washed three times with Hanks’ balanced salt solution and the remaining adherent cells were used as freshly isolated monocytes or cultured in Medium-199 containing penicillin/streptomycin with or without various factors for the days indicated. The monocyte content of this preparation is >92% as determined by flow cytometry using mAb CD68.
MPO Activity Assay
Monocytes and monocyte-derived macrophages were harvested with buffer A [(10 mmol/L phosphate buffer containing phenylmethylsulfonyl fluoride (1 mmol/L), leupeptin (100 μmol/L), pepstatin A (1 μmol/L), and cetyltrimethylammonium bromide (0.5%) pH = 7.0]. The samples were sonicated at 4°C. DNA content was measured in the total cell lysate. 27 The cell lysate was centrifuged at 400 × g for 10 minutes and the supernatant was used for MPO activity assays and Western blotting. Peroxidase activity in the cell lysate was measured by the guaiacol peroxidation method 2 and MPO activity was calculated from NaN3-inhibitable peroxidase activity using purified MPO as a standard. In situ endogenous peroxidase activity in atherosclerotic tissue was examined by diaminobenzidine or tetramethylbenzidine as the reducing substrate. Atheromatous carotid plaques and nondiseased aortas were homogenized in buffer A and centrifuged at 3000 × g for 10 minutes. In the supernatant, NaN3-inhibitable peroxidase activity was measured by tetramethylbenzidine peroxidation method and protein concentration was determined by bicinchoninic acid kit (Pierce, Rockford, IL).
Western Blotting
Human nondiseased aorta and atheromatous carotid arteries were homogenized in buffer A and centrifuged at 400 × g for 10 minutes. The supernatant was collected as the total arterial extract. MPO in the cell lysate, and the total arterial extract were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted onto polyvinylidene difluoride membranes (Bio-Rad, Richmond, CA) with a semidry blotting apparatus. Nonspecific binding was reduced by blocking buffer (5% nonfat dry milk, 2% normal serum, 0.1% Tween20/PBS), and the primary (1:250 anti-MPO and 1:250 anti-GM-CSF) and peroxidase-conjugated secondary (1:4,000 anti-rabbit-IgG and 1:5,000 anti-mouse-IgG from Jackson ImmunoResearch, West Grove, PA) antibodies were diluted in the blocking buffer. Blots were incubated in chemiluminescence reagent (DuPont-New England Nuclear Life Science, Boston, MA) and visualized by exposure to X-ray film.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) Analysis of MPO and GM-CSF-mRNA Expression
Total RNA was extracted from HL-60 cells, arterial tissues, monocytes, and macrophages. Total RNA (5 μg) was used for cDNA synthesis, and the reverse transcriptase reaction mixture (2 μl) was used for PCR reaction with a DNA thermal cycler for 40 cycles (30 seconds at 92°C, 30 seconds at 56°C, and 2 minutes at 72°C). Identical reactions were also set up in the absence of reverse transcriptase for control experiments and these control reactions yielded the negative results, indicating no genomic contamination (data not shown). Primers: G3PDH sense 5′-ACCACAGTCCATGCCATCAC-3′, antisense 5′-TCCACCACCCTGTTGCTGTA-3′; MPO sense 5′-GACACCTCGTTGGTGCTGAG-3′, antisense 5′-TCGCAGTTGACGCCAGTGC-3′; GM-CSF sense 5′-CCTGAACCTGAGTAGAGACAC-3′, antisense 5′-TCCCATTCTTCTGCCATGCCT-3′. The amplified PCR products (G3PDH, 452 bp; MPO, 674 bp; GM-CSF, 484 bp) were electrophoresed in 1.5% agarose gels and visualized by ethidium bromide staining.
Assay of HOCl Production and MPO Degranulation from Macrophages
HOCl production from macrophages was determined as reported previously. 28 GM-CSF-treated macrophages were stimulated by various stimuli [phorbol myristate acetate (PMA), opsonized zymosan, A23187, norepinephrine, interleukin-1β (IL-1β), tumor necrosis factor-α , interferon-γ, lysophosphatidylcholine (lysoPC), CD40L, or cholesterol crystals] in Krebs-Henseleit buffer solution (pH = 7.4) containing taurine (20 mmol/L) at 37°C for 2 hours. The concentration of HOCl-mediated product, taurine chloramine, in the incubation medium was determined by Ellman’s Reagent. MPO levels in the culture medium were determined by MPO-specific enzyme-linked immunosorbent assay (Calbiochem, La Jolla, CA). Cells were harvested by PBS-ethylenediaminetetraacetic acid to measure DNA content. 27
Statistical Analysis
Data in figures represent the means ± SD of the indicated number of samples. The mean values for more than three groups were compared by analysis of variance. The difference between two mean values was analyzed with the unpaired Student’s t-test. A value of P < 0.05 was considered statistically significant.
Results
MPO-Positive Macrophages Vary at Different Stages of Lesion Evolution in Human Atherosclerosis
Recent studies have demonstrated the presence of enzymatically active MPO in human atherosclerosis. 2 Advanced atheromatous plaques (type Va, n = 25) consistently expressed abundant MPO immunoreactivity in intimal mononuclear cells (Figure 1, A and B) ▶ , and human atherosclerotic lesions expressed MPO variably depending on the stage of disease (Table 1 ▶ and Figure 1A ▶ ). Advanced atheromatous lesions contained MPO-positive cells particularly in the fibrous cap, near microvessels, and in the lipid core (Figure 1B) ▶ . Arteries with diffuse intimal thickening (n = 14), fatty streaks (n = 14), and fibromuscular plaques (n = 17) generally exhibited little MPO immunoreactivity, although some of these specimens did contain MPO-containing cells (Table 1 ▶ and Figure 1A ▶ ). Advanced fibromuscular plaques (type Vc) contained MPO-positive cells in the subendothelial space and deep intimal region (Figure 1B) ▶ . Most MPO-containing cells were not lipid-laden foam cells, but some lipid-laden foam cells were also MPO-positive. Arteries that showed no signs of disease (n = 7) all lacked MPO immunoreactivity (data not shown).
Figure 1.
