

Abstract
Selected bile acids activate a nuclear factor-kappa B (NF-κB)-dependent survival signaling cascade in cultured hepatocytes. These data suggest that in cholestasis where liver tissue bile acid concentrations are increased, NF-κB should be activated and inhibition of NF-κB should potentiate liver injury. Our aims were to test these two predictions. Cholestasis was obtained by common bile duct ligation in mice. NF-κB activation was demonstrated in nuclear extracts by the electrophoretic mobility gel shift assay from 3-day bile duct-ligated (BDL) mice but not in controls. Immunohistochemistry for NF-κB demonstrated nuclear localization in hepatocytes of BDL mice consistent with its activation in this liver cell type. Electrophoretic mobility gel shift assay and immunohistochemistry for NF-κB in BDL tumor necrosis factor-receptor 1 knockout mice demonstrated hepatocyte NF-κB activation, suggesting that tumor necrosis factor-α was not responsible for the activation of this transcription factor. Liver injury was assessed in BDL mice after administration of the adenovirus 5 inhibitor of kappa B superrepressor (Ad5IκBsr) to inhibit NF-κB. TUNEL-positive cells and serum alanine aminotransferase values were increased at least threefold in mice treated with the Ad5IκBsr versus the empty virus. Liver histology also demonstrated increased liver injury in the BDL mice treated with the Ad5IκBsr. In conclusion, NF-κB is activated in hepatocytes during obstructive cholestasis and functions to reduce liver injury.
Cholestasis, defined as an impairment in bile formation, is a feature of many chronic human liver diseases. 1 Cholestasis is associated with retention of normal bile constituents, including bile acids, within the liver. 2 The retention and accumulation of bile acids within the liver during cholestasis is thought to exacerbate liver injury. 3,4 Indeed, the importance of bile acid-induced liver injury is demonstrated in children with subtype 2 of the progressive familial intrahepatic cholestasis syndromes. 5 These children have mutations in the cannalicular transport protein for bile acid secretion into bile and develop progressive liver disease due to the inability to excrete bile acids from the hepatocyte. 5 Thus, the mechanisms by which bile acids modulate liver injury in cholestasis are of clinical and scientific importance.
Numerous studies have now shown that bile acid concentrations that occur during cholestasis induce hepatocyte apoptosis, thus providing a cellular mechanism for bile acid-mediated liver injury. 6 Not all bile acids are toxic, however, and minor changes in bile acid structure alter their potential cytotoxicity. For example, the glycine conjugate of chenodeoxycholate induces hepatocyte apoptosis in vitro, whereas the taurine conjugate does not. 7 Previous concepts suggested that bile acid toxicity correlated with relative hydrophobicity, with hydrophobic bile acids being cytotoxic and hydrophilic bile acids nontoxic. However, we could not establish a relationship between bile acid-induced apoptosis and their relative hydrophobicity. 7 More recently we have demonstrated in isolated cell systems that selective hydrophobic bile acids such as taurochenodeoxycholate activate a phosphatidylinositol 3-kinase (PI3-kinase)-dependent survival pathway, which prevent their otherwise inherent toxicity. 8 In this study, we also identified the transcription factor nuclear factor-kappa B (NF-κB) as one of the downstream targets of the taurochenodeoxycholate-stimulated PI3-kinase activity. Inhibition of NF-κB resulted in hepatocyte apoptosis by taurochenodeoxycholate. 8 Attenuation of bile acid-induced apoptosis by NF-κB is consistent with other studies demonstrating that NF-κB also reduces hepatocyte apoptosis by tumor necrosis factor-α (TNF-α) and during liver regeneration. 9
The activation of NF-κB by bile acids in vitro results in two predictions relevant to cholestatic liver diseases where liver tissue bile acid concentrations are increased. First, NF-κB should be activated in the liver during cholestasis. Second, inhibition of NF-κB should accentuate cholestasis-associated liver injury. The overall objective of the current study was to test these two hypotheses. Our specific aims were to address the following questions using the bile duct-ligated (BDL) mouse as a model of cholestasis: Is NF-κB activated in hepatocytes? Does inhibition of NF-κB result in increased apoptosis of hepatocytes? Is increased apoptosis caused by inhibiting NF-κB associated with an exacerbation of liver injury? Collectively, the results of the current study demonstrate that NF-κB is activated during cholestasis and functions to reduce liver injury. These data suggest that the rate and progression of human cholestatic liver injury may depend on the activation status of NF-κB.
