

Abstract
The aims of this study were to investigate the role of cathepsin K in the pathology of amyloidosis by demonstrating its presence in multinucleated giant cells (MGCs) adjacent to amyloid deposits, and determining its ability to degrade amyloid fibril proteins in vitro. The study was performed using autopsy and biopsy specimens from patients with AA or AL amyloidosis. In six (55%) patients with AA amyloidosis and seven (58%) patients with AL amyloidosis, variable numbers of CD68-immunoreactive MGCs were found adjacent to amyloid deposits. In each case strong cytoplasmic immunostaining for cathepsin K was found in MGCs; immunostaining of amyloid deposits was present in five (45%) patients with AA amyloidosis and three (25%) patients with AL amyloidosis. In vitro degradation experiments showed that recombinant cathepsin K completely degraded AA amyloid fibril proteins at pH 5.5 as shown by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting. Less effective degradation took place at pH 7.4 and there was no degradation in the presence of a general cysteine protease inhibitor (E64) or in the absence of cathepsin K. This is the first study to show that cathepsin K is expressed in MGCs adjacent to amyloid deposits and to demonstrate its ability to degrade amyloid fibril proteins.
Amyloidoses are characterized by local, organ-limited, or generalized proteinaceous deposits of autologous origin. Approximately 72 to 75% of all generalized amyloidoses are either AA or AL amyloidosis 1 and the fibril proteins of both diseases originate from an autologous physiological precursor protein. The acute phase protein serum amyloid A (SAA) is the precursor of the AA fibril protein deposited in AA amyloidosis, a disorder that is commonly associated with chronic inflammatory conditions. Immunoglobulins and their fragments are the precursors of the fibril proteins deposited in AL amyloidosis, a condition that may be secondary to plasmacytoma.
Macrophages (M℘) are commonly found in many different amyloid diseases, including AA and AL amyloidosis, and it has been proposed that they are involved in amyloidogenesis. They synthesize a broad range of proteases that may process the precursor protein to generate the fibril protein, and they may also be involved in the degradation of the deposits. 2-4 Multinucleated giant cells (MGCs) are considered to be a specific phenotype of M℘ and they are also, although less commonly, found in amyloid deposits and often show an intimate spatial relationship to the deposits. 5-9 Earlier animal studies provided evidence that MGCs might be involved in the resorption of amyloid deposits. 10
We have previously found that MGCs, like osteoclasts, selectively express cathepsin K. The expression of cathepsin K in MGCs was not restricted to a specific disease and was found in tuberculosis, sarcoidosis, sarcoid-like lesions, and foreign body reactions (Röcken C, Bühling F, unpublished observation). Cathepsin K belongs to the papain family of cysteine proteases and has unique properties. 11,12 In adults it is expressed in osteoclasts and their mononuclear precursors, in bronchial epithelium, bile duct epithelium, M℘, and smooth muscle cells of atherosclerotic arteries, in synoviocytes of patients suffering from rheumatoid arthritis, and also in breast carcinoma cells. 13-18 It is not expressed in M℘ under normal conditions. 13 Cathepsin K is the most potent mammalian elastase yet described and it has been shown to possess unique collagenolytic activity. This activity does not depend on destabilization of the triple helix, but rather it cleaves type I and II collagen at the ends (telopeptide) and at multiple sites within the native triple helix. 12,19 Other proteinases with collagenolytic activity cleave collagen at the telopeptides only (eg, cathepsins L, B, and S) or within the molecule at a single site (eg, MMP-1, -2, -8, and -13). 12 Cathepsin K also shows gelatinolytic activity in the pH range 4.0 to 7.0 and it may function both intracellularly and extracellularly. 19 The stability of cathepsin K is modulated by glycosaminoglycans; in the presence of chondroitin 4-sulfate there is an increase in the hydrolysis of soluble and insoluble collagen by cathepsin K. 20 The role of cathepsin K in bone turnover is well established; mutations in the cathepsin K gene lead to pycnodysostosis and cathepsin K knock-out mice develop osteopetrosis. 21-23 As yet there has been little investigation into the putative role of cathepsin K in other diseases.
Our previous observations have shown that cathepsin K is expressed not only by osteoclasts, but also by MGCs in general. Therefore we performed a study to determine the presence of cathepsin K in MGCs adjacent to amyloid deposits and to investigate its ability to degrade fibril proteins in vitro, hence indicating a possible involvement in the pathology of amyloidoses.
Materials and Methods
Materials
Chondroitin 4-sulfate, chondroitin 6-sulfate, dermatan sulfate, and heparan sulfate were purchased from Sigma (Deisenhofen, Germany). Serum amyloid A-rich high-density lipoprotein was obtained from Calbiochem-Novabiochem GmbH (Bad Soden, Germany). Recombinant human cathepsin K was produced as previously described. 24
Case Selection
Fifty-three archived, formalin-fixed, and paraffin-embedded autopsy and biopsy specimens from 23 patients with histologically proven AA or AL amyloidosis were used in this study. Age, sex distribution, and basic diseases are summarized in Table 1 ▶ . The classification of amyloid was based on immunohistochemistry and clinical history as described elsewhere. 1,25
Table 1.
Age, Sex Distribution, and Basic Diseases among the Cases with Amyloidosis
| AA amyloidosis | AL amyloidosis | |
|---|---|---|
| Number of cases | 11 | 12 |
| Age (years, range) | 57 (35–70) | 64 (50–86) |
| Sex (m : f) | 7:4 | 4:8 |
| Underlying disease (n) | Rheumatoid arthritis (6) | Plasmacytoma (4) |
| Rosai-Dorfman disease (1) | Immunocytoma (1) | |
| Chronic interstitial pneumonia (1) | MALT lymphoma (1) | |
| Unknown (3) | Primary amyloidosis (6) |
Eleven patients had suffered from AA amyloidosis; specimens of both liver and kidney were available from nine of these patients, from the spleen in eight patients, and the thyroid in one patient. Variable amounts of amyloid were present in each organ as vascular and interstitial deposits.