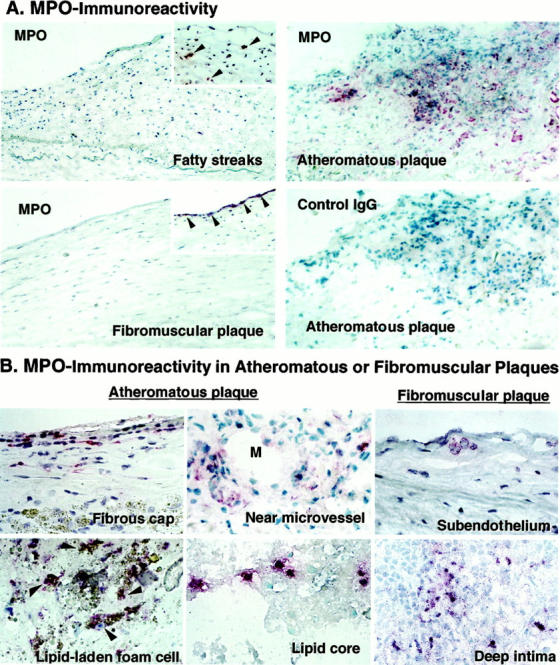
Expression of MPO immunoreactivity in various stages of human atherosclerotic arteries and localization in advanced plaques. Frozen sections were incubated with pAb-MPO and MPO immunoreactivity was visualized with the alkaline-phosphatase ABC method (red). A: MPO immunoreactivity localized in early to advanced human atherosclerosis (arrowheads). Atheromatous plaques exhibited abundant MPO, but lacked staining by nonimmune rabbit IgG used as a negative control. Arteries with fatty streaks, and fibromuscular plaques usually exhibited little MPO (large panels), but some samples contained MPO in the intima (insets). Original magnification, ×200. These results are representative of 14 fatty streaks, 17 fibromuscular plaques, and 25 atheromatous plaques. B: MPO-containing cells were localized in all regions of advanced plaques, especially in fibrous cap, near microvessel (M, indicates lumen of microvessel), and lipid core of atheromatous plaques. Some of fibromuscular plaques contained MPO-containing cells in the subendothelial space and deep intima. Some of the lipid-laden foam cells were also MPO-positive in atheroma (arrowheads). Original magnification, ×400.
Table 1.
Semiquantitative Analysis of MPO Expression in Various Stages of Human Atherosclerosis
| Artery type | n | MPO-positive sample (%) | MPO-positive cells/LPF |
|---|---|---|---|
| Nondiseased arteries | 7 | 0 (0%) | 0 ± 0 |
| Type I (Diffuse intimal thickening) | 14 | 3 (21%) | 1 ± 0 |
| Type II (Fatty streaks) | 14 | 10 (71%) | 3 ± 1 |
| Type Va (Atheromatous plaque) | 25 | 25 (100%) | 48 ± 5* |
| Type Vc (Fibromuscular plaque) | 17 | 6 (35%) | 5 ± 3 |
| Type VI (Rupture of atheromatous plaque) | 8 | 8 (100%) | 118 ± 25† |
| Type VI (Erosion of fibromuscular plaque) | 7 | 6 (86%) | 69 ± 26‡ |
MPO immunoreactivity was evaluated in human nondiseased arteries and various stages of human atherosclerotic arteries. Human arteries were classified by the American Heart Association histological criteria. To semiquantify the MPO expression, we counted the number of MPO-positive cells in the low-power field (LPF, ×100) in every lesion by two independent observers and MPO-positive cells/LPF is shown as mean ± SE.
*P < 0.01 versus type Vc.
†P < 0.01 versus type Va.
‡P < 0.01 versus type Vc.
Human Atherosclerotic Lesions Contain Two Phenotypes of Macrophages
To determine unambiguously which cells within human atheroma express MPO, we performed double immunostaining using anti-MPO antibody and cell-type specific antibodies (CD68 for macrophages and CD66b for neutrophils). Intimal cells of human atherosclerotic arteries did not express the neutrophil marker CD66b (data not shown). The MPO-positive cells in atherosclerotic lesions were mononuclear cells and reacted with CD68, identifying these cells as macrophages (Figure 2 ▶ , arrowheads). Fatty streaks contained many CD68-positive intimal mononuclear cells but few MPO-containing cells (Figure 2 ▶ , left). In contrast, many cells in advanced atheromatous plaques were positive for both MPO and CD68, although some CD68-positive macrophages lacked MPO (Figure 2 ▶ , right). Thus, human atherosclerotic lesions contained two phenotypes of macrophages, one MPO-positive and the other MPO-negative.
Figure 2.

MPO-positive macrophages and MPO-negative macrophages in human atherosclerosis. Fatty streaks and atheromatous plaques were examined by double immunostaining with pAb-MPO (red) and mAb-CD68 (blue). Bottom: A high magnification of each of the top panels. MPO-positive cells were CD68-positive mononuclear cells in both fatty streaks and atheromatous plaques (purple, arrowheads). Fatty streaks contained many intimal CD68-positive mononuclear cells but few MPO-positive cells. In atheromatous plaques, many MPO-positive cells were CD68-positive macrophages (purple, arrowheads), although some CD68-positive macrophages lacked MPO (blue). Original magnification, ×100 (top), ×400 (bottom). These results are representative of six fatty streaks and 10 atheromatous plaques.
MPO-Containing Macrophage Accumulation in the Culprit Lesions of Acute Coronary Syndromes
Most myocardial infarctions are caused by thrombosis on the substrate of a disrupted atherosclerotic plaque. 22,23 Major mechanisms of plaque disruption include fracture of the fibrous cap 22 and superficial erosion of the endothelial cells lining the artery wall. 21 We therefore studied culprit lesions of both types in coronary arteries from victims of sudden cardiac death. Sites of plaque rupture in the fibrous caps (n = 8) and superficial erosions (n = 7) in coronary arteries from victims of sudden cardiac death exhibited abundant MPO-containing mononuclear cells (Figure 3 ▶ and Table 1 ▶ ). We found significantly increased numbers of MPO-positive macrophages at sites of eroded or ruptured plaques (Table 1 ▶ , P < 0.01). In eroded or ruptured plaques, few neutrophils (CD66b-positive cells) localized at the sites of erosion or rupture, although the occlusive thrombi do contain occasional neutrophils (data not shown).