Materials and Methods
Animals and Surgical Procedures
The care and use of animals for these studies were reviewed and approved by the Institutional Animal Care and Use Committee at the Mayo Foundation. Six- to 12-week-old male C57B6 and tumor necrosis factor-receptor 1 (TNF-R1) knockout mice (Jackson Labs, Bar Harbor, ME) were used for these studies. Under ether anesthesia using a nose cone containing ether-soaked tissue, a laparotomy was performed via a midline abdominal incision. The common bile duct was identified, isolated, and double-ligated using 5-0 silk (Ethicon, Somerville, NJ). The common bile duct was next transected between the ligatures. The abdominal incision was closed in layers using 3-0 chromic gut sutures (Ethicon). Sham operations were performed as above by identifying but not ligating the common bile duct.
Adenovirus 5 Inhibitor of κB Superrepressor (Ad5IκBsr)
The recombinant replication-deficient adenovirus 5 inhibitor of κB (Ad5IκB), containing an IκB in which serines 32 and 36 are mutated to alanines (generous gift of D. A. Brenner, University of North Carolina at Chapel Hill, Chapel Hill, NC), and Ad5ΔE1, an empty adenovirus for control experiments, were grown and purified as described previously. 8,9
Experimental Protocol
On postoperative day 2, 0.22 ml of 1.6 × 10 9 plaque-forming units (pfu)/ml of Ad5IκB or Ad5ΔE1, which was stored in 10% glycerol and diluted in saline immediately before use, were injected into the tail veins under ether anesthesia. Mice were sacrificed on postoperative day 3 under deep ether anesthesia. Blood samples (0.4 ml) were obtained from infrahepatic vena cava using a 28 gauge needle attached to a 1-ml syringe. Immediately after the venipuncture for blood sampling, the portal vein was cannulated using a 20 gauge angiocath, and the liver was flushed with phosphate-buffered saline (PBS; pH 7.4, 25°C) containing 137 mmol/L NaCl, 2.7 mmol/L KCl, 8 mmol/L Na2HPO4 · 7H2O, and 1.5 mmol/L KH2PO4. Blood was flushed out of the liver by transecting the infrahepatic inferior vena cava. The liver was cut into small pieces (approximately 1 × 1 × 0.5 cm) and used for the experiments described below.
Electrophoretic Mobility Gel Shift Assay (EMSA)
Nuclear protein extracts from liver tissue were prepared using NE-PER nuclear and cytoplasmic extraction reagents, CER, (Pierce, Rockford, IL) according to the manufacturer’s instructions. Briefly, liver tissue was homogenized in 4 ml of PBS and centrifuged for 10 minutes at 500 × g at 4°C. All subsequent steps were performed at 4°C. After removing the supernatant, the pellet was resuspended in 500 μl CER I buffer including protease inhibitors (250 mg/ml benzamidine, 2 mg/ml aprotinin, 2 mg/ml leupeptin, 0.2 mol/L phenylmethylsulfonyl fluoride), and incubated on ice for 10 minutes. Then, 27.5 μl CER II buffer was added to the sample, vortexed, and centrifuged at 16,000 × g for 5 minutes. The supernatant, containing the cytosolic proteins, was removed and stored at −80°C. The pellet was resuspended in 250 μl of NER buffer including protease inhibitors, vortexed every 10 minutes for 40 minutes, and then centrifuged at 16,000 × g for 10 minutes. Nuclear proteins (the supernatant) were used immediately or stored at −