Twelve patients had suffered from AL amyloidosis. The immunohistochemical classification showed that the deposits consisted in five patients of λ-light chain and in three of κ-light chain. 26,27 The amyloid deposits of four patients stained for amyloid P component only; two of these patients had suffered from a plasmacytoma whereas the other two had no disease commonly associated with either AA or AL amyloidosis. These cases were classified as myeloma-associated or primary AL amyloidosis, respectively, as described in detail elsewhere. 26 Specimens of both liver and kidney were available from eight of these patients, from the spleen in six patients, and from the tonsil, breast, lymph node, and lung each in one patient. Large interstitial and occasional vascular deposits of amyloid were found in every case.
Additionally, unfixed amyloid-containing tissue was available from a single patient with generalized AA amyloidosis and this was used for the preparation of amyloid fibril proteins and degradation experiments with recombinant cathepsin K as described below.
Two of the 31 patients have previously been published in detail as case reports. 28,29
Light Microscopy
Deparaffinized serial sections from formalin-fixed tissue were used to immunolocalize MGCs and cathepsin K. The presence of amyloid was demonstrated by the appearance of green birefringence from alkaline alcoholic Congo Red staining under polarized light. 30 Immunostaining was performed using monoclonal antibodies directed against AA amyloid (monoclonal; dilution, 1:500), and CD68 (1:5,000), as well as polyclonal antibodies directed against λ-light chain (1:7,500), amyloid P component (1:1,600; all Dakopatts, Hamburg, Germany), and cathepsin K (1:1,000). 14 Before immunostaining the specimens were pretreated with 10 mmol/L ethylenediaminetetraacetic acid (EDTA) (2 × 10 minutes, 450 W microwave oven; CD68, amyloid P component) and 0.5 U/ml protease I (16 minutes; λ-light chain; Ventana, Strasbourg, France). Immunoreaction was visualized with the avidin biotin complex method applying a Vectastain ABC alkaline phosphatase kit (distributed by Camon, Wiesbaden, Germany) or UltraTech HRP streptavidin-biotin universal detection system (Immunotech, France). Neufuchsin and 3,3-diaminobenzidine-tetrahydrochloride, respectively, served as chromogens. The specimens were counterstained with hematoxylin.
The specificity of immunostaining was controlled using specimens containing known classes of amyloid (AA amyloid), using positive controls recommended by the manufacturers (remaining antibodies), using normal rabbit serum instead of primary antibody, and by omitting the primary antibodies.
Immunostaining was evaluated using an Axioskop microscope from Zeiss (Jena, Germany).
Electron Microscopy
For electron microscopy specimens from a tonsil containing AL amyloid and multiple MGCs were fixed in a mixture of 2% formalin/2.5% glutaraldehyde (pH 7.2, overnight, 4°C) and then in 3.125% glutaraldehyde (7 hours, 4°C). Following standard procedures of tissue processing for electron microscopy, the specimens were finally embedded in Lowicryl using the K4M kit (Plano, Wetzlar, Germany). Specimens for postembedding immunoelectron microscopy were not postfixed with OsO4. Polymerization took place throughout 24 hours at −30°C and was initiated by UV light. Semithin sections (1 μm) were stained with toluidine blue. Ultrathin sections (80 to 120 nm) were mounted on copper grids and counterstained with 3% aqueous uranyl acetate (30 minutes, room temperature) and contrasted with 1% aqueous lead citrate (15 minutes, room temperature).
For postembedding immunoelectron microscopy, ultrathin sections (120 nm) were mounted on formvar-coated nickel grids (200 mesh, Plano). Immunoelectron microscopy was performed in triplicate as described previously. 25 The specificity of immunostaining was controlled by omitting the primary antibody. The sections were air-dried and examined using a Zeiss EM900 electron microscope.
Preparation of Amyloid Fibril Proteins
Amyloid fibril proteins were prepared as described by Skinner and colleagues. 31 Briefly this was as follows: ∼6 g of amyloidotic tissue, which had been stored at −80°C, was homogenized in 60 ml of aqueous 0.9% NaCl solution for 30 seconds using an Ultra-Turrax. The homogenate was centrifuged at 2,500 × g for 30 minutes at 4°C. The supernatant was discarded and the pellet was resuspended in 60 ml of 0.9% NaCl for 30 seconds, centrifuged, resuspended for 15 seconds in 60 ml of sodium citrate buffer (0.05 mol/L sodium citrate, 0.01 mol/L Tris, pH 8.0), and centrifuged again. Subsequently the supernatant was discarded and the pellet was resuspended in 60 ml of 0.9% NaCl solution for 15 seconds and centrifuged. The last washing step was repeated until the optical density (OD 280) of the supernatant was <0.1. Thereafter the pellet was resuspended in 50 ml of deionized H2O (dH2O) and centrifuged for 200 minutes at 2,500 × g (4°C). This step was repeated three times. The supernatants collected from the last three washing steps contained amyloid fibril and precursor proteins (AFPPS) and they were stored in aliquots at −20°C until further use (see below). The final pellet had a whitish top layer and also contained both amyloid fibril and precursor proteins (AFPPP) as demonstrated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. This was also stored at −20°C until further use.
Degradation Experiments
In vitro degradation experiments with recombinant cathepsin K were performed as follows. Samples from the pellet containing amyloid fibril and precursor proteins (AFPPP) were dissolved in water by heating for 30 minutes at 100°C. Samples from the supernatant containing amyloid fibril and precursor proteins (AFPPS) were concentrated in a speed-vac without heating. Both AFPPP or AFPPS were then dispersed at a concentration of 5 mg/ml in 100 mmol/L sodium acetate buffer (substituted with 5 mmol/L EDTA, 2.5 mmol/L dithiothreitol, pH 5.5) or 50 mmol/L Tris-HCl buffer (substituted with 5 mmol/L EDTA, 2.5 mmol/L dithiothreitol, pH 7.4). Degradation was performed at 37°C and was started by the addition of cathepsin K (0.8 nmol/L). The reaction was stopped by the addition the cysteine protease inhibitor E-64 (200 μmol/L). Omission of cathepsin K and incubation in the presence of 200 μmol/L E64 served as a control. All experiments were performed in triplicate.
In situ degradation experiments were performed using frozen sections (8 μm) of unfixed amyloidotic tissue. Specimens were shortly hydrated in 100 mmol/L of sodium acetate buffer (substituted with 5 mmol/L EDTA, 2.5 mmol/L dithiothreitol, pH 5.5). Degradation was performed at 37°C and was started by the addition of cathepsin K (0.8 nmol/L) in 100 mmol/L sodium acetate buffer. The reaction was stopped by jet-washing in 50 mmol/L Tris-HCl buffer and the specimens were fixed in 3% p-formaldehyde (substituted with 5% CaCl2) for 5 minutes. Immunostaining was performed as described above. Omission of cathepsin K and incubation in the presence of 200 μmol/L E64 served as a control.
SDS-PAGE and Western Blotting
Proteins were resolved in 16.5% polyacrylamide gels according to Schägger and von Jagow 32 and visualized by staining with Coomassie blue or silver stain. 33 Using Western blotting, proteins on unstained polyacrylamide gels were transferred onto a polyvinylidene difluoride membrane (Immobilon-PSQ, pore size 0.1 μm; Millipore, Bedford, MA) using the tank-blotting system from Bio-Rad Laboratories (München, Germany) according to the manufacturer’s instructions. Transferred proteins were visualized by Coomassie blue staining. Immunostaining of transferred proteins was performed using the Vectastain ABC alkaline phosphatase kit (Camon) and antibodies directed against AA fibril protein (monoclonal, clone mc1) and cathepsin K. Immunostaining was visualized with BCIP/NBT (BioTrend Chemikalien GmbH, Köln, Germany). N-terminal amino acid sequencing was performed by WITA GmbH (Teltow, Germany).
Results
Multinucleated Giant Cells
In six (55%) patients with AA amyloidosis and seven (58%) patients with AL amyloidosis, a variable number of multinucleated CD68-immunoreactive foreign body-type giant cells was found adjacent to amyloid deposits (Figure 1) ▶ . Some of these had a vacuolated cytoplasm and asteroid bodies. Cases of AL amyloidosis occasionally showed intracellular immunostaining for λ-light chain as described previously. 29
Figure 1.
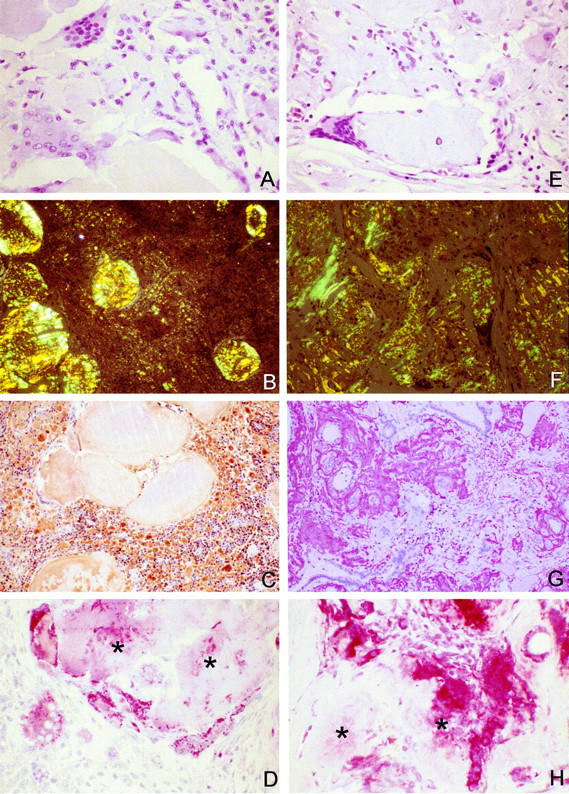
MGCs adjacent to deposits of AL amyloid in the tonsil (A–D) and AA amyloid in the thyroid (E–H) of two patients who had suffered from a plasmacytoma and rheumatoid arthritis, respectively. Cathepsin K was found in the cytoplasm of MGCs as well as within amyloid deposits (asterisks). H&E stain (A and E). Congo Red staining in polarized light (B and F). Immunostaining with anti-λ-light chain (C), anti-AA amyloid (G), and anti-cathepsin K (D and H); hematoxylin counterstain. Original magnifications: ×100 (A, B, D, and H), ×76 (C and E–G).
Immunolocalization of Cathepsin K
Cathepsin K was detected in 41 (77%) of 53 specimens from 22 (96%) patients. Immunostaining of variable intensity was found in thyroid epithelium, bile duct epithelium, renal tubular epithelium, polymorphonuclear cells, smooth muscle cells of vessel walls, and fibroblasts (not shown). Except for thyroid epithelium, none of these cells showed a spatial relationship to amyloid deposits. However, strong immunostaining for cathepsin K was found in MGCs adjacent to amyloid deposits in six (55%) patients with AA amyloidosis and seven (58%) with AL amyloidosis. The number of immunoreactive MGCs varied and some were immunonegative (Figure 1) ▶ . Immunostaining of amyloid deposits was present in five (45%) patients with AA amyloidosis and three (25%) patients with AL amyloidosis and it was mainly confined to deposits adjacent to or surrounded by MGCs (Figure 1) ▶ . AA amyloidosis showed cathepsin K-immunoreactive MGCs in the spleen (four patients), liver (three patients), and kidney (two patients). AL amyloidosis showed cathepsin K-immunoreactive MGCs in the liver of two patients, and in the kidney, spleen, tonsil, breast, lymph node, and lung each of one patient. No differences were found between amyloid deposits of λ- and κ-light chain origin.
No immunostaining was found using normal rabbit serum instead of primary antibody, or after omission of the primary antibody.
Electron Microscopy
Electron microscopy of ultrathin sections from a tonsil containing AL amyloid showed rigid nonbranching amyloid fibrils of 10 to 12 nm diameter and indefinite length, and MGCs with abundant vesicular storage granules. Postembedding immunolabeling with the antibody directed against cathepsin K yielded immunostaining that was closely spatially related to amyloid fibrils (Figure 2) ▶ . Additional intracellular immunostaining was found in the storage granules of MGCs (Figure 2) ▶ .
Figure 2.
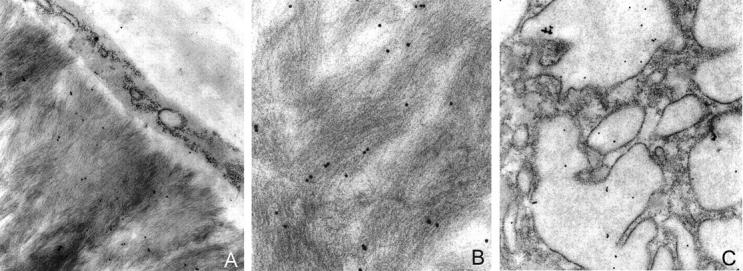
Postembedding immunolabeling using the antibody directed against cathepsin K yielded immunostaining that was directly spatially related to amyloid fibrils (A and B). Additional intracellular immunostaining was found in the storage granules of MGCs (C). Original magnification: ×30,000 (A and C), ×64,000 (B).
In Situ Degradation Experiments
Unfixed frozen sections of spleen with green birefringent AA amyloid deposits were incubated for 4 hours with 0.8 nmol/L cathepsin K at pH 5.5, both with and without E64. Incubation without cathepsin K served as a further negative control. The results of in situ proteolysis were controlled by immunohistochemistry using antibodies directed against AA amyloid and amyloid P component. Without protease pretreatment the amyloid deposits showed immunostaining of the margins only (Figure 3) ▶ . The staining pattern obtained with the antibody directed against AA amyloid changed after pretreatment with cathepsin K in that immunostaining was also observed in the center of the deposits (Figure 3) ▶ . Co-incubation with E64 reversed this effect of cathepsin K. Immunostaining for amyloid P component was unaffected by cathepsin K pretreatment and was predominantly confined to the margins of the deposits (Figure 3) ▶ .
Figure 3.
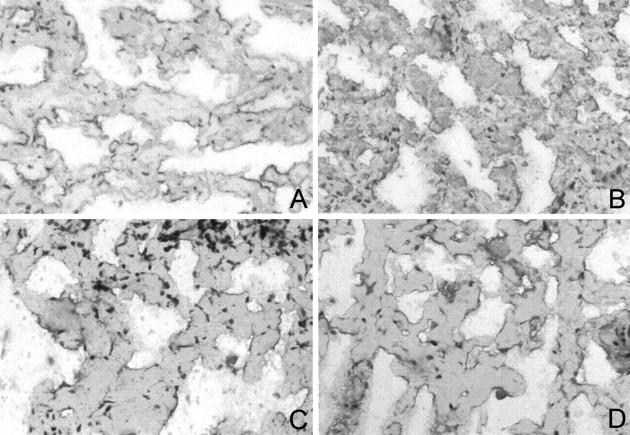
Unfixed frozen sections of a spleen with AA amyloid deposits were incubated with cathepsin K at pH 5.5 in the presence (A and C) and absence (B and D) of E64. In the presence of E64 amyloid deposits showed immunostaining for AA amyloid at the margins only (A). In the absence of E64 immunostaining was also present in the center of the deposits (B). Immunostaining for amyloid P component was not affected by cathepsin K pretreatment and it was predominantly confined to the margins of the deposits (C and D). Immunostaining with anti-AA amyloid (A and B) and anti-amyloid P component (C and D); hematoxylin counterstain. Original magnification, ×100.
In Vitro Degradation Experiments
In vitro degradation experiments used amyloid fibril and precursor proteins (AFPP) prepared from the spleen of a patient who had suffered from generalized AA amyloidosis. 28 After SDS-PAGE and Western blotting (see below) two fibril proteins of ∼5.8 and 6.9 kd were identified (Figure 4) ▶ . N-terminal sequencing showed that both fibril proteins had identical N-terminal ends (SFFSFL) homologous to the N-terminal region of serum amyloid A starting at position 2. In addition, AFPPS and AFPPP contained trace amounts of precursor protein (SAA) and several bands that were larger than 12 kd as shown by SDS-PAGE and Western blotting (Figure 4) ▶ . These bands were interpreted as oligomeric fibril proteins because they disappeared after prolonged heat-denaturation and the amount of detectable fibril proteins increased accordingly (not shown).
Figure 4.

AFPP was prepared from a spleen of a patient who had suffered from generalized AA amyloidosis. 28 After SDS-PAGE and Western blotting (see below) two fibril proteins of ∼5.8 and 6.9 kd were identified (lanes 1–4). In addition, AFPP contained trace amounts of precursor protein (SAA) and several bands that were larger than 12 kd. AFPP in the pellet (AFPPP; lanes 1 and 2) and in the supernatant (AFPPS; lanes 3 and 4) as obtained during the water-wash procedure described by Skinner and colleagues. 31 SAA-rich high-density apolipoproteins (lanes 5 and 6). MW, Molecular weight standards. SDS-PAGE and Coomassie blue staining (lanes 1, 3, and 5). Western blotting with anti-AA amyloid (lanes 2, 4, and 6).
In vitro degradation experiments revealed that recombinant human cathepsin K is able to degrade amyloid fibril and precursor proteins, both AFPPP and AFPPS, completely. No differences were observed between AFPPP and AFPPS. Degradative activity was optimal at pH 5.5 (Figure 5) ▶ . After 10 minutes of incubation, multiple additional bands ranging from 4 to >49 kd were detected by SDS-PAGE and Western blotting (Figure 5) ▶ . The number of detectable bands decreased throughout time and after 4 hours both AFPPP and AFPPS were completely degraded. Proteolytic activity at pH 7.4 lasted for just 10 minutes (Figure 5) ▶ ; the 5.8- and 6.9-kd bands of the fibril proteins disappeared and, as observed at pH 5.5, multiple bands were found spread out over the entire lane. However, this pattern showed no further changes after 0.5, 2, 4, and 8 hours of incubation (Figure 5) ▶ . The proteolytic activity at pH 7.4 was not prolonged by the addition of 0.15% (w/v) solutions of any of the following to the incubation medium: chondroitin 4-sulfate, chondroitin 6-sulfate, dermatan sulfate, or heparan sulfate. The degradation profile as demonstrated by SDS-PAGE was identical to degradation in the absence of glycosaminoglycans (GAGs; not shown). Cathepsin K also completely degraded native SAA, as present in SAA-rich high-density lipoproteins (not shown) at pH 5.5 and 7.4. Again at pH 7.4 degradation lasted for just a short period of time and was not influenced by the addition of GAGs.
Figure 5.
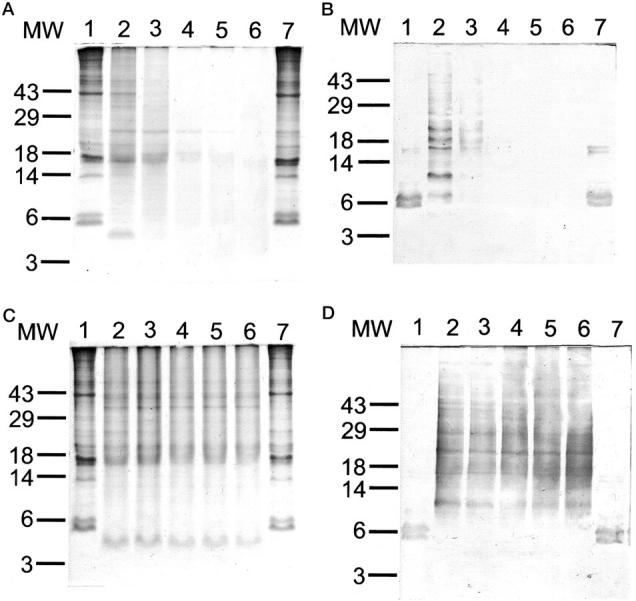
In vitro degradation experiments revealed that recombinant human cathepsin K is able to completely degrade amyloid fibril and precursor proteins. In vitro degradation was performed at pH 5.5 (A and B) and pH 7.4 (C and D) for 10 minutes (lane 2), 30 minutes (lane 3), 2 hours (lane 4), 4 hours (lane 5), and 8 hours (lane 6). Incubation for 8 hours without cathepsin K (lane 1) and with cathepsin K + E64 (lane 7) served as controls. SDS-PAGE and Coomassie blue staining (A and C); Western blotting with anti-AA amyloid (B and D).
Degradation at pH 5.5 and pH 7.4 was completely inhibited by the addition of E64 (Figure 5) ▶ . Unfixed tissue samples were not available for in vitro degradation experiments with AL amyloid.
Discussion
In almost all patients with AA amyloidosis and >95% of patients with AL amyloidosis the deposits enclosed fragments of the precursor proteins, which in the case of AA amyloidosis are known to be generated by proteolysis. 34,35 Different hypotheses may explain the presence of proteolytic fragments within amyloid deposits: 1) Proteolysis of a nonamyloidotic precursor protein may liberate the fibril protein hence facilitating amyloidogenesis. This mechanism occurs in Aβ amyloidosis, which is commonly associated with Alzheimer’s disease, and in which proteolysis may be a rate-limiting step of amyloidogenesis. As yet there is no evidence to suggest that this mechanism accounts for AA and AL amyloidosis because both precursor proteins have been shown to be potentially amyloidogenic. 35-37 2) Proteolysis of an amyloidogenic precursor protein may occur either before or after fibrillogenesis. Proteolysis before amyloid formation may have different effects; depending on the cleavage site it may enhance fibrillogenesis by generating fibril proteins that are more prone to self-assembly than the precursor protein, or it may prevent fibrillogenesis by degrading the precursor protein in a region that is most important for self assembly such as the N-terminal end of SAA. 38 Previous studies 39,40 have shown that cleavage at the N-terminal end of SAA prevents the formation of amyloid, whereas the fibril protein always has an intact N-terminal end and lacks between 18 and 60 amino acids at the C-terminus only. This was exemplified here using amyloid fibril preparations from a patient with generalized AA amyloidosis that contained two amyloid fibril proteins of 5.8- and 6.9-kd size and intact N-terminals, whereas the precursor protein has a molecular weight of ∼12 kd (Figure 4) ▶ . Proteolysis after amyloid has formed may have no effect or it may facilitate lateral growth of the fibril or, alternatively, it may lead to the degradation of amyloid.
This investigation is the first to show that cathepsin K is expressed by MGCs adjacent to amyloid deposits and also that it is present extracellularly with a close spatial relationship to amyloid fibrils. Based on these findings we assume that cathepsin K, under certain conditions, may be involved in the pathology of AA and AL amyloidosis. MGCs represent a specific response to the presence of amyloid deposits rather than being the cause of amyloid formation because several patients studied here had amyloid deposits without MGCs. Cathepsin K is found only in association with MGCs and therefore its expression is not a prerequisite for amyloidogenesis to occur, but it may exert an effect on the degradation of amyloid deposits. To test this hypothesis a series of degradation experiments was performed.
In vitro degradation experiments using crude amyloid fibril extracts demonstrated that cathepsin K is an adequate fibril protein-degrading protease. This cysteine protease completely degraded crude fibril extracts within 8 hours, irrespective of whether the fibril protein was present as a monomer or as an oligomer (Figure 5) ▶ and degradation occurred even at pH 7.4. These experiments show that cathepsin K is able to degrade amyloid fibril proteins extracellularly at a physiological pH albeit for a short period of time. It has been shown by others that the activity of cathepsin K is influenced by GAGs. In the presence of chondroitin 4-sulfate the hydrolysis of soluble and insoluble collagen by cathepsin K is increased mainly by stabilizing the protease. Chondroitin 6-sulfate, dermatan sulfate, and heparan sulfate had no effect. 20 We were unable to show that these GAGs had any effect on proteolytic activity at neutral pH. This may be due partly to the fact that GAGs co-purify with amyloid fibrils when using the water-wash procedure of Skinner and colleagues 31 and hence proteolytic activity of cathepsin K at neutral pH may be because of the presence of GAGs. GAGs have been shown to accumulate in all amyloids studied 41,42 and they may be a structural component of the fibril 43 and exert an effect on fibrillogenesis. 41 The composition of GAGs associated with amyloid deposits varies between different amyloid diseases, patients, and organs studied. For example, the relative amount of chondroitin sulfate among amyloid-associated GAGs may range from 25 to 70% with the amount of other GAGs, such as heparan sulfate or dextran sulfate, changing accordingly. 44 Some of the GAGs are bound to a protein backbone as proteoglycan. A variable amount and composition of GAGs within amyloid deposits may influence the proteolytic activity of cathepsin K. Using SAA-rich high-density lipoproteins (that are initially devoid of GAG) instead of AFPP as a substrate, we were unable to detect any effect of GAGs on the proteolytic activity of cathepsin K at pH 7.4. This finding is in agreement with our previous report showing that the GAG-enhancing effect on the proteolysis of protein substrates is primarily restricted to acidic pH conditions. 20
In situ experiments using frozen sections containing intact AA amyloid fibrils showed that cathepsin K is most likely to change the antigenicity of the fibril protein by retrieving epitopes. These are retained in native material but not in formalin-fixed specimens because, in contrast to immunostaining of frozen sections, staining was homogeneous on formalin-fixed and paraffin-embedded specimens (Figure 1) ▶ . This antigen retrieval, as observed on frozen sections, may indicate proteolytic attack of the amyloid fibril protein itself because immunostaining for amyloid P component was unchanged. Amyloid P component is probably a structural constituent of amyloid fibrils and present in the deposits of every amyloid disease studied so far. 45
The deposition of amyloid is not irreversible. Progression of generalized amyloidosis can be delayed or stopped by treatment of the underlying disease, and deposits may also regress. 10,46-48 Identification of proteases that are involved in this degradation process and specifying their modes of action may help in the development of new treatments, particularly in situations in which treatment of the underlying disease has shown only little if any benefit, eg, in primary AL amyloidosis. We have demonstrated here that cathepsin K is a possible candidate; among M℘ it is a unique protease in that it is expressed only by MGCs and epithelioid cells (Röcken C, Bühling F, unpublished observation). Also, it is the most potent mammalian elastase yet described and has unique collagenolytic activity and the potential to degrade AA amyloid fibril proteins, as shown in this study. Additional evidence for the putative significance of MGCs in amyloidosis comes from earlier animal studies performed by Wright and colleagues, 10 who have found an association between amyloid resorption and the presence of MGCs. However, further studies are required to investigate the mechanisms by which M℘ occurring in amyloid deposits can be stimulated to become MGCs that synthesize and secrete cathepsin K.
Acknowledgments
The authors thank Mrs. Mansfeld, Mrs. Koçalkova, and Mrs. Röseler for their excellent and skillful assistance.
Footnotes
Address reprint requests to Dr. Christoph Röcken, Institute of Pathology, Otto-von-Guericke University, Leipziger Str. 44, D-39120 Magdeburg, Germany. E-mail: christoph.roecken@medizin.uni-magdeburg.de.
Supported by grants from the Deutsche Forschungsgemeinschaft (grants RO 1173/3-1 and SFB 387/B-7), Bonn Bad-Godesberg, Germany; and the Wilhelm Vaillant-Stiftung, München, Germany.
References
- 1.Röcken C, Schwotzer E, Linke RP, Saeger W: The classification of amyloid deposits in clinicopathological practice. Histopathology 1996, 29:325-335 [DOI] [PubMed] [Google Scholar]
- 2.Shirahama TS, Cohen AS: Intralysosomal formation of amyloid fibrils. Am J Pathol 1975, 81:101-116 [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi M, Yokota T, Kawano H, Gondo T, Ishihara T, Uchino F: Ultrastructural evidence for intracellular formation of amyloid fibrils in macrophages. Virchows Arch A Pathol Anat Histopathol 1989, 415:411-419 [DOI] [PubMed] [Google Scholar]
- 4.Röcken C, Kisilevsky R: Binding and endocytosis of murine high density lipoprotein from healthy (HDL) and inflamed donors (HDLSAA) by murine macrophages in vitro. A light- and electronmicroscopic investigation. Amyloid 1997, 4:259-273 [Google Scholar]
- 5.Michaels L, Hyams VJ: Amyloid in localised deposits and plasmacytomas of the respiratory tract. J Pathol 1979, 128:29-38 [DOI] [PubMed] [Google Scholar]
- 6.Yokoo H, Nakazato Y: Primary localized amyloid tumor of the breast with osseous metaplasia. Pathol Int 1998, 48:545-548 [DOI] [PubMed] [Google Scholar]
- 7.Yamada M, Hatakeyama S, Yamamoto E, Kimura Y, Tsukagoshi H, Yokota T, Uchino F: Localized amyloidosis of the uterine cervix. Virchows Arch A Pathol Anat Histopathol 1988, 413:265-268 [DOI] [PubMed] [Google Scholar]
- 8.Sidoni A, Alberti PF, Bravi S, Bucciarelli E: Amyloid tumours in the soft tissues of the legs. Case report and review of the literature. Virchows Arch 1998, 432:563-566 [DOI] [PubMed] [Google Scholar]
- 9.Olsen KE, Sletten K, Sandgren O, Olsson H, Myrvold K, Westermark P: What is the role of giant cells in AL-amyloidosis. Amyloid 1999, 6:89-97 [DOI] [PubMed] [Google Scholar]
- 10.Wright JR, Ozdemir AI, Matsuzaki M, Binette P, Calkins E: Amyloid resorption: possible role of multinucleated giant cells. The apparent failure of penicillamine treatment. Johns Hopkins Med J 1972, 130:278-288 [PubMed] [Google Scholar]
- 11.Turk B, Turk V, Turk D: Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors. Biol Chem 1997, 378:141-150 [PubMed] [Google Scholar]
- 12.Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaisse JM: The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem 1998, 273:32347-32352 [DOI] [PubMed] [Google Scholar]
- 13.Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, Lee-Rykaczewski E, Coleman L, Rieman D, Barthlow R, Hastings G, Gowen M: Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem 1996, 271:12511-12516 [DOI] [PubMed] [Google Scholar]
- 14.Bühling F, Gerber A, Häckel C, Krüger S, Kohnlein T, Brömme D, Reinhold D, Ansorge S, Welte T: Expression of cathepsin K in lung epithelial cells. Am J Respir Cell Mol Biol 1999, 20:612-619 [DOI] [PubMed] [Google Scholar]
- 15.Häckel C, Krüger S, Bühling F, Broemme D, Franke K, Schuetze A, Roese I, Roessner A: Expression of cathepsin K in the human embryo and fetus. Dev Dyn 1999, 216:89-95 [DOI] [PubMed] [Google Scholar]
- 16.Hummel KM, Petrow PK, Franz JK, Muller-Ladner U, Aicher WK, Gay RE, Brömme D, Gay S: Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J Rheumatol 1998, 25:1887-1894 [PubMed] [Google Scholar]
- 17.Littlewood-Evans AJ, Bilbe G, Bowler WB, Farley D, Wlodarski B, Kokubo T, Inaoka T, Sloane J, Evans DB, Gallagher JA: The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma. Cancer Res 1997, 57:5386-5390 [PubMed] [Google Scholar]
- 18.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P: Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest 1998, 102:576-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kafienah W, Brömme D, Buttle DJ, Croucher LJ, Hollander AP: Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem J 1998, 331:727-732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Hou WS, Brömme D: Collagenolytic activity of cathepsin K is specifically modulated by cartilage-resident chondroitin sulfates. Biochemistry 2000, 39:529-536 [DOI] [PubMed] [Google Scholar]
- 21.Gelb BD, Shi GP, Chapman HA, Desnick RJ: Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996, 273:1236-1238 [DOI] [PubMed] [Google Scholar]
- 22.Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, Bertoncello I, Drake F, Zavarselk S, Tellis I, Hertzog P, Debouck C, Kola I: Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res 1999, 14:1654-1663 [DOI] [PubMed] [Google Scholar]
- 23.Lazner F, Gowen M, Pavasovic D, Kola I: Osteopetrosis and osteoporosis: two sides of the same coin. Hum Mol Genet 1999, 8:1839-1846 [DOI] [PubMed] [Google Scholar]
- 24.Linnevers CJ, McGrath ME, Armstrong R, Mistry FR, Barnes MG, Klaus JL, Palmer JT, Katz BA, Brömme D: Expression of human cathepsin K in Pichia pastoris and preliminary crystallographic studies of an inhibitor complex. Protein Sci 1997, 6:919-921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Röcken C, Roessner A: An evaluation of antigen retrieval procedures for immunoelectron microscopical classification of amyloid deposits. J Histochem Cytochem 1999, 47:1385-1394 [DOI] [PubMed] [Google Scholar]
- 26.Müller D, Roessner A, Röcken C: Distribution pattern of matrix metalloproteinases (MMP-1, -2, -3, and -9), tissue inhibitors of matrix metalloproteinases (TIMP-1 and -2), and α 2-macroglobulin in cases of generalized AA and AL amyloidosis. Virchows Arch 2000, 437:521-527 [DOI] [PubMed] [Google Scholar]
- 27.Röcken C, Bässler R, Stix B, Kronsbein H, Roessner A: Coincidence of breast cancer and amyloidosis of the breast: a report of two cases. Path Res Pract 2000, 196:355 [Google Scholar]
- 28.Röcken C, Wieker K, Grote HJ, Müller G, Franke A, Roessner A: Rosai-Dorfman disease and generalized AA amyloidosis. A case report. Hum Pathol 2000, 31:621-624 [DOI] [PubMed] [Google Scholar]
- 29.Röcken C, Hegenbarth V, Schmitz M, Stix B, Schade G, Mohnert A, Roessner A: Plasmacytoma of the tonsil with secondary AL amyloidosis and osseous metaplasia. Virchows Arch 2000, 436:336-344 [DOI] [PubMed] [Google Scholar]
- 30.Puchtler H, Sweat F, Levine M: On the binding of Congo red by amyloid. J Histochem Cytochem 1962, 10:355-364 [Google Scholar]
- 31.Skinner M, Shirahama TS, Cohen AS, Deal CL: The association of amyloid p-component (AP) with amyloid fibril: an updated method for amyloid fibril protein isolation. Preparat Biochem 1983, 12:461-476 [DOI] [PubMed] [Google Scholar]
- 32.Schägger H, Jagow G: Tricine-sodium dodecyl sulphate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 1987, 166:368-379 [DOI] [PubMed] [Google Scholar]
- 33.Merril LR, Goldman D, Sedman SA, Ebert MH: Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid proteins. Science 1981, 211:1437-1438 [DOI] [PubMed] [Google Scholar]
- 34.Husby G, Marhaug G, Dowton B, Sletten K, Sipe JD: Serum amyloid A (SAA): biochemistry, genetics and the pathogenesis of AA amyloidosis. Amyloid 1994, 1:119-137 [Google Scholar]
- 35.Solomon A, Weiss DT: Protein and host factors implicated in the pathogenesis of light chain amyloidosis (AL amyloidosis). Amyloid 1995, 2:269-279 [Google Scholar]
- 36.Yamada T, Kluve-Beckerman B, Liepnieks JJ, Benson MD: Fibril formation from recombinant human serum amyloid A. Biochim Biophys Acta 1994, 1226:323-329 [DOI] [PubMed] [Google Scholar]
- 37.Yakar S, Kaplan B, Livneh A, Martin B, Miura K, Ali-Khan Z, Shtrasburg S, Pras M: Direct evidence for SAA deposition in tissues during murine amyloidogenesis. Scand J Immunol 1994, 40:653-658 [DOI] [PubMed] [Google Scholar]
- 38.Westermark GT, Engström U, Westermark P: The N-terminal segment of protein AA determines its fibrillogenic property. Biochem Biophys Res Commun 1992, 182:27-33 [DOI] [PubMed] [Google Scholar]
- 39.Yamada T, Kluve-Beckerman B, Liepnieks JJ, Benson MD: In vitro degradation of serum amyloid A by cathepsin D and other acid proteases: possible protection against amyloid fibril formation. Scand J Immunol 1995, 41:570-574 [DOI] [PubMed] [Google Scholar]
- 40.Yamada T, Liepnieks J, Benson MD, Kluve-Beckerman B: Accelerated amyloid deposition in mice treated with the aspartic protease inhibitor, pepstatin. J Immunol 1996, 157:901-907 [PubMed] [Google Scholar]
- 41.Kisilevsky R, Fraser P: Proteoglycans and amyloid fibrillogenesis. The Nature and Origin of Amyloid Fibrils. 1996, :pp 58-72 John Wiley & Sons Ltd., Chichester [DOI] [PubMed] [Google Scholar]
- 42.Magnus JH, Stenstad T: Proteoglycans and the extracellular matrix in amyloidosis. Amyloid 1997, 4:121-134 [Google Scholar]
- 43.Inoue S, Kisilevsky R: A high resolution ultrastructural study of experimental murine AA amyloid. Lab Invest 1996, 74:670-683 [PubMed] [Google Scholar]
- 44.Stenstad T, Magnus JH, Kolset SO, Cornwell GGI, Husby G: Macromolecular properties of glycosaminoglycans in primary AL amyloid fibril extracts of lymphoid tissue origin. Scand J Immunol 1991, 34:611-617 [DOI] [PubMed] [Google Scholar]
- 45.Pepys MB, Booth DR, Hutchinson WL, Gallimore JR, Collins PM, Hohenester E: Amyloid P component. A critical review. Amyloid 1997, 4:274-295 [Google Scholar]
- 46.Hawkins PN, Richardson S, Macsweeney JE, King AD, Vigushin DM, Lavender JP, Pepys MB: Scintigraphic quantification and serial monitoring of human visceral amyloid deposits provide evidence for turnover and regression. Q J Med 1993, 86:365-374 [PubMed] [Google Scholar]
- 47.Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O, Wallin BG, Seymour A, Richardson S, Hawkins PN, Pepys MB: Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993, 341:1113-1116 [DOI] [PubMed] [Google Scholar]
- 48.Tan SY, Irish A, Winearls CG, Brown EA, Gower PE, Clutterbuck EJ, Madhoo S, Lavender JP, Pepys MB, Hawkins PN: Long term effect of renal transplantation on dialysis-related amyloid deposits and symptomatology. Kidney Int 1996, 50:282-289 [DOI] [PubMed] [Google Scholar]