Figure 3.
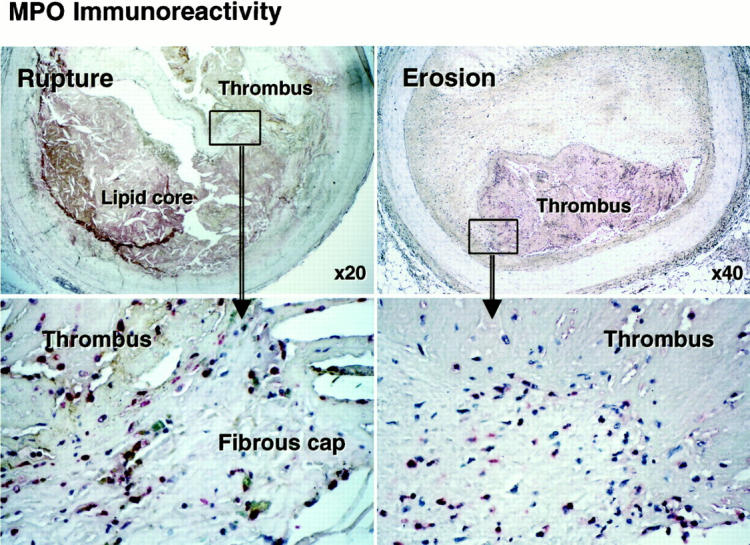
MPO immunoreactivity in ruptured and eroded coronary plaques in patients with acute coronary syndromes. Paraffin sections of ruptured or eroded plaques were incubated with pAb-MPO and MPO immunoreactivity was visualized with the alkaline-phosphatase ABC method (red), MPO-positive macrophages (red) were prominent at rupture sites in atheroma and at sites where fibromuscular plaques had eroded in victims of sudden cardiac death. Bottom: High-power views (original magnifications, ×400) of each upper rectangle area. These results are representative of six eroded and eight ruptured plaques.
GM-CSF Selectively Regulates MPO Expression during Monocyte-Macrophage Differentiation in Vitro
We used freshly isolated and cultured human monocytes to explore mechanisms that might regulate MPO expression in macrophages. Culturing adherent monocytes in medium supplemented with human serum (5%) caused an initial rapid loss of both MPO activity and protein that was almost complete by 3 days as reported previously. 29 Similar results were observed when serum concentrations varied from 5 to 20% (data not shown). GM-CSF and, to a lesser extent, IL-3 preserved MPO activity during the 7-day culture period, whereas M-CSF was inactive in this regard (Figure 4A) ▶ . Retention of MPO activity depended on GM-CSF concentration (10 to 500 U/ml) (Figure 4B) ▶ and the GM-CSF-treated macrophages (but not human serum-treated macrophages or M-CSF-treated macrophages) contained MPO protein (Figure 4C) ▶ . These results indicate that GM-CSF selectively regulates MPO levels during differentiation of circulating monocytes into macrophages. RT-PCR detected MPO mRNA in cultured HL-60 cells but not in human atherosclerotic tissue, monocytes, or monocyte-derived macrophages (data not shown).
Figure 4.

GM-CSF, but not M-CSF, modulates macrophage phenotype to express MPO in vitro. MPO activity (guaiacol peroxidation method) and MPO protein were examined in cellular lysate. A: GM-CSF (100 U/ml) and IL-3 (10 ng/ml), but not M-CSF (100 U/ml), preserved MPO activity in monocyte-derived macrophages during 7 days in culture (n = 3). IL-1β (10 ng/ml), tumor necrosis factor-α (10 ng/ml), PDGF (10 ng/ml), interferon-γ (1,000 U/ml), PMA (100 nmol/L), lysoPC (15 μmol/L), and CD40L (2 μg/ml). Each factor was added to the medium on the 0, second, fourth, and sixth day in culture. B: The effect of GM-CSF on MPO activity depended on its concentration (10 to 500 U/ml) (n = 3). C: Extracts of cultured macrophages containing 2 μg of DNA were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. GM-CSF-treated macrophages, but not human serum-treated macrophages and M-CSF-treated macrophages expressed MPO protein. Each result represents three independent experiments.
Advanced Atheroma Contain GM-CSF and MPO-Positive Macrophages Localize in the GM-CSF-Expressing Intima
Advanced atheromatous plaques (type Va) consistently exhibited considerable GM-CSF immunoreactivity in the intima (n = 15) (Figure 5A) ▶ . The advanced carotid artery atheroma, but not nondiseased aorta, expressed GM-CSF protein and GM-CSF mRNA (Figure 5, B and C) ▶ . MPO-positive macrophages localized in the GM-CSF-expressing atherosclerotic intima (Figure 5D) ▶ . Depending on the stage of disease, human atherosclerotic lesions also variably expressed GM-CSF in the intima; arteries with fatty streaks (n = 8), and fibromuscular plaques (n = 12) varied but generally exhibited little immunoreactive GM-CSF (data not shown). We furthermore found that the areas of immunoreactive GM-CSF and the number of MPO-positive macrophages correlated significantly in human atherosclerotic lesions (R 2 = 0.84, P < 0.001 by linear regression analysis) (Figure 5E) ▶ .
Figure 5.
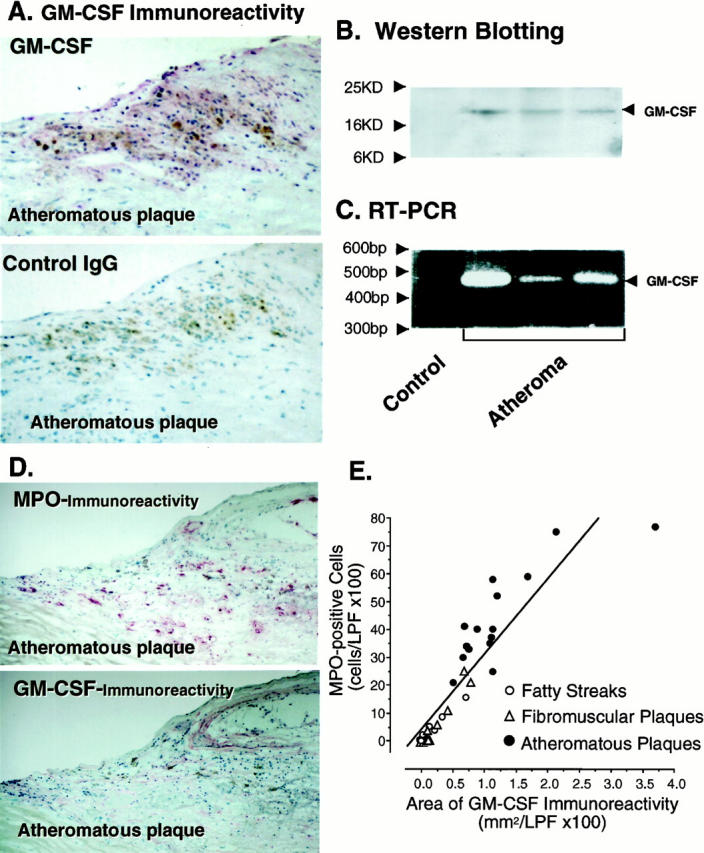
Human atherosclerotic arteries express GM-CSF and MPO-positive macrophages localize in the GM-CSF-expressing atherosclerotic intima. Frozen sections were incubated with mAb-GM-CSF and GM-CSF immunoreactivity was visualized with the alkaline-phosphatase ABC method (red). A: Advanced atheromatous plaques expressed substantial GM-CSF immunoreactivity in the intima, but lacked staining by nonimmune mouse IgG1 used as a negative control. These results are representative of those obtained by study of 15 atheromatous plaques. Original magnification, ×200. B: Each total tissue homogenate of nondiseased aorta and advanced atheromatous carotid arteries (100 μg of protein/lane) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and GM-CSF protein was determined by Western blotting. C: GM-CSF mRNA expression was determined by RT-PCR analysis as described in Materials and Methods. The advanced atheromatous carotid arteries, but not nondiseased aorta, expressed GM-CSF protein and mRNA. D: MPO and GM-CSF immunoreactivity in the adjacent sections of atheromatous plaques (red). The MPO-positive macrophages localized in the GM-CSF-expressing intima of atheroma. These results are representative of those obtained by study of 15 atheromatous plaques. Original magnification, ×200. E: MPO-positive cells were counted in the low-power field (×100), and GM-CSF-immunoreactive areas were selected and quantitated by computer-assisted image analysis. (open circle, fatty streaks; open triangle, fibromuscular plaques, filled circle, atheromatous plaques). The areas of immunoreactive GM-CSF and the number of MPO-positive macrophages correlated significantly in human atherosclerotic lesions (R2=0.84, P < 0.001).
MPO, Endogenous Peroxidase Activity, and HOCl-Modified Protein Co-Localize in Advanced Atheromatous Plaques
In advanced atheromatous plaques, MPO immunoreactivity and endogenous peroxidase activity co-localized intracellularly in the intima and extracellularly in the lipid core (Figure 6A ▶ , n = 12). Extracts of human advanced atheroma (n = 5), but not of nondiseased aortae (n = 4), contain MPO activity (277 ± 143 mU/mg protein; range, 77 to ∼825; n = 5). Advanced human atheromatous plaques typically have a large central hypocellular lipid-rich region. 30,31 This lipid core probably plays a key role in the biomechanical destabilization of plaques and in thrombogenesis. 17,22,23,32 Lipid cores of advanced atherosclerotic lesions, especially lipid cores with gruel, contained both MPO immunoreactivity and endogenous peroxidase activity (Figure 6A) ▶ . The in situ endogenous peroxidase activity was inhibited by the heme poison NaN3 and by a polyclonal antibody to MPO (data not shown). We also found that the endogenous peroxidase activity and immunoreactive HOCl-modified proteins co-localized in the intima of advanced atheroma (Figure 6B) ▶ . Specifically, thin fibrous caps, zones surrounding the necrotic lipid core, and fragmented ECM in atheroma contain HOCl-modified protein epitopes (Figure 6B) ▶ . The MPO-poor fibromuscular plaques examined (n = 17) contained few HOCl-modified proteins (data not shown). HOCl-modified protein immunoreactivity was prominent at the sites of ruptured fibrous caps (n = 8) or eroded plaques (n = 7) of acute coronary syndromes (Figure 6C) ▶ .
Figure 6.
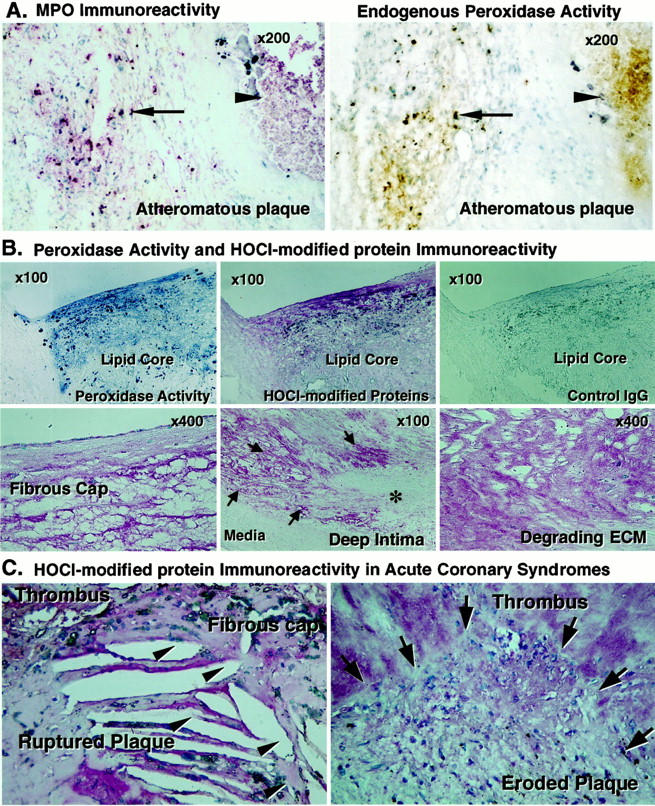
MPO, endogenous peroxidase activity, and HOCl-modified proteins co-localized in advanced atheroma and HOCl-modified proteins abound in culprit lesions of acute coronary syndromes. A: Frozen sections of advanced atheromatous plaques were incubated with pAb-MPO and MPO immunoreactivity was visualized with the alkaline-phosphatase ABC method (red), and endogenous peroxidase activity was examined using the diaminobenzidine peroxidation method (brown). The adjacent sections of atheromatous plaques show that MPO immunoreactivity (left) and endogenous peroxidase activity (right) co-localized intracellularly in the intima and extracellularly in the lipid core (arrow, cellular MPO; arrowhead, lipid core MPO). These results are representative of 12 atheromatous plaques. B: Frozen (top, adjacent sections) and paraffin sections of advanced atheromatous plaques (bottom) were incubated with mAb-HOCl-modified proteins and the immunoreactivity was visualized with the alkaline-phosphatase ABC method (red). Endogenous peroxidase activity was examined using the tetramethylbenzidine peroxidation method (blue). The endogenous peroxidase activity and immunoreactive HOCl-modified proteins co-localized in the intima of advanced atheroma, but the lesions lacked staining by nonimmune mouse IgG2b used as a negative control (top, n = 8). The thin fibrous cap (left),areas of matrix fragmentation near the lipid core (middle, arrows; degrading area), and fragmented ECM in atheroma (right), but not the area of intact ECM (asterisk) and media (middle), contain HOCl-modified protein epitopes. These results are representative of 12 atheromatous plaques. C: Paraffin sections of ruptured or eroded coronary plaques were incubated with mAb-HOCl-modified proteins and HOCl-modified protein immunoreactivity was visualized with the alkaline-phosphatase ABC method (red). Shown is substantial staining of HOCl-modified protein immunoreactivity at the site of ruptured fibrous cap and erosion. (arrowhead, ruptured site; arrow, eroded site). These results are representative of six eroded and eight ruptured plaques.
Macrophages Release HOCl and MPO in Response to Pathophysiological Stimuli
To investigate the possibility that MPO-containing macrophages may mediate oxidative stress and trigger inflammatory events in atherosclerotic lesions, we examined the behavior of macrophages treated with GM-CSF in vitro. The cells chlorinated extracellular taurine in response to PMA and to opsonized zymosan (Figure 7) ▶ and PMA provoked MPO release from macrophages, presumably by degranulation (medium MPO levels: vehicle, 1.0 ± 0.2; PMA, 21.2 ± 3.1 ng/ml; n = 3; P < 0.01). Pathophysiological stimuli known to be present in human atherosclerotic plaques, CD40L, 33 lysoPC, 34 or cholesterol crystals, 30 induced HOCl-release from GM-CSF-treated macrophages (Figure 7) ▶ and MPO release from macrophages in vitro (medium MPO levels: vehicle, 1.0 ± 0.2; CD40L, 17.5 ± 2.6; cholesterol crystals, 12.1 ± 1.8; lysoPC, 14.8 ± 2.2 ng/ml; n = 3; P < 0.01).
Figure 7.

MPO-positive macrophages in vitro produce HOCl in response to atherogenic stimuli. Bar graph indicates HOCl production from GM-CSF (100 U/ml)-treated macrophages on day 7 measured by taurine chloramine generation. PMA (300 nmol/L), opsonized zymosan (OZ) (0.2 mg/ml), lysoPC (15 μmol/L), CD40L (5 μg/ml), or cholesterol crystals (1 mg/ml) induced HOCl production in macrophages, but A23187 (1 μmol/L), norepinephrine (10 μmol/L), IL-1β (10 ng/ml), tumor necrosis factor-α (50 ng/ml), and interferon-γ (1,000 U/ml) had no effect (*, P < 0.01 versus vehicle, n = 3).
Discussion
This study functionally identified a distinct phagocytic population, MPO-containing macrophages, in lesions of human atherosclerosis at various stages, particularly those provoking the acute coronary syndromes. MPO-containing macrophages produce HOCl, a specific MPO-derived reactive oxygen species, in response to pathophysiological stimuli known to be present in human atheroma and HOCl-modified proteins accumulated at ruptured or eroded sites of human coronary atheroma. Such cells may therefore influence the stability of atherosclerotic plaque and hence the thrombotic complications of atheroma by augmenting oxidative stress. Our results also suggest that GM-CSF can promote accumulation of MPO-positive macrophage in human atheroma. Macrophages in fatty streaks have little MPO, perhaps because such lesions contain scant GM-CSF. These observations help to explain why fatty streaks seldom, if ever, disrupt and provoke thrombosis, as do advanced atheroma where MPO-positive macrophages abound. This observation also supports the notion that expression of pro-oxidative enzymes can modulate the evolution of atherosclerotic lesions.
Acute coronary syndromes involve the remodeling of arterial ECM and the erosion of plaques or rupture of the fibrous cap. 19-23 Recently, several studies have implicated lesional macrophages in ECM degradation because of production of matrix metalloproteinases (MMPs), 35,36 inhibitors of metalloproteinases (the TIMPs), and because the MMP pro-enzymes require extracellular activation. 37 Therefore, increased MMP synthesis alone may not suffice for tissue destruction; activation of pro-MMPs and a shift in the MMP-TIMP balance would need to prevail at sites of pathological ECM degradation. The present study demonstrates that MPO-positive macrophages generate HOCl and that the fragmented ECM in atheroma contains HOCl-modified protein. HOCl can inactivate TIMP-1 38 and HOCl can activate pro-MMPs, 39,40 that are expressed in human atheroma. 35,36 Thus, the MPO-containing macrophages could play a special role in ECM degradation by acceleration of proteinase cascade through oxidative stress. Our observation that MPO-containing macrophages and HOCl-modified proteins localize in the fibrous cap and HOCl itself can solubilize collagen 41,42 also suggest that these macrophages may contribute to structural weakening of the fibrous cap and plaque rupture independent of proteolysis. MPO-positive macrophages localized in subendothelial space in some advanced fibromuscular plaques and HOCl itself can degrade proteoglycan, 43 decrease adhesiveness of subendothelial ECM to endothelial cells, 44 and cause endothelial cell retraction and death. 45 Each of these functions may promote local endothelial desquamation that produces superficial erosion of the intima, an important cause of coronary thrombosis in fibromuscular plaques. We also found significantly increased numbers of these MPO-positive macrophages and presence of HOCl-modified proteins in the culprit lesions of acute coronary syndromes. Taken together, these observations indicate that the lesional MPO-containing macrophages may participate in the development of acute coronary syndromes.
Morphologically, so-called “vulnerable” or unstable plaques have a thin fibrous cap and a large lipid core (atheromatous plaque, type Va), and so-called “stable” plaques have a thick fibrous cap, abundant ECM, and smooth muscle cells with low lipid content (fibromuscular plaque, type Vc). 19,32,46 We found many MPO-containing macrophages in the advanced atheromatous plaques (“unstable plaques”) and observed that macrophages of this phenotype accumulated locally at sites of rupture in coronary plaques. Some fibromuscular plaques with morphological features of stability also locally contained numerous MPO-containing macrophages in the subendothelial space. Eroded coronary fibromuscular plaques have similar levels of MPO-containing macrophages, as do atheromatous plaques, suggesting that accumulated MPO-containing macrophages locally destabilized the fibromuscular plaques. Thus, the present observations support a novel concept that local accumulation of MPO-containing macrophages in plaques could influence atheroma stability irrespective of the dominant plaque morphology, and in that manner govern a plaque’s propensity to provoke thrombosis.
Although most atheroma exhibit features of chronic inflammation, 47 the actual triggers of the acute local inflammation that leads to clinical symptoms remain unclear. For MPO-positive macrophages to realize their pro-inflammatory capacity in the arterial wall, they must release MPO into the extracellular space. The stimuli for and mechanisms of this degranulation remain unknown. We demonstrate here that CD40L, lysoPC, or cholesterol crystals, stimuli found in human atheroma, 30,33,34 can trigger the release of MPO and HOCl production from MPO-positive macrophages in vitro. Thus, the activation of MPO-containing macrophages and subsequent degranulation might contribute to the transition to acute complications of atheroma. The present findings that PMA (a potent PKC activator) induced the HOCl production from MPO-positive macrophages in vitro suggest possible signaling pathways.
Our data underscore macrophage heterogeneity. 17,18,20,24 We identified two phenotypes of macrophages in human atherosclerotic lesions, one MPO-positive and the other MPO-negative. Previously, Owen and colleagues 48 and Akiyama and colleagues 49 reported the existence of two distinct monocyte populations, high MPO and low MPO monocytes, in circulating human blood. The present observations demonstrate a similar dichotomy of macrophage phenotype in human atherosclerosis in vivo. MPO-containing macrophages in plaques might arise from the high MPO monocytes in circulating blood through selective survival and differentiation if local environmental factors maintained their phenotype. We demonstrate here that GM-CSF, but not M-CSF, preserves MPO activity in human monocyte-derived macrophages in vitro. We also show that MPO-positive macrophages localize in the GM-CSF-expressing atherosclerotic intima and localize abundant GM-CSF within the atheroma. These observations suggest a specific role of GM-CSF in the regulation of macrophage phenotype in human atherosclerosis. Alternatively, the MPO-positive macrophages might arise from preferential recruitment into atheroma. Human atheromata contain several chemokines, including monocyte chemoattractant protein-1, IP-10, Mig, SDF, I-TAC, or IL-8. 50-52 Circulating mononuclear cells have heterogeneous chemokine receptor expression. 53 The notion that this heterogeneity might selectively recruit high MPO monocytes into atherosclerotic arteries requires further investigation.
We demonstrated that advanced human atheromatous plaques expressed GM-CSF-protein and mRNA in the present study. We have not yet formally identified the cell types that express GM-CSF in the arterial wall. However, all types of atheroma-associated cells, endothelial cells, macrophages, smooth muscle cells, and T-lymphocytes can produce GM-CSF. 54,55 Moreover, bioactive GM-CSF can bind to extracellular proteoglycans and retain biological activity. 56 Thus, GM-CSF is synthesized locally in the artery wall and stored extracellularly in proteoglycan-rich plaques. Elucidation of the mechanisms that regulate GM-CSF expression in atherosclerosis will require further study.
The underlying mechanisms of MPO expression in human atherosclerosis remain uncertain. However, the absence of detectable MPO mRNA in atheroma suggests that transcription of MPO gene and de novo synthesis of MPO protein in the arterial wall contribute little to MPO expression in atherosclerosis. MPO is a stable and highly cationic protein that binds readily to negatively charged ECM components such as glysaminoglycans and proteoglycans. This property may permit accumulation and persistence of biologically active MPO protein in the atherosclerotic plaques.
Many lines of evidence indicate production of HOCl by MPO in human atherosclerosis. 3-6 We show here the presence of HOCl-modified proteins within advanced atheroma and particularly in culprit lesions of acute coronary syndromes. These findings support the pathological significance of HOCl production in atherogenesis. We cannot ascertain the actual local in vivo concentrations of hypochlorous anion produced in atherosclerotic lesions. The present study showed that cultured MPO-positive macrophages produce ∼7.5 pmol HOCl/10 3 cells/hour, and the ruptured plaques contain 118 ± 25 MPO-positive macrophages/low-power field. HOCl can rapidly react with next target molecules at the site of its generation. Hypochlorous anion within the extracellular space may reach approximately millimolar concentrations during moderate inflammation. 8
In conclusion, we provide evidence that a distinct phenotype of macrophages that expresses MPO localized in human atherosclerosis and the culprit lesions of acute coronary syndromes. We identify GM-CSF as an endogenous mediator that may favor accumulation of macrophages of this phenotype in atheroma. Such pro-inflammatory macrophages could play a particular role in the pathogenesis of human atherosclerosis and acute coronary syndromes by contributing to oxidative stress.
Acknowledgments
We thank Dr. Marilyn R. Kehry for her kind gift of recombinant CD40 ligand (CD40L). We also thank Dr. Maria Muszynski, Eugenia Shvartz, and Elissa Simon-Morrissey for their excellent technical assistance, Dr. Midori Sugiyama for helpful discussion, and Karen Williams for her editorial expertise.
Footnotes
Address reprint requests to Peter Libby, M. D., Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, 221 Longwood Ave., LMRC-307, Boston, MA 02115. E-mail: plibby@rics.bwh.harvard.edu.
Supported by grants from National Heart, Lung, and Blood Institute (HL34636 to P. L. and AG12293 and AG15013 to J. W. H.).
Portions of this work were presented in preliminary form at the Annual Scientific Session of the American Heart Association in 1997 (Circulation 1997;96: I-417) and 1998 (Circulation 1998;98: I-315).
S. S. is the recipient of Banyu Fellowship Awards in Lipid Metabolism and Atherosclerosis from Banyu Pharmaceutical Co., Ltd. and the Merck Company Foundation. J. W. H. is an Established Investigator of the American Heart Association.
References
- 1.Ross R: Atherosclerosis: an inflammatory disease. N Engl J Med 1999, 340:115-126 [DOI] [PubMed] [Google Scholar]
- 2.Daugherty A, Dunn JL, Rateri DL, Heinecke JW: Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest 1994, 94:437-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hazell LJ, Arnold L, Flowers D, Waeg G, Malle E, Stocker R: Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J Clin Invest 1996, 97:1535-1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazen SL, Heinecke JW: 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest 1997, 99:2075-2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leeuwenburgh C, Rasmussen JE, Hsu FF, Mueller DM, Pennathur S, Heinecke JW: Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J Biol Chem 1997, 272:3520-3526 [DOI] [PubMed] [Google Scholar]
- 6.Malle E, Waeg G, Schreiber R, Grone EF, Sattler W, Grone H-J: Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in atherosclerotic lesions. Colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem 2000, 267:4495-4503 [DOI] [PubMed] [Google Scholar]
- 7.Klebanoff SJ: Phagocytic cells: products of oxygen metabolism. Inflammation: Basic Principles and Clinical Correlates. 1988, :pp 391-444 Raven Press, New York [Google Scholar]
- 8.Weiss SJ: Mechanisms of disease: tissue destruction by neutrophils. N Engl J Med 1989, 320:365-376 [DOI] [PubMed] [Google Scholar]
- 9.Weiss SJ, Klein R, Slivka A, Wei M: Chlorination of taurine by human neutrophils; evidence for hypochlorous acid generation. J Clin Invest 1982, 70:598-607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison JE, Schultz J: Studies on the chlorinating activity of myeloperoxidase. J Biol Chem 1976, 251:3520-3526 [PubMed] [Google Scholar]
- 11.Heinecke JW, Li W, Francis GA, Goldstein JA: Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of protein. J Clin Invest 1993, 91:2866-2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savenkova MI, Mueller DM, Heinecke JW: Tyrosyl radical generated by myeloperoxidase is a physiological catalyst for the initiation of lipid peroxidation in low density lipoprotein. J Biol Chem 1994, 269:20394-20400 [PubMed] [Google Scholar]
- 13.Biasucci LM, D’Onofrio G, Liuzzo G, Zini G, Monaco C, Caligiuri G, Tommasi M, Rebuzzi AG, Maseri A: Intracellular neutrophil myeloperoxidase is reduced in unstable angina and acute myocardial infarction, but its reduction is not related to ischemia. J Am Coll Cardiol 1996, 27:611-616 [DOI] [PubMed] [Google Scholar]
- 14.Steinberg DS, Lewis A: Conner Memorial Lecture. Oxidative modification of LDL and atherosclerosis. Circulation 1997, 95:1062-1071 [DOI] [PubMed] [Google Scholar]
- 15.Heinecke JW: Mechanisms of oxidative damage of low density lipoprotein in human atherosclerosis. Curr Opin Lipidol 1997, 8:268-274 [DOI] [PubMed] [Google Scholar]
- 16.Podrez EA, Schmitt D, Hoff HF, Hazen SL: Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest 1999, 103:1547-1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Libby P, Clinton SK: The role of macrophages in atherogenesis. Curr Opin Lipidol 1993, 4:355-363 [Google Scholar]
- 18.Raines EW, Rosenfeld ME, Ross R: The role of macrophages. Fuster V Ross R Topol EJ eds. Atherosclerosis and Coronary Artery Disease. 1996, :pp 539-555 Lippincott-Raven, Philadelphia [Google Scholar]
- 19.Libby P: Molecular bases of the acute coronary syndromes. Circulation 1995, 91:2844-2850 [DOI] [PubMed] [Google Scholar]
- 20.Libby P, Gen Y-J, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT: Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol 1996, 7:330-335 [DOI] [PubMed] [Google Scholar]
- 21.van der Wal AC, Becker AE, van der Loos CM, Das PK: Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89:36-44 [DOI] [PubMed] [Google Scholar]
- 22.Falk E: Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J 1983, 50:127-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis MJ: The Paul Dudley White Lecture 1995. Stability and instability: two faces of coronary atherosclerosis. Circulation 1996, 94:2013-2020 [DOI] [PubMed] [Google Scholar]
- 24.van der Wal AC, Das PK, Tigges AJ, Becker AE: Macrophage differentiation in atherosclerosis. Am J Pathol 1992, 141:161-168 [PMC free article] [PubMed] [Google Scholar]
- 25.Koeffler HP, Ranyard J, Pertcheck M: Myeloperoxidase: its structure and expression during myeloid differentiation. Blood 1985, 65:484-491 [PubMed] [Google Scholar]
- 26.Malle E, Hazell L, Stocker R, Sattler W, Esterbauer H, Waeg G: Immunologic detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arterioscler Thromb Vasc Biol 1995, 15:982-989 [DOI] [PubMed] [Google Scholar]
- 27.Labarca C, Paigen K: A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 1980, 102:344-352 [DOI] [PubMed] [Google Scholar]
- 28.Kettle AJ, Winterbourn CC: Assays for the chlorination activity of myeloperoxidase. Packer, L. Oxygen Radicals in Biological Systems, Part-C. Methods Enzymol 1994, 233:502-512 [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa A, Nathan CF, Cohn ZA: Hydrogen peroxide metabolism in human monocytes during differentiation in vitro. J Clin Invest 1981, 68:1243-1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stary HC, Chandler B, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW: A definition of advanced types of atherosclerotic lesions and histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Atherosclerosis, American Heart Association. Circulation 1995, 92:1355-1374 [DOI] [PubMed] [Google Scholar]
- 31.Guyton JR, Klemp KF: Development of the lipid-rich core in human atherosclerosis. Arterioscler Thromb Vasc Biol 1996, 16:4-11 [DOI] [PubMed] [Google Scholar]
- 32.Lee RT, Libby P: The unstable atheroma. Arterioscler Thromb Vasc Biol 1997, 17:1859-1867 [DOI] [PubMed] [Google Scholar]
- 33.Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy J-Y, Pober JS, Libby P: Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implication for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci USA 1997, 94:1931-1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Portman OW, Alexander M: Lysophosphatidylcholine concentrations and metabolism in aortic intima plus inner media: effect of nutritionally induced atherosclerosis. J Lipid Res 1969, 10:158-165 [PubMed] [Google Scholar]
- 35.Galis ZS, Sukhova GK, Lark MW, Libby P: Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 1994, 94:2493-2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown DL, Hibbs MS, Kearney M, Loushin C, Isner JM: Identification of 92-kD gelatinase in human coronary atherosclerotic lesions: association of active enzyme synthesis with unstable angina. Circulation 1995, 91:2125-2131 [DOI] [PubMed] [Google Scholar]
- 37.Murphy G, Willenbrock F, Crabbe T, O’Shea M, Ward R, Atkinson S, O’Connell J, Docherty A: Regulation of matrix metalloproteinase activity. Ann NY Acad Sci 1994, 732:31-41 [DOI] [PubMed] [Google Scholar]
- 38.Shabani F, McNeil J, Tippett L: The oxidative inactivation of tissue inhibitor of metalloproteinase-1 (TIMP-1) by hypochlorous acid (HOCl) is suppressed by anti-rheumatic drugs. Free Radic Res 1998, 28:115-123 [DOI] [PubMed] [Google Scholar]
- 39.Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE: Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc Natl Acad Sci USA 1990, 87:364-368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peppin GJ, Weiss SJ: Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc Natl Acad Sci USA 1986, 83:4322-4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davies JM, Horwitz DA, Davies KJ: Potential roles of hypochlorous acid and N-chloramines in collagen breakdown by phagocytic cells in synovitis. Free Radic Biol Med 1993, 15:637-643 [DOI] [PubMed] [Google Scholar]
- 42.Daumer KM, Khan AU, Steinbeck MJ: Chlorination of pyridinium compounds: possible role of hypochlorite, N-chloramines, and chlorine in the oxidation of pyridinoline cross-links of articular cartilage collagen type II during acute inflammation. J Biol Chem 2000, 275:34681-34692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klebanoff SJ, Kinsella MG, Wight TN: Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am J Pathol 1993, 143:907-917 [PMC free article] [PubMed] [Google Scholar]
- 44.Vissers MCM, Thomas C: Hypochlorous acid disrupts the adhesive properties of subendothelial matrix. Free Radic Biol Med 1997, 23:401-411 [DOI] [PubMed] [Google Scholar]
- 45.Tatsumi T, Fliss H: Hypochlorous acid and chloramines increase endothelial permeability: possible involvement of cellular zinc. Am J Physiol 1994, 267:H1597-H1607 [DOI] [PubMed] [Google Scholar]
- 46.Davies MJ, Richardson PD, Woolf N, Katz DR, Mann J: Risk of thrombosis in human atherosclerotic plaques: role of extracellular lipid, macrophage, and smooth muscle cell content. Br Heart J 1993, 69:377-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Libby P, Ross R: Cytokines and growth regulatory molecules. Fuster V Ross R Topol EJ eds. Atherosclerosis and Coronary Artery Disease. 1996, :pp 585-594 Lippincott-Raven, Philadelphia [Google Scholar]
- 48.Owen CA, Campbell MA, Boukedes SS, Stockley RA, Campbell EJ: A discrete subpopulation of human monocytes expresses a neutrophil-like proinflammatory (P) phenotype. Am J Physiol 1994, 267:L775-L785 [DOI] [PubMed] [Google Scholar]
- 49.Akiyama Y, Miller PJ, Thurman GB, Neubauer RH, Oliver C, Favilla T, Beman JA, Oldham RK, Stevenson HC: Characterization of human blood monocyte subset with low peroxidase activity. J Clin Invest 1983, 72:1093-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelken NA, Coughlin SR, Gordon D, Wilcox JN: Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest 1991, 88:1121-1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang N, Tabas I, Winchester R, Ravalli S, Rabbani LE, Tall A: Interleukin 8 is induced by cholesterol loading of macrophages and expressed by Macrophages foam cells in human atheroma. J Biol Chem 1996, 271:8837-8842 [DOI] [PubMed] [Google Scholar]
- 52.Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, Luster AD: Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest 1999, 104:1041-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baggiolini M, Dewald B, Moser B: Human chemokines: an update. Annu Rev Immunol 1997, 15:675-705 [DOI] [PubMed] [Google Scholar]
- 54.Nimer SD, Uchida H: Regulation of granulocyte-macrophage colony-stimulating factor and interleukin 3 expression. Stem Cells 1995, 13:324-335 [DOI] [PubMed] [Google Scholar]
- 55.Plenz G, Koenig C, Severs NJ, Robenek H: Smooth muscle cells express granulocyte-macrophage colony-stimulating factor in the undiseased and atherosclerotic human coronary artery. Arterioscler Thromb Vasc Biol 1997, 17:2489-2499 [DOI] [PubMed] [Google Scholar]
- 56.Gordon MY, Riley GP, Watt SM, Greaves MF: Compartmentalization of a haematopoietic growth factor (GM-CSF) by glycosaminoglycans in the bone marrow microenvironment. Nature 1987, 326:403-405 [DOI] [PubMed] [Google Scholar]