

Abstract
Glomeruloid bodies are a defining histological feature of glioblastoma multiforme and some other tumors and vascular malformations. Little is known about their pathogenesis. We injected a nonreplicating adenoviral vector engineered to express vascular permeability factor/vascular endothelial growth factor-164 (VPF/VEGF164) into the ears of athymic mice. This vector infected local cells that strongly expressed VPF/VEGF164 mRNA for 10 to 14 days, after which expression gradually declined. Locally expressed VPF/VEGF164 induced an early increase in microvascular permeability, leading within 24 hours to edema and deposition of extravascular fibrin; in addition, many pre-existing microvessels enlarged to form thin-walled, pericyte-poor, “mother” vessels. Glomeruloid body precursors were first detected at 3 days as focal accumulations of rapidly proliferating cells in the endothelial lining of mother vessels, immediately adjacent to cells expressing VPF/VEGF164. Initially, glomeruloid bodies were comprised of endothelial cells but subsequently pericytes and macrophages also participated. As they enlarged by endothelial cell and pericyte proliferation, glomeruloid bodies severely compromised mother vessel lumens and blood flow. Subsequently, as VPF/VEGF164 expression declined, glomeruloid bodies devolved throughout a period of weeks by apoptosis and reorganization into normal-appearing microvessels. These results provide the first animal model for inducing glomeruloid bodies and indicate that VPF/VEGF164 is sufficient for their induction and necessary for their maintenance.
Glomeruloid bodies (GBs) are structures that form in a number of different types of tumors and malformations and are so named because of their resemblance to renal glomeruli. 1 They are of two general types depending on whether they are primarily epithelial or vascular in nature. 1,2 Vascular GBs, the subject of this report, are one of the defining histological characteristics of glioblastoma multiforme brain tumors 2,3 and are found, although less commonly, in gastrointestinal carcinomas 4 and thymomas; 5 they have also been described in cutaneous vascular tumors and malformations. 6,7 Vascular GBs have not been well characterized and their pathogenesis is primarily unknown.
To elucidate the steps and mechanisms of pathological and physiological angiogenesis, we recently engineered adenoviral vectors to express angiogenic cytokines and have used these as vehicles for expressing these cytokines in mice and rats. 1 Because of its prominent role in both angiogenesis and vasculogenesis, we chose vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) as the cytokine for initial study 1,8,9 In the course of these studies, we noted that typical GB formed in intimate association with infected cells that expressed VPF/VEGF. 1 Therefore, this system provided an excellent animal model for investigating the pathogenesis of GB formation. The present study was undertaken to elucidate the origins, composition, and fate of VPF/VEGF164-induced vascular GBs.
Materials and Methods
Animals and Adenoviral Vectors
Four- to 6-week-old female athymic nude mice on two backgrounds were used for these studies with equivalent results: BALB/c ByJ hfh11nu (Jackson Laboratory, Bar Harbor, ME) and Nu/Nu (National Cancer Institute, Bethesda, MD).
Nonreplicating adenoviral vectors engineered to express murine VPF/VEGF164 (adeno-vpf/vegf164) or LacZ (adeno-lacZ) were prepared as previously described under the direction of Dr. Richard C. Mulligan in the Harvard Institute of Human Genetics. 1 In brief, appropriate coding sequences were inserted into the pMDM transcriptional cassette consisting of a complete immediate early cytomegalovirus promoter and intron- and poly A-containing sequences derived from the human β-globin gene. 10,11 Vectors were purified using a double cesium chloride-banding procedure. Immediately before injection into mice, vectors were desalted using Quick Spin, High Capacity G-50 Sephadex columns (Boehringer Mannheim, Indianapolis, IN) and diluted in phosphate-buffered saline (PBS) and glycerol.
Vectors were injected intradermally into mouse ears with a 30-gauge needle. Virus (5 × 10 7 or 1 × 10 8 pfu) or vehicle alone (Hanks’ balanced salt solution) was administered in a volume of 10 μl. Animals were sacrificed by CO2 narcosis at different times (0, 4, 18, and 24 hours and 3, 7, 10, 14, 21, 28, 35, and 90 days). Ears were either embedded in OCT compound (Miles Diagnostics, Elkhart, IN) and frozen in liquid nitrogen for preparation of cryostat sections or were fixed in paraformaldehyde-glutaraldehyde and processed for 1-μm Epon sections 12 or electron microscopy. 13 All studies were performed under protocols approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Antibodies and Other Reagents
Primary antibodies and their sources are listed in Table 1 ▶ . 14-31
Table 1.
Overview Regarding Expression of Cell-Specific Markers, Basal Lamina and Extracellular Matrix Components in Normal Vessels and in Glomeruloid Bodies (GBs) at Different Stages of Their Evolution
| Antibody and source | Normal microvessels 0 days | Primitive GB 3–4 days | Mature GB 7–10 days | Devolving GB 14–21 days | End stage >28 days |
|---|---|---|---|---|---|
| Endothelium | |||||
| CD31; Pharmingen, San Diego CA (mAb) 14,15 | + | + | + | + | + |
| VE cadherin; Pharmingen (mAb) 16 | + | − | − | −/+ | + |
| VEGFR-2 protein; Drs. Brekken/Thorpe (pAb) | + | + | + | + | + |
| Neuropilin; Oncogene, Cambridge, MA (pAb) | − | − | − | −/+ | − |
| Pericytes | |||||
| NG2; Chemicon, Temecula CA (mAb) 17,18 | −/+ | − | + | + | + |
| α-SMA; Sigma, St. Louis MO (mAb clone 1A4) 19 | + | − | −/+ | + | + |
| PDGF β-receptor; Santa Cruz (mAb clone 958) 20 | −/+ | −/+ | + | + | + |
| Smooth muscle myosin; Sigma (mAb clone hSM-V) 21 | −/+ | − | − | −/+ | −/+ |
| α and γ-actin; DAKO (mAb clone HHF) 22,23 | −/+ | − | − | −/+ | −/+ |
| Calponin; Sigma (mAb clone hCP) 24,25 | −/+ | − | − | −/+ | −/+ |
| Desmin; Chemicon (mAb) | −/+ | − | − | −/+ | −/+ |
| Macrophages | |||||
| F4/80; Serotec, Raleigh NC (mAb) | − | − | + | + | − |
| HLA class II; DAKO (mAb clone CR3/43) 26 | − | − | + | + | − |
| Basal Lamina | |||||
| Entactin; Chemicon mAb (mAb) 27 | + | + | + | + | + |
| Perlecan; mAb Chemicon (mAb) 28 | + | + | + | + | + |
| Laminin; mAb Chemicon (mAb) 29 | + | + | + | + | + |
| Collagen IV; Chemicon (mAb) 30 | + | + | + | + | + |
| Extracellular matrix | |||||
| Fibrillar collagen, Masson trichrome stain (Sigma) | + | − | − | −/+ | + |
| Fibrin, anti-fibrinogen; DAKO (pAb) 31 | − | − | + | + | −/+ |
+, all cells/structures positive; −/+, focal expression; −, no detectable expression. mAb, monoclonal antibody; pAb, polyclonal antibody.
Secondary antibodies including biotinylated F(ab)2 fragments of rabbit anti-mouse immunoglobulin, biotinylated F(ab)2 fragments of swine anti-rabbit immunoglobulin, and mouse IgG were purchased from DAKO (Carpinteria, CA). The Accustain trichrome stain kit, normal serum (rabbit, mouse, goat, and swine), and IgG (rat, rabbit, and mouse) were purchased from Sigma Chemical Co. (St. Louis, MO). The biotinylated rabbit anti-rat IgG, biotinylated horse anti-mouse IgG, Texas Red Avidin D, goat anti-rabbit IgG Texas Red conjugate, and rabbit anti-rat IgG fluorescein conjugate were purchased from Vector Laboratories (Burlingame, CA). Goat anti-mouse IgG fluorescein conjugate was purchased from Becton-Dickinson (Mountain View, CA), and the TdT apoptosis kit from R&D Systems (Minneapolis, MN).
All antibodies were diluted in solution 9 (PBS, pH 7.4, 0.1% bovine serum albumin, 150 mmol/L tranexamic acid, 20 μg/ml aprotinin (3 to 7 TIU/mg), 1.8 mmol/L ethylenediaminetetraacetic acid, and 2 mmol/L iodoacetic acid). Optimal antibody concentrations were determined by serial dilution.
Detection of Mouse Antigens with Mouse Monoclonal Antibodies
Six-μm serial cryostat sections were fixed in ice-cold acetone for 10 minutes and rinsed in PBS. IgG binding sites were blocked with normal rabbit serum diluted 1/5 in solution 9 further supplemented with 2% 3-omega fatty acid (Sigma) for 1 hour at room temperature. Complex formation between the primary and biotinylated secondary reagents was performed by appropriately diluting primary antibodies in solution 9 together with rabbit anti-mouse F(ab)2 IgG at a final concentration of 4 μg/ml for 1 hour at 37°C. Thereafter, to block residual binding sites for mouse IgG, mouse serum was added to a final concentration of 1/200, and incubated for 1 hour at 37°C. Sections were then incubated with the monoclonal antibody-F(ab)2-mouse serum complex for 1 hour at room temperature, rinsed, and incubated with 3% H2O2 in methanol for 5 minutes to deplete endogenous peroxidase. After rinsing, staining was performed with the avidin-biotin complex method, using the Vectastain ABC elite kit (Vector Laboratories) with diaminobenzidine as the peroxidase reporter. Sections were counterstained lightly with Mayer’s hematoxylin, dehydrated, and mounted in Permount.
Detection of Mouse Antigens with Rat or Rabbit Antibodies
Frozen sections were fixed, rinsed, and blocked as above. They were then incubated with primary antibody for 1 hour, rinsed in PBS, and depleted of endogenous peroxidase as above. After incubation with biotinylated rabbit anti-rat or goat anti-rabbit IgG, 7.5 μg/ml, for 30 minutes, staining was performed with the Vectastain ABC elite kit.
In Situ Hybridization
Tissues were fixed in RNase-free 4% paraformaldehyde in PBS, pH 7.4, for 4 hours at 4°C and were transferred to 30% sucrose in PBS overnight at 4°C before embedding in OCT compound. Cryostat sections were hybridized with antisense and sense (control), single-stranded, 35S-labeled RNA probes to murine VPF/VEGF, VEGFR-1, VEGFR-2, Ang-1, Ang-2, Tie-1, and Tie-2 as previously described. 32,33
Double Staining and Confocal Imaging
One hundred-μm cryostat sections were fixed in 100% acetone at 4°C for 20 minutes and rehydrated in PBS containing 0.2% Tween 40. Sections were blocked with normal goat serum containing 2% 3-omega fatty acid for 1 hour at room temperature. Sections were then incubated for 2 hours with rabbit anti-mouse NG2, rinsed, and incubated with a second primary antibody, biotinylated rat anti-mouse CD31, for 2 hours. After rinsing, sections were incubated with avidin-coupled fluorescein isothiocyanate (FITC) for 2 hours. Sections were then washed 5× in distilled water and mounted with Vectashield (Vector Laboratories).
Perfusion Studies
To determine whether the GBs that had formed in mouse ears were perfused with blood, mice were injected intravenously with 4 mg of TRITC-dextran (MW, 70,000) and 4 mg of FITC-dextran (MW, 2,000,000) in 0.9% NaCl. Fifteen minutes later mice were sacrificed, ears were fixed in a 7:3 (vol:vol) mixture of absolute ethanol and 10% formalin for 4 hours at room temperature, and were then processed for paraffin embedding. 12 Forty-micron optical sections were evaluated in a Bio-Rad MRC-1024 confocal microscope equipped with an argon/krypton laser. Sections were digitized, filtered with edge definition and median filters, and viewed as compiled images.
Alternatively, anesthetized mice were perfused through the left ventricle with heparin-saline and then with ∼20 ml of a 1:4 dilution of Sumi black ink (Yasutomo and Co., South San Francisco, CA). Ears were fixed in paraformaldehyde-glutaraldehyde, dehydrated in alcohols, cleared in methyl salicylate, split, and mounted for microscopic analysis in Permount.
Cell Proliferation
Mice were injected intravenously with 40 μCi of 3H-thymidine. Two hours later, ears were fixed and processed for 1-μm Epon sections and autoradiography. 12
Statistical Analyses
Statistical analyses were performed with the Student’s t-test.
Results
Primitive GBs (3 to 4 Days)
Adeno-vpf/vegf164 induced a characteristic sequence of events in the ears of nude mice as previously reported. 1 Microvessels were rendered hyperpermeable to plasma proteins with consequent extravasation of fibrinogen and deposition of an extravascular fibrin-rich provisional matrix. In addition, many microvessels enlarged greatly to become thin-walled, pericyte-poor mother vessels (Figure 1, a–f ▶ ; Figure 2 ▶ , a and b; Figure 3 ▶ ; Figure 4, b and d ▶ ; and Figure 5, a and b ▶ ). Mother vessels were evident as early as 18 hours and increased in size and number through ∼3 to 4 days, after which they evolved into vascular structures of several different types, one of which was GB.
Figure 1.
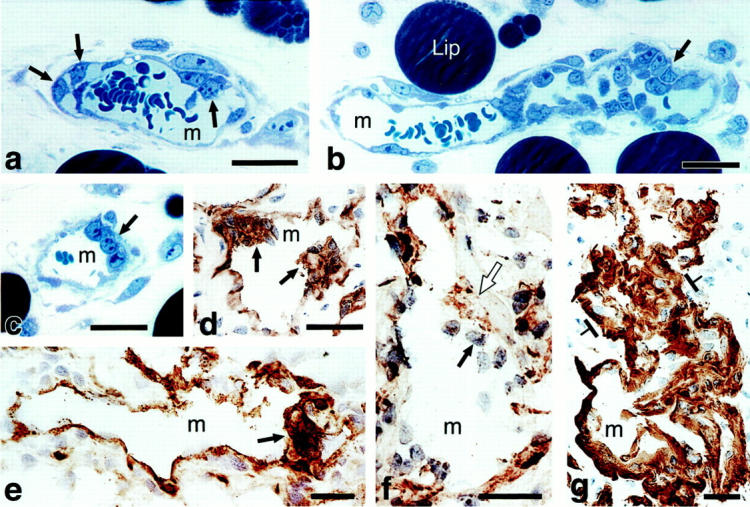
Primitive GBs developing in mother vessels 3 to 4 days after injection of adeno-vpf/vegf into ears of athymic mice. a–c: One-micron-thick, Giemsa-stained Epon sections illustrating focal accumulations of large, primitive cells (arrows) in endothelial cell lining of mother vessels that subsequently develop into GBs. d–g: Immunohistochemical staining demonstrates that primitive precursor cells (black arrows) bear endothelial cell markers (CD-31) (d), relatively increased staining for VEGFR-2 (e), but lack pericyte markers (α-SMA) (f). White arrow in f indicates α-SMA-positive pericytes just peripheral to primitive GBs (black arrow). Staining for basement membrane proteins, decreased or lost during the course of mother vessel formation, 1 is now increased in intensity (entactin) (g). m, mother vessels; lip, osmophilic, lipid-filled cell. Scale bars, 25 μm.
Figure 2.
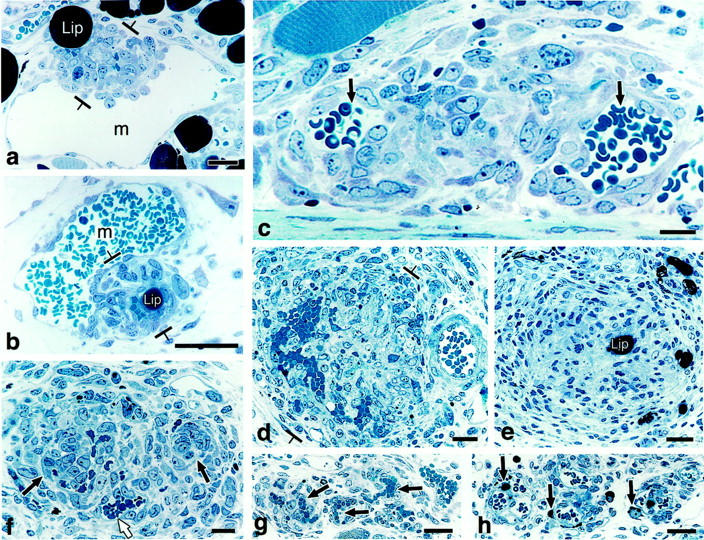
Course of GB development and devolution in 1-μm Giemsa-stained, Epon sections. a and b: Primitive GBs develop as focal nodules (between brackets) as the result of cell proliferation in the wall of a mother vessel (m) and extend both into the lumen and out into the extravascular connective tissue. Note intimate association with lipid-containing cells (lip). c–e: Maturing GBs encroach on mother vessels, reducing their single large lumens into two or more much smaller lumens (arrows) or obliterating them altogether (e). Note centrally placed, osmophilic, lipid-containing cell (lip) in e. f: Devolving GB has begun to reorganize into normal-appearing microvessels. One newly formed microvessel has acquired a lumen containing red blood cells (white arrow) whereas lumens are just beginning to form in two others (black arrows). g and h: End-stage GB is transformed into relatively normal microvessels (black arrows in g). Residual lipid-containing cells in h (black arrows), much smaller than at earlier stages of GB evolution (compare with a, b, and e), are immediately adjacent to each of three newly formed microvessels. Scale bars, 25 μm.
Figure 3.
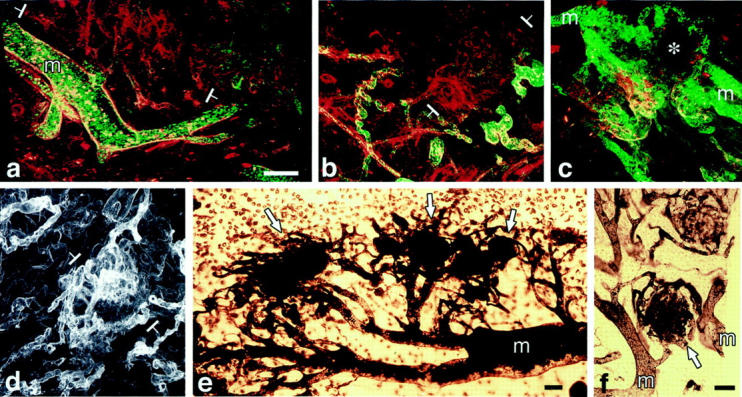
a–d: Confocal microscopy of mature GBs. Those in a–c are from mice injected intravenously with both FITC-D (MW, 2,000,000) and TRITC-D (MW, 70,000) 15 minutes before sacrifice. In a and b, GB (white brackets) are perfused with TRITC-D (red), but not with the larger FITC-D (green) that is confined to mother vessels (m) and other larger vessels that feed GBs. In c, mother vessels (two of which are labeled) stain with both dextrans but a GB (asterisk) does not, indicating lack of perfusion with either FITC-D or TRITC-D. d: Immunofluorescent staining of a GB with the basement membrane marker, entactin, illustrates multiple afferent and efferent vessels supplying a GB (white brackets). e and f:. Whole mounts of ear from a mouse perfused with black ink. Note multiple mature GBs (white arrows) supplied by multiple afferent and efferent vessels connected to mother vessels (m). In f, a GB links two mother vessels. Scale bars, 50 μm.
Figure 4.
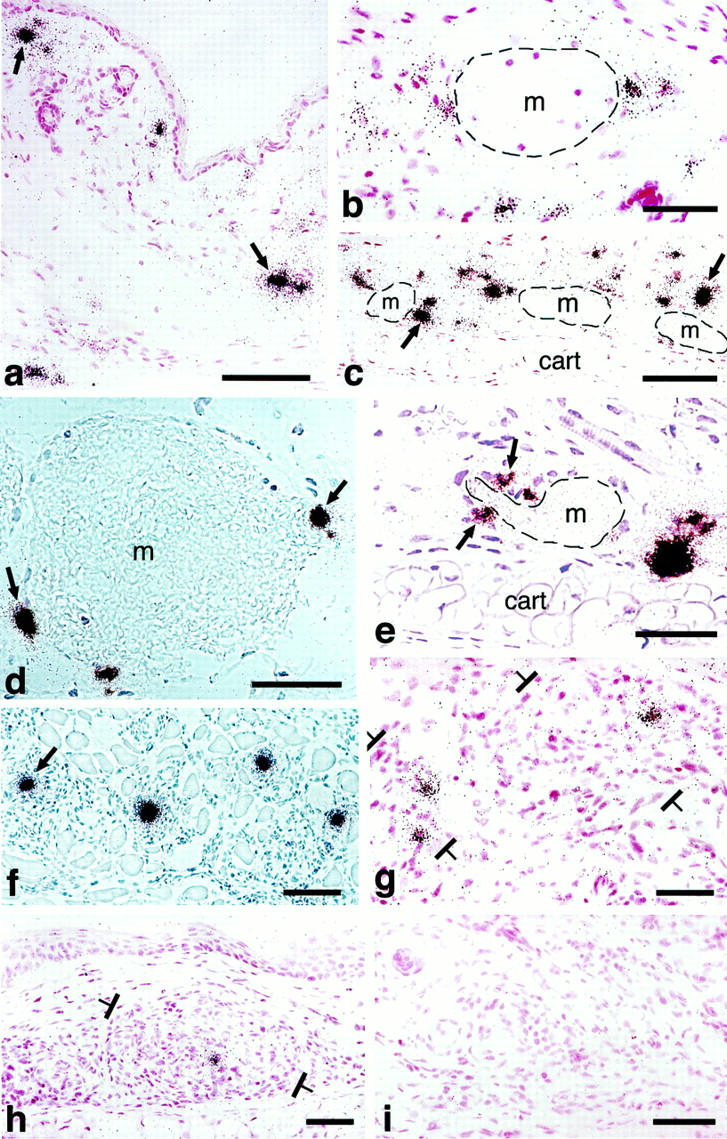
In situ hybridization illustrating cellular VPF/VEGF mRNA expression at various times after injection of adeno-vpf/vegf into the ears of athymic mice. a and b: At 18 hours, scattered cells in dermis (some indicated by black arrows) express large amounts of VPF/VEGF mRNA. b: Some of the positive cells are immediately adjacent to a mother vessel (m). c–e: At 4 days, numerous VPF/VEGF-positive cells are observed deep in the dermis just above the ear cartilage (cart), the location where GBs most commonly form. Note strongly positive cells immediately adjacent to mother vessels (m). e: VPF/VEGF-expressing cells are in immediate proximity to a primitive GB (arrows), developing in a deep dermal mother vessel (m) adjacent to ear cartilage (cart). f: Eight-day reaction illustrating VPF/VEGF-expressing cells in centers of several maturing GBs (one indicated with an arrow). g: At 14 days, several VPF/VEGF-expressing cells persist within GBs (brackets) but average intensity is somewhat reduced in comparison with 8 days. h: Further reduction in numbers and staining intensity of VPF/VEGF-expressing cells at 21 days. A residual positive cell is placed centrally within a GB (brackets). i: By 35 days, VPF/VEGF mRNA was no longer detectably expressed and GBs had begun to devolve into normal microvessels. d and f: Phase contrast microscopy; all others, bright field. Scale bars, 25 μm.
Figure 5.

In situ hybridization for expression of VPF/VEGF receptors in adeno-vpf/vegf injected skin. a and b: VEGFR-1 mRNA expression in vascular endothelium of mother vessels (m) at 4 days. E, epidermis. c and d: VEGFR-2 mRNA expression in GBs (brackets) at 10 days (c) and 21 days (d). Scale bars, 25 μm.
Primitive GBs were first recognized at 3 days as focal accumulations of a few large, poorly differentiated cells in the endothelial lining of developing or fully formed mother vessels (Figure 1, a–f ▶ , and Tables 1 and 2 ▶ ▶ ). Primitive GBs arose in immediate proximity to adeno-vpf/vegf164-infected cells that expressed abundant VPF/VEGF mRNA (Figure 4; b, d, and e ▶ ). They were derived from a rapidly dividing cell population such that on days 4 and 5, respectively, 22 ± 14% and 18 ± 8% (mean ± SD) labeled with 3H thymidine (Table 2) ▶ .
Table 2.
Proliferation and Apoptosis of Cells Comprising Glomeruloid Bodies (GBs) at Various Stages of Their Evolution and Devolution
| Normal microvessels 0 days | Primitive GB 3–4 days | Mature GB 7–10 days | Devolving GB 14–21 days | End stage >28 days | |
|---|---|---|---|---|---|
| Proliferation | 0 | ++ | ++ | ± | 0 |
| Apoptosis | 0 | 0 | 0 | + | 0 |
*Proliferation was assessed in 1-μm Epon sections for incorporation of 3H-thymidine. Cells with ≥4 grains were considered positive. Apoptosis was assessed with the TUNEL assay. 0, no labeled cells; ±, occasional labeled cells; +, 1 to 10% of cells labeled; ++, >10% of cells labeled.
All of the cells comprising the earliest GBs were CD31-positive (Figure 1d) ▶ . They also stained more intensely for VEGFR-2 (Figure 1e) ▶ , but less intensely for VE-cadherin (not shown), than adjacent mother-vessel endothelial cells that were not involved in GB formation. None stained with pericyte (Figure 1f) ▶ or macrophage (not shown) markers. Pericytes were, however, identified immediately abluminal to developing GBs by staining with antibodies to pericyte markers such as α-smooth muscle actin (α-SMA) (Figure 1f) ▶ . During the course of mother-vessel formation, staining for basal lamina proteins was reduced focally and in some cases circumferentially. 1 However, by 3 to 4 days, basal lamina staining had returned to normal and even higher than normal levels (Figure 1g) ▶ .
Mature GBs (7 to 10 Days)
The cells comprising primitive GB continued to proliferate actively (eg, 14 ± 7% of cells labeled with 3H thymidine on day 8) and became organized into cell nests of increasing size. Whereas on day 4 after adeno-vpf/vegf164 injection the number of cells comprising GBs in random 1-μm sections was 21 ± 14, by day 8 that number had increased to 50 ± 23 (P = 0.0002).
As they increased in number, the cells comprising GBs projected into the lumens of mother vessels and also extended outward into the surrounding extravascular matrix (Figure 2, a to e) ▶ . As a result, they encroached on and compressed the mother vessels from which they had arisen, eventually dividing their lumens into much smaller channels that were only marginally perfused with blood (Figure 2, c and d ▶ ; Figure 3, a–c ▶ ). By day 8 it was often difficult to tell that GBs had arisen from mother vessels. However, in whole-mount preparations the close relationship between mother vessels and GBs could still be discerned (Figure 3, d–f) ▶ . GBs were sometimes found to link adjacent mother vessels (Figure 3f) ▶ .
The great majority of cells comprising mature GBs continued to express endothelial cell markers (Table 1) ▶ ; ie, they expressed VEGFR-1 and -2 mRNAs (Figure 5) ▶ and CD-31 (Figure 6a) ▶ and VEGFR-2 proteins (not shown), but not neuropilin (not shown). However, cells having ultrastructural characteristics of pericytes (Figures 7 and 8) ▶ ▶ and staining for pericyte markers (Figure 6c) ▶ were now also present. By immunohistochemistry, these cells were strongly positive for NG2, the murine analogue of the high molecular weight melanoma-associated antigen, and the platelet-derived growth factor (PDGF)-β receptor. However, they expressed lower concentrations of proteins associated with contractile functions (α-SMA (compare Figure 6, b and c ▶ ), calponin, desmin, and smooth muscle myosin) than pericytes of surrounding, normal microvessels or of microvessels of normal skin (not shown). By immunohistochemistry pericytes appeared to be admixed randomly with endothelial cells. However, by electron microscopy, it was clear that pericytes were always situated peripherally to small cell clusters of endothelial cells with small or no evident vascular lumens (Figures 7 and 8) ▶ ▶ . Occasional endothelial cell-pericyte contacts were identified by electron microscopy (not shown). Monocytes and macrophages also began to appear as determined by staining with antibodies to F4/80 (Figure 6d) ▶ and HLA DR2 (not shown) and by electron microscopy (Figure 7 ▶ and Figure 8 ▶ ); macrophages tended to concentrate at the interface between GBs and the surrounding connective tissue matrix (Figure 6d) ▶ . Prominent lipid-filled cells were often found adjacent to or centrally placed within GBs (Figure 1b ▶ ; Figure 2, a, b, and e ▶ ; Figure 7 ▶ ; and Figure 8 ▶ ). Occasional eosinophils and neutrophils were also present, but not nerves or mast cells. As at earlier times, basal lamina proteins were deposited between the cells comprising GBs in abnormal thickened multilayers (Figure 6f ▶ and Figure 8 ▶ , inset).
Figure 6.
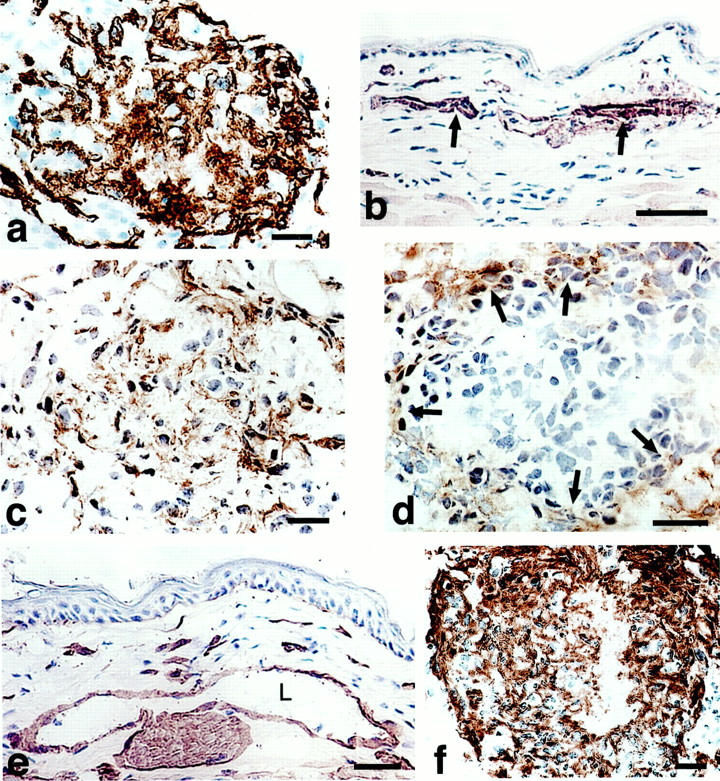
Immunohistochemical characterization of mature GBs (7 to 10 days). a: CD-31-positive cells (putative endothelial cells) continue to predominate. b and c: α-SMA staining of pericytes in vessels of normal skin (arrows in b) and in a mature GB (c) where they are intermingled with endothelial cells. d: F40/80-positive cells (macrophages) are present at the GB periphery (arrows). e and f: Antibodies to entactin (and other basement membrane proteins, not shown) stain basement membranes of vessels and nerves in normal skin (e) and are abundantly deposited but in a disorganized manner in mature GBs (f). L, vascular lumen. Scale bars, 25 μm.
Figure 7.
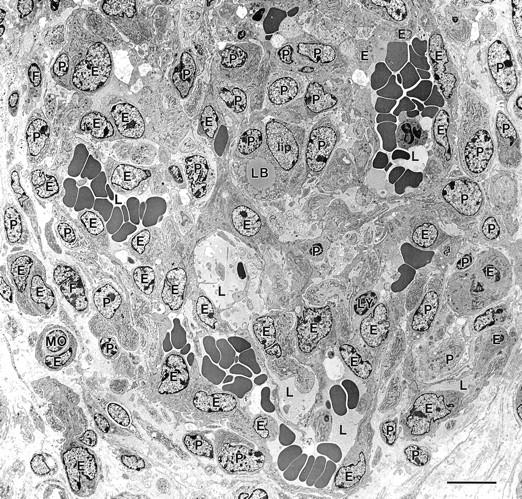
Electron micrograph of a maturing GB. GB appears as a nodular structure with a centrally placed lipid-containing cell (lip) and comprised of several different cell types: E, endothelial cells; P, pericytes; lip, lipid-containing cell with a cytoplasmic lipid body, LB; MO, monocyte; F, fibroblasts. Cells were identified by examination of this same field at higher magnification, using standard electron microscopic criteria. 59,60 Multiple small vascular lumens (L), many but not all containing red blood cells, are lined by endothelial cells; lesser numbers of pericytes are scattered in between. Scale bar, 10 μm.
Figure 8.
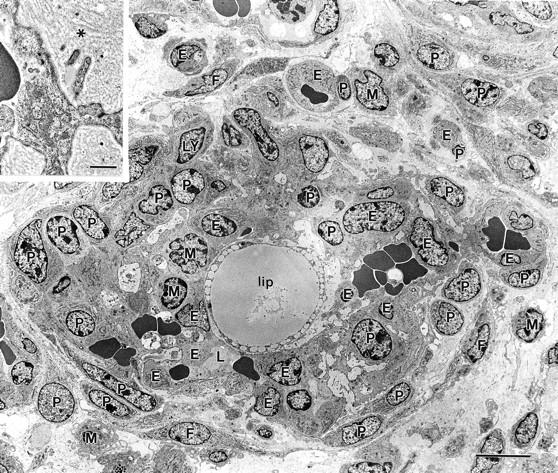
Electron micrograph illustrating a somewhat more mature GB, again centered about a large lipid-containing cell (lip). Several small erythrocyte-containing vascular lumens persist (one marked L). Cell code is the same as that in Figure 7 ▶ with the addition of the following: M, macrophages; Ly, lymphocytes. Individual cells were identified by standard electron microscopic criteria after examining this same field at higher magnification. 59,60 Inset illustrates multiple layers of basal lamina (asterisk) deposited between luminal endothelial cells and pericytes, a finding also typical of the GBs of glioblastomas. 3 Scale bar, 10 μm; inset bar, 1 μm.
Devolving GBs (14 to 21 Days)
By day 14 GBs began to devolve into a series of smaller microvascular units, each comprised of centrally placed endothelial cells surrounded by pericytes (Figure 9) ▶ . Endothelial cell-pericyte orientations now more clearly resembled those of normal microvessels. Individual pericytes were at first commonly positioned between adjacent clumps of endothelial cells, but, throughout time, each endothelial cell cluster came to acquire its own mantle of pericytes. Endothelial cells once again began to express VE cadherin (not shown) and vascular lumens reappeared centrally in endothelial cell clusters (Figure 2f ▶ and Figure 9, c–e ▶ ). Concomitant with these events, basal lamina proteins were deposited between endothelial cells and pericytes in a distribution typical of normal vessels (Figure 9, e and f) ▶ . In this manner, individual, normal-appearing microvessels were formed and began to spin off from the residual GB mass (Figure 9, g–i) ▶ . Macrophages decreased markedly in number. GB devolution was accompanied by significant apoptosis, evident on day 14 and maximal at day 21 (Figure 10) ▶ . In parallel, numerous epidermal cells underwent apoptosis and the epidermis, which had become hyperplastic in the course of GB formation, gradually returned to normal thickness.
Figure 9.
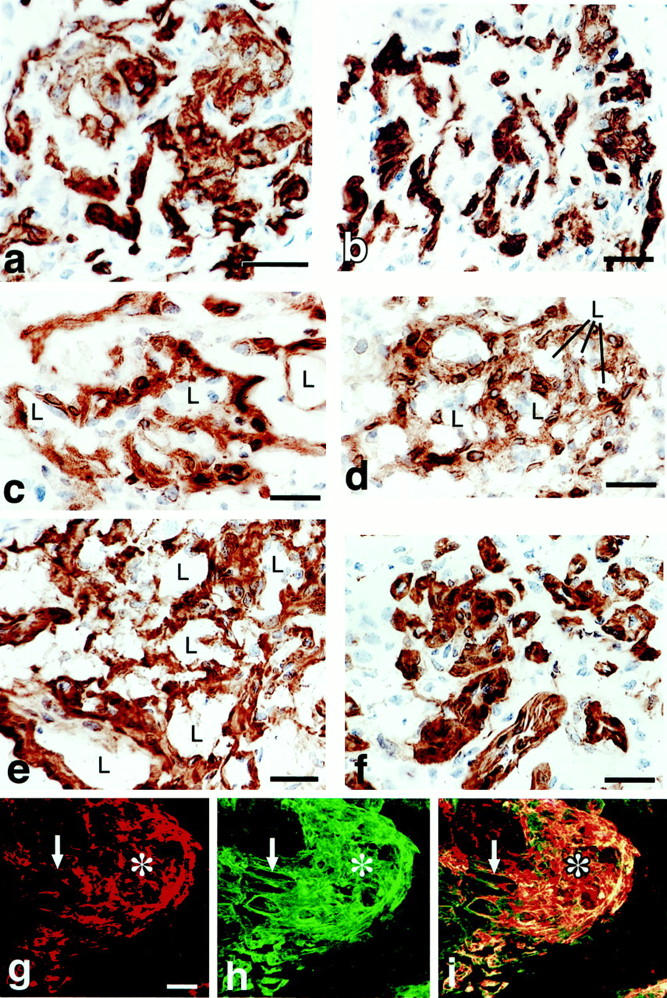
Early (a, c, and e) and later stages (b, d, and f) of GB devolution into daughter microvessels. a and b: CD-31 staining demonstrates endothelial cells forming discrete clusters that are separated from each other by nonstaining cells (for the most part pericytes). c and d: Pericytes stained with antibodies to NG2 envelop endothelial cells that have begun to form lumens (L). e and f: Entactin staining for basement membrane. g–i: A GB undergoing devolution into daughter microvessels is illustrated by double-fluorescent staining for NG2-expressing pericytes (red, g), basement membrane entactin (green, h), and composite (i). Individual daughter vessels (white arrows) have separated from the main GB cell mass (asterisk). Scale bars, 12.5 μm (a–f) and 25 μm (g–i).
Figure 10.
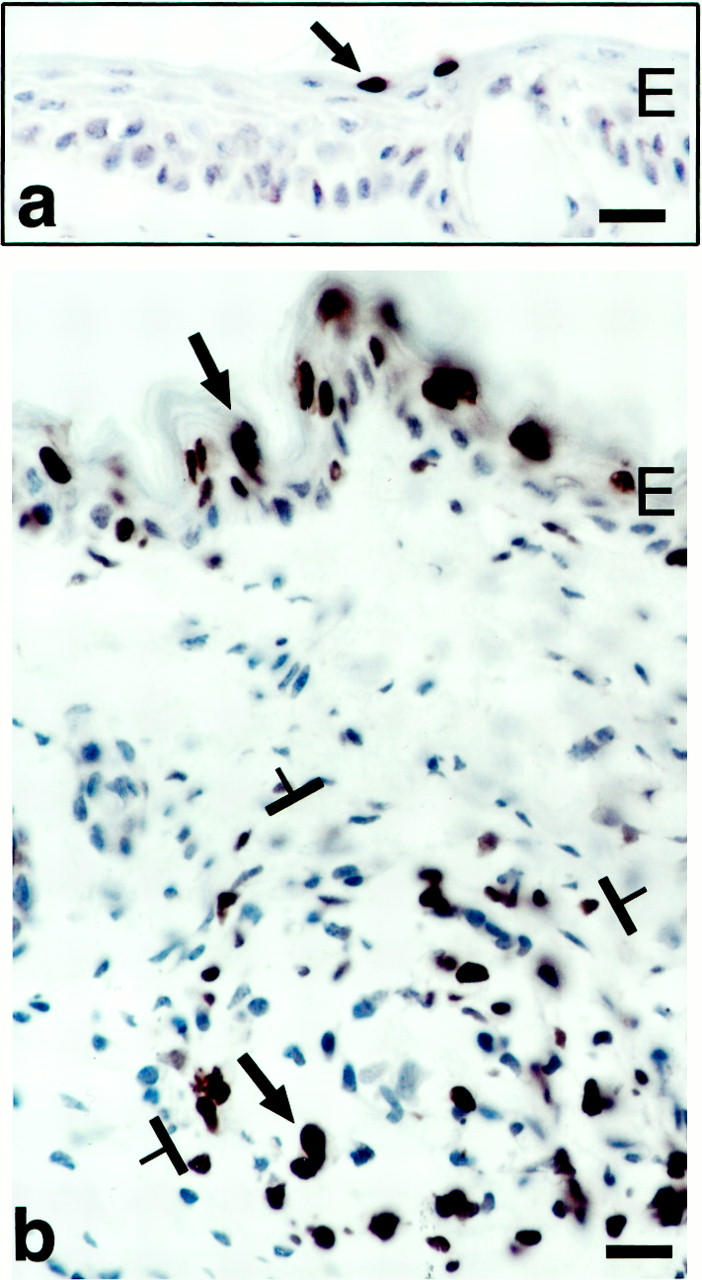
TUNEL staining to demonstrate apoptotic cells (some indicated by arrows). a: Low-level apoptosis in normal mouse ear epidermis. b: Extensive apoptosis in the formerly hyperplastic epidermal ear skin and in a GB (brackets) at 21 days after ear injection with adeno-vpf/vegf. E, epidermis. Scale bars, 25 μm.
End-Stage GBs (28 to 90 Days)
During this period, GBs disappeared altogether as distinct entities and were replaced by multiple small daughter microvessels. As newly formed microvessels formed and began to separate from GBs, a subpopulation of pericytes appeared that, in addition to expressing the PDGF-β receptor (not shown) and NG2 (Figure 11b) ▶ , also strongly expressed α-SMA (Figure 11a) ▶ , γ-actin, desmin, smooth muscle myosin, and calponin (not shown), a complement of proteins that comprise the cell’s contractile machinery. Pericytes expressing this set of proteins were observed at points of residual contact between adjacent developing daughter vessels (Figure 11, a and b) ▶ . Fibrillar collagen was also deposited in these same locations (Figure 11c) ▶ , progressively replacing the fibrin provisional matrix and further separating individual daughter microvessels from each other. Four to 5 weeks after injection normal-appearing daughter capillaries and venules were evenly dispersed (not shown), much as in normal ear skin.
Figure 11.

a and b: Immunohistochemical staining for pericytes in devolving GB. Pericytes (arrows) strongly express α-SMA (a) and NG2 (b) at residual points of interconnection between newly formed daughter vessels. c: Trichrome stain illustrates deposition of fibrillar collagen around newly formed daughter vessels. L, vascular lumens. Scale bars, 12.5 μm.
Expression of VPF/VEGF164, VEGF Receptors, and Other Cytokines/Receptors
In situ hybridization was used to identify the cells that were infected by adeno-vpf/vegf164 and to investigate the kinetics of VPF/VEGF164 expression. From 18 hours to 4 days cells expressing abundant VPF/VEGF mRNA were present in the dermis, especially in the deep dermis, adjacent to the ear cartilage (Figure 4, a–e) ▶ . Many of these heavily labeled cells were in immediate proximity to developing or fully developed mother vessels (Figure 4, b–e) ▶ , ie, in a position normally occupied by pericytes. However, the technologies we used did not allow us to determine whether these VPF/VEGF164-expressing cells were in fact pericytes. At 3 days, strongly VPF/VEGF164-expressing cells were found adjacent to mother vessels in the immediate vicinity of developing GBs (Figure 4e) ▶ , and at subsequent intervals strongly labeled cells were found in the centers of mature GBs (Figure 4f) ▶ . Curiously, prominent lipid-filled cells were also commonly observed in the centers of mature GBs (Figure 2, a, b, and e ▶ , Figure 7 ▶ , and Figure 8 ▶ ). Unfortunately, we were unable to determine whether these lipid-filled cells were the cells expressing large amounts of VPF/VEGF164 mRNA.
Because adenoviral vectors are not incorporated into chromosomal DNA, the transgenes they introduce are only expressed for a limited period of time. The numbers of VPF/VEGF mRNA-expressing cells and the intensity of expression as judged by grains per cell remained constant for ∼2 weeks, after which both the frequency and intensity of labeling of individual cells declined gradually until by 21 days only occasional, weakly positive cells remained (Figure 4, g–i) ▶ .
VEGFR-1 and VEGFR-2 expression followed similar kinetics, becoming detectable in mother vessel endothelial cells at 18 hours and more strongly at 4 days (Figure 5) ▶ . Thereafter, GBs continued positive for the mRNAs of both receptors, declining after 2 weeks and falling to low levels by 21 days.
Cells in GB expressed Ang-1 and Ang-2 mRNAs weakly from 10 to 21 days and expression declined thereafter. Tie-1 and Tie-2 mRNAs were weakly expressed in GBs as early as 3 days, strongly at 10 days, and weakly thereafter through 21 days.
Discussion
Infection of mouse ear skin with an adenoviral vector engineered to express VPF/VEGF164 stimulated local generation of vascular GBs typical of those found in glioblastoma multiforme, certain other human tumors, and vascular malformations. 2-7,34-36 GBs were first recognized at 3 days after adeno-vpf/vegf164 injection as focal collections of large, primitive cells in the endothelial cell lining of mother vessels. These cells expressed endothelial cell markers and were intimately associated with adeno-vpf/vegf-infected cells that expressed large amounts of VPF/VEGF mRNA. Primitive GBs grew rapidly in size as a consequence of cell proliferation and extended inwards as well as outwards into the extravascular connective tissue where they formed nodules that were centered around VPF/VEGF164-expressing cells. The nature of the VPF/VEGF164-expressing cells was not determined with certainty; initially many infected cells had the distribution of pericytes but centrally placed cells in developing and mature GBs often contained large amounts of lipid. With further expansion, GBs severely compromised the mother vessels in which they had arisen, reducing originally large single lumens into multiple, much smaller channels. As a result, blood flow through GBs was greatly reduced and sometimes shut down entirely. Initially, all of the cells comprising GBs bore endothelial cell markers, but, by 7 to 10 days, pericytes appeared and came to envelop small clusters of endothelial cells; subsequently, macrophages also appeared but were primarily confined to the periphery, at the interface of maturing GBs with the surrounding extracellular matrix. After ∼14 days, VPF/VEGF mRNA expression declined and, concomitantly, GBs devolved by a process that involved both apoptosis and reorganization of endothelial cells and pericytes into daughter microvessels typical of those found in normal ear skin. The entire sequence of events is diagrammed schematically in Figure 12 ▶ . Taken together, our experiments provide the first animal model for generating vascular GBs, provide important insights into the mechanisms responsible for GB formation and devolution, and demonstrate that VPF/VEGF164 is both sufficient for their generation and necessary for their maintenance.
Figure 12.
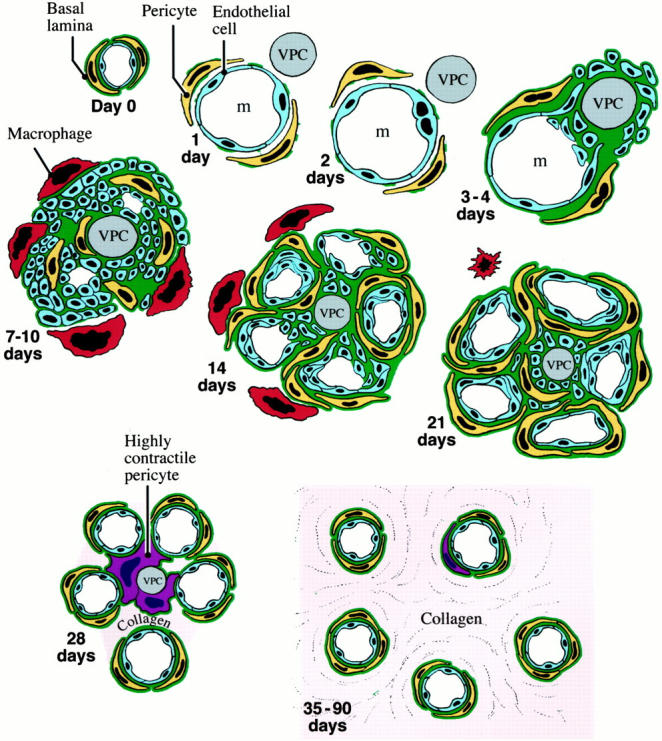
Schematic drawing of GB evolution and devolution. Day 0: Normal microvessel lined by endothelial cells, pericytes, and basal lamina. Day 1: One day after injection of adeno-vpf/vegf precursor microvessel has enlarged greatly as the result of pericyte detachment and basal lamina degradation to form a mother vessel (m). A VPF/VEGF-expressing cell (VPC) lies immediately adjacent. Day 2: Proliferation of mother vessel endothelium first noted. Days 3 to 4: Proliferating endothelial cells extend outside the mother vessel and cluster around a VPC. Basal lamina components are deposited in excessive and disorganized multilayers. Pericytes begin to proliferate. Days 7 to 10: GB expansion as the result of cell division has compromised original mother vessel lumen, reducing it here into two smaller channels. Pericytes are apparently intermingled among endothelial cells. Macrophages accumulate at GB periphery. Day 14: Earliest stages of GB devolution as VPF/VEGF expression by VPC is on the wane. Pericytes are more numerous and surround clusters of endothelial cells that have now formed obvious vascular lumens. Day 21: Later stage of GB devolution with greatly reduced VPF/VEGF expression and better definition of individually forming microvessels. Day 28: Newly formed microvessels have begun to separate from each other but some remain linked by pericytes that strongly express contractile proteins such as α-SMA. Small amounts of fibrillar collagen are deposited. Days 35 to 90: GBs are completely replaced by normal-appearing microvessels interspersed between moderate amounts of newly deposited fibrillar collagen.
The GBs induced in mouse skin with adeno-vpf/vegf164 closely resembled those found in glioblastoma multiforme in structure and composition. 2,3,34-36 At one time it was thought that the GB of human glioblastomas were comprised entirely of endothelial cells. However, more recent studies have demonstrated the presence of cells that express pericyte markers, HMW-MAA and α-SMA. 34,35 Thus, both pericytes and endothelium contribute importantly to the GBs of high-grade human gliomas and to those induced here in normal mouse tissues with an adenoviral vector. Whether the GBs that develop in human glioblastomas also arise from mother vessels remains to be determined.
High concentrations of VPF/VEGF164 were apparently necessary to induce and sustain GBs. In our mouse model, GBs formed in immediate anatomical proximity to adeno-vpf/vegf164-infected cells that strongly expressed VPF/VEGF mRNA. Glioblastomas express larger amounts of VPF/VEGF than most other human tumors 3,36,37 ; however, glioblastomas also express many other cytokines and growth factors that might reasonably be implicated in GB formation. Therefore, the finding that overexpression of VPF/VEGF164 alone leads to GB formation is significant and allows us to postulate that the GBs found in glioblastomas, in other human tumors, and in vascular malformations also result from local overexpression of VPF/VEGF. Further correlative evidence in support of VPF/VEGF’s role in the etiology of GB formation came from later events in our mouse adenoviral vector model. As VPF/VEGF164 expression declined, GBs began to devolve. Of course no such decline in VPF/VEGF expression would be expected in glioblastomas as these tumors continue to express VPF/VEGF and generate GBs throughout their course.
The cells that were first recognized as primitive GBs were located amid other endothelial cells lining mother vessels and expressed endothelial markers (CD31, VEGFR-1, VEGFR 2). Together these findings are consistent with an origin from pre-existing mother-vessel endothelium. However, the markers that these cells expressed are not entirely specific for endothelium and are also represented on hematopoietic cell precursors. 38-40 Therefore, a blood or bone marrow source for some or all of these precursor cells cannot be excluded. Subsequently, pericytes also contributed to GBs and their origins were also not established with certainty. The likeliest possibility, however, is that they developed from mother-vessel pericytes because they first appeared admixed among endothelial cells; there was no evidence to suggest that they differentiated from primitive mesenchymal or other dermal cells.
The stimuli responsible for pericyte participation in GBs are uncertain. One possibility is that VPF/VEGF induced the proliferation of local pericytes by stimulating endothelial cells to express PDGF-B chain, 41 a potent pericyte mitogen. 42,43 PDGF BB is also chemotactic for pericytes 44 and is thought to be important for pericyte differentiation and their coating of newly formed vessels during embryogenesis. 44-47 However, recent findings raise the alternative possibility that VPF/VEGF acted directly on pericytes. In addition to its selective action on endothelial cells, VPF/VEGF interacts with macrophages and certain populations of smooth muscle cells that express biologically active VPF/VEGF receptors. 8,9,48 VEGFR-1 has also been found to be expressed on cultured pericytes, and under certain conditions (hypoxia), VPF/VEGF is chemotactic for pericytes. 49-51 These findings raise the possibility that VPF/VEGF stimulated pericyte migration and proliferation directly.
Beginning around day 14, GBs began a process of devolution that was characterized by significant apoptosis. Endothelial cells, pericytes, and macrophages all declined in number, paralleling a decline in local VPF/VEGF164 expression. VPF/VEGF is an important survival factor for vascular endothelium, 47,52 particularly for immature vessels that have not acquired a pericyte coat. The endothelial cell apoptosis observed may therefore have resulted from a combination of decreased local VPF/VEGF and insufficient pericyte coating. Keratinocytes express VPF/VEGF, 53 but have not been reported to express VPF/VEGF receptors. Most likely the apoptotic events observed reflect a series of complex paracrine relationships among the various cell types that were upset by decreased VPF/VEGF. Similar paracrine stimulatory loops are thought to maintain GBs in glioblastoma multiforme 34,36,37,54,55 . These findings may have interesting implications for anti-angiogenesis therapies designed to suppress VPF/VEGF expression in glioblastomas. Our studies would suggest that decreased local VPF/VEGF expression would initiate GB devolution, and it is uncertain whether such devolution with replacement by normal microvessels would be of advantage to the tumor or to the host.
As GBs devolved, they were gradually replaced by normal-appearing microvessels that at first were closely joined together and only later separated from one another. A subpopulation of pericytes that expressed proteins involved in contractile events may have had a role in positioning and separating these newly formed daughter vessels from each other. Pericytes that expressed α-SMA, γ-actin, and so forth, were strategically positioned at points of interconnection between developing daughter vessels, suggesting a possible role in the final separation of daughter vessels from each other. This would be consistent with other examples in which pericytes expressing a similar profile of contractile machinery have important roles in tissue remodeling. 56-58
In conclusion, injection of an adenoviral vector engineered to express VPF/VEGF164 induced the formation of typical vascular GBs in normal ear skin. These GBs developed in mother vessels from recruitment and proliferation of endothelial cells and pericytes in intimate contact with VPF/VEGF-expressing cells. As local VPF/VEGF expression began to decline at ∼2 weeks, GBs gradually devolved, a process characterized by apoptosis and reorganization of remaining endothelial cells and pericytes into normal-appearing microvessels.
Acknowledgments
We thank Drs. R. Brekken and P. Thorpe for the gift of their antibody to VEGFR-2, Dr. Richard Mulligan and the Harvard Institute of Human Genetics for adeno-vpf/vegf, Dr. George Yancopoulos for probes to ang-1, ang-2, Tie-1, and Tie-2, Mr. Steven Moskowitz for preparation of the figures and the schematic, and Ms. Katherine Pyne for printing the electron micrographs.
Footnotes
Address reprint requests to Harold F. Dvorak, M.D., Department of Pathology, Beth Israel Deaconess Medical Center, Boston, MA 02215. E-mail: hdvorak@caregroup.harvard.edu.
Supported in part by U. S. Public Health Service grants CA-50453 and HL-59316 (to H. F. D.), AI-33372 and AI-44066 (to A. M. D.), by a contract from the National Foundation for Cancer Research (to H. F. D.), and by a grant from the Swedish Cancer Foundation, Konung Gustaf V:s 80 årsfond (to C. S.).
Present address of C. S.: Dept. of Medical Biochemistry and Microbiology, Uppsala University, BMC, Box 575, SE-751 23, Uppsala, Sweden.
References
- 1.Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF: Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest 2000, 80:99-115 [DOI] [PubMed] [Google Scholar]
- 2.Rojiani AM, Dorovini-Zis K: Glomeruloid vascular structures in glioblastoma multiforme: an immunohistochemical and ultrastructural study. J Neurosurg 1996, 85:1078-1084 [DOI] [PubMed] [Google Scholar]
- 3.McLendon R, Enterline D, Tien R, Thorstad W, Bruner J: Tumors of central neuroepithelial origin. Bigner D McLendon R Bruner J eds. Russell and Rubinstein’s Pathology of Tumors of the Nervous System, ch 9. 1998, :pp 307-571 Oxford University Press, New York [Google Scholar]
- 4.Ohtani H: Glomeruloid structures as vascular reaction in human gastrointestinal carcinoma. Jpn J Cancer Res 1992, 83:1334-1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaker H, Dragoje S, Laissue JA, Otto HF: Pericardial involvement by thymomas. Entirely intrapericardial thymoma and a pericardial metastasis of thymoma with glomeruloid vascular proliferations. Pathol Oncol Res 1999, 5:160-163 [DOI] [PubMed] [Google Scholar]
- 6.McKee P: Pathology of the Skin with Clinical Correlations. 1996:pp 16.57-16.66 Mosby International, London
- 7.Strutton G: Vascular Tumors, Skin Pathology Ch 38. Edited by D Weedon. Edinburgh, Churchill Livingstone, 1997, pp 821–854
- 8.Brown L, Detmar M, Claffey K, Nagy J, Feng D, Dvorak A, Dvorak H: Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. Goldberg I Rosen E eds. Regulation of Angiogenesis. 1997, :pp 233-269 Birkhauser Verlag, Basel [DOI] [PubMed] [Google Scholar]
- 9.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 10.Ory DS, Neugeboren BA, Mulligan RC: A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA 1996, 93:11400-11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML: Construction of adenovirus vectors through Cre-lox recombination. J Virol 1997, 71:1842-1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy J, Morgan E, Herzberg K, Manseau E, Dvorak A, Dvorak H: Pathogenesis of ascites tumor growth. Angiogenesis, vascular remodeling and stroma formation in the peritoneal lining. Cancer Res 1995, 55:376-385 [PubMed] [Google Scholar]
- 13.Dvorak A: Procedural guide to specimen handling for the ultrastructural pathology service laboratory. J Electron Microsc Tech 1987, 6:255-301 [Google Scholar]
- 14.Baldwin HS, Shen HM, Yan HC, Delisser HM, Chung A, Mickanin C, Trask T, Kirschbaum NE, Newman PJ, Albeda SM, Buck CA: Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development 1994, 120:2539-2553 [DOI] [PubMed] [Google Scholar]
- 15.Parums DV, Cordell JL, Micklem K, Heryet AR, Gatter KC, Mason DY: JC70: a new monoclonal antibody that detects vascular endothelium associated antigen on routinely processed tissue sections. J Clin Pathol 1990, 43:752-757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lampugnani MG, Resnati M, Raiteri M, Pigott R, Piscane A, Houen G, Ruco LP, Dejana E: A novel endothelial specific membrane protein is a marker of cell-cell contacts. J Cell Biol 1992, 118:1511-1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine JM, Nishiyama A: The NG2 chondroitin sulfate proteoglycan: a multifunctional proteoglycan associated with immature cells. Perspect Dev Neurobiol 1996, 3:245-259 [PubMed] [Google Scholar]
- 18.Burg MA, Pasqualini R, Arap W, Rouslahti E, Stallcup WB: NG2 proteoglycan binding peptides target tumor neovasculature. Cancer Res 1999, 59:2869-2874 [PubMed] [Google Scholar]
- 19.Skalli O, Ropraz P, Trzeciak A, Benzonona G, Gilessen D, Gabbiani G: A monoclonal antibody against alpha smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 1986, 103:2787-2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heldin C-H, Bäckström G, Östman A, Hammacher A, Rönnstrand L, Rubin K, Nistér M, Westermark B: Binding of different dimeric forms of PDGF to human fibroblasts: evidence for two separate receptor types. EMBO J 1988, 7:1387-1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longtine JA, Pinkus GS, Fujiwara K, Corson JM: Immunohistochemical localization of smooth muscle myosin in normal human tissues. J Histochem Cytochem 1985, 33:179-184 [DOI] [PubMed] [Google Scholar]
- 22.Gown AM, Tsukada T, Ross R: Human atherosclerosis. II. Immunocytochemical analysis of the cellular composition of human atherosclerotic lesions. Am J Pathol 1986, 125:191-207 [PMC free article] [PubMed] [Google Scholar]
- 23.Tsukada T, McNutt MA, Ross R, Gown AM: HHF35, a muscle actin-specific monoclonal antibody. II. Reactivity in normal, reactive, and neoplastic human tissues. Am J Pathol 1987, 127:389-402 [PMC free article] [PubMed] [Google Scholar]
- 24.Frid MG, Shekhonin BV, Koteliansky VE, Glukhova MA: Phenotypic changes of human smooth muscle cells during development: late expression of heavy caldesmon and calponin. Dev Biol 1992, 153:185-193 [DOI] [PubMed] [Google Scholar]
- 25.Lazard D, Sastre X, Frid MG, Glukhova MA, Thiery JP, Koteliansky VE: Expression of smooth muscle-specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue. Proc Natl Acad Sci USA 1993, 90:999-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith ME, Holgate CS, Williamson JM, Grigor I, Quirke P, Bird CC: Major histocompatibility complex class II antigen expression in B and T cell non-Hodgkin’s lymphoma. J Clin Pathol 1987, 40:34-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ljubimov AV, Afanasjeva AV, Litvinova LV, Senin VM: Basement membrane components produced by a mouse ascites teratocarcinoma TB24. Analysis with monoclonal and polyclonal antibodies. Exp Cell Res 1986, 165:530-540 [DOI] [PubMed] [Google Scholar]
- 28.Couchman JR, Ljubimov AV: Mammalian tissue distribution of a large heparan sulfate proteoglycan detected by monoclonal antibodies. Matrix 1989, 9:311-321 [DOI] [PubMed] [Google Scholar]
- 29.Desjardins M, Bendayan M: Heterogenous distribution of type IV collagen, entactin, heparan sulfate proteoglycan, and laminin among renal basement membranes as demonstrated by quantitative immunocytochemistry. J Histochem Cytochem 1989, 37:885-897 [DOI] [PubMed] [Google Scholar]
- 30.Andujar MB, Magloire H, Hartmann DJ, Herbage D, Ville G, Prunieras M: Early mouse molar development: cellular changes and distribution of fibronectin, laminin and type IV collagen. Differentiation 1985, 30:111-122 [DOI] [PubMed] [Google Scholar]
- 31.Clemmensen I: Three new E-antigenic fibrinogen fractions found in a commercial plasmin preparation. Science Tools LKB Instr J 1973, 20:7-8 [Google Scholar]
- 32.Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Dvorak HF, Senger DR: Increased expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in kidney and bladder carcinomas. Am J Pathol 1993, 143:1255-1262 [PMC free article] [PubMed] [Google Scholar]
- 33.Brown LF, Dezube BJ, Tognazzi K, Dvorak HF, Yancopoulos GD: Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi’s sarcoma and cutaneous angiosarcoma. Am J Pathol 2000, 156:2179-2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wesseling P, Schlingemann RO, Rietveld FJ, Link M, Burger PC, Ruiter DJ: Early and extensive contribution of pericytes/vascular smooth muscle cells to microvascular proliferation in glioblastoma multiforme: an immuno-light and immuno-electron microscopic study. J Neuropathol Exp Neurol 1995, 54:304-310 [DOI] [PubMed] [Google Scholar]
- 35.Haddad SF, Moore SA, Schelper RL, Goeken JA: Vascular smooth muscle hyperplasia underlies the formation of glomeruloid vascular structures of glioblastoma multiforme. J Neuropathol Exp Neurol 1992, 51:488-492 [DOI] [PubMed] [Google Scholar]
- 36.Plate KH, Breier G, Weich HA, Mennel HD, Risau W: Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer 1994, 59:520-529 [DOI] [PubMed] [Google Scholar]
- 37.Plate KH, Breier G, Weich HA, Risau W: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359:845-848 [DOI] [PubMed] [Google Scholar]
- 38.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM: Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275:964-967 [DOI] [PubMed] [Google Scholar]
- 39.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM: Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 1999, 85:221-228 [DOI] [PubMed] [Google Scholar]
- 40.Shi Q, Rafii S, Wu MH, Wijelath ES, Yu C, Ishida A, Fujita Y, Kothari S, Mohle R, Sauvage LR, Moore MA, Storb RF, Hammond WP: Evidence for circulating bone marrow-derived endothelial cells. Blood 1998, 92:362-367 [PubMed] [Google Scholar]
- 41.Arkonac BM, Foster LC, Sibinga NE, Patterson C, Lai K, Tsai JC, Lee ME, Perrella MA, Haber E: Vascular endothelial growth factor induces heparin-binding epidermal growth factor-like growth factor in vascular endothelial cells. J Biol Chem 1998, 273:4400-4405 [DOI] [PubMed] [Google Scholar]
- 42.D’Amore PA, Smith SR: Growth factor effects on cells of the vascular wall: a survey. Growth Factors 1993, 8:61-75 [DOI] [PubMed] [Google Scholar]
- 43.Sundberg C, Ljungström M, Lindmark G, Gerdin B, Rubin K: Microvascular pericytes express platelet-derived growth factor β-receptors in human healing wounds and colorectal adenocarcinoma. Am J Pathol 1993, 143:1377-1388 [PMC free article] [PubMed] [Google Scholar]
- 44.Hirschi KK, Rohovsky SA, D’Amore PA: PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol 1998, 141:805-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Betsholtz C, Raines EW: Platelet-derived growth factor: a key regulator of connective tissue cells in embryogenesis and pathogenesis. Kidney Int 1997, 51:1361-1369 [DOI] [PubMed] [Google Scholar]
- 46.Hellstrom M, Kal M, Lindahl P, Abramsson A, Betsholtz C: Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126:3047-3055 [DOI] [PubMed] [Google Scholar]
- 47.Benjamin LE, Hemo I, Keshet E: A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125:1591-1598 [DOI] [PubMed] [Google Scholar]
- 48.Brown LF, Detmar M, Tognazzi K, Abu-Jawdeh G, Iruela-Arispe ML: Uterine smooth muscle cells express functional receptors (flt-1 and KDR) for vascular permeability factor/vascular endothelial growth factor. Lab Invest 1997, 76:245-255 [PubMed] [Google Scholar]
- 49.Grosskreutz CL, Anand-Apte B, Duplaa C, Quinn TP, Terman BI, Zetter B, D’Amore PA: Vascular endothelial growth factor-induced migration of vascular smooth muscle cells in vitro. Microvasc Res 1999, 58:128-136 [DOI] [PubMed] [Google Scholar]
- 50.Takagi H, King GL, Aiello LP: Identification and characterization of vascular endothelial growth factor receptor (Flt) in bovine retinal pericytes. Diabetes 1996, 45:1016-1023 [DOI] [PubMed] [Google Scholar]
- 51.Yamagishi S, Yonekura H, Yamamoto Y, Fujimori H, Sakuri S, Tanaka N, Yamamoto H: Vascular endothelial growth factor acts as a pericyte mitogen under hypoxic conditions. Lab Invest 1999, 79:501-509 [PubMed] [Google Scholar]
- 52.Benjamin L, Golijanin D, Itin A, Pode D, Keshet E: Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999, 103:159-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, van de Water L: Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med 1992, 176:1375-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakayama Y, Sueishi K, Oka K, Kono S, Tomonaga M: Stromal angiogenesis in human glioma: a role of platelet-derived endothelial cell growth factor. Surg Neurol 1998, 49:181-187 [DOI] [PubMed] [Google Scholar]
- 55.Vassbotn FS, Östman A, Langeland N, Holmsen H, Westermark B, Heldin C-H, Nister M: Activated platelet-derived growth factor autocrine pathway drives the transformed phenotype of a human glioblastoma cell line. J Cell Phys 1994, 158:381-389 [DOI] [PubMed] [Google Scholar]
- 56.Herman IM, D’Amore PA: Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol 1985, 101:43-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sundberg C, Ivarsson M, Gerdin B, Rubin K: Pericytes as collagen producing cells in excessive dermal scarring. Lab Invest 1996, 74:452-466 [PubMed] [Google Scholar]
- 58.Ivarsson M, Sundberg C, Farrokhnia N, Pertoft H, Rubin K, Gerdin B: Recruitment of type I collagen producing cells from the microvasculature in vitro. Exp Cell Res 1996, 229:336-349 [DOI] [PubMed] [Google Scholar]
- 59.Dvorak A: Hemangiopericytoma. Blood 1998, 91:1255 [Google Scholar]
- 60.Ghadially F: Ultrastructural Pathology of the Cell and Matrix. 1982. Butterworths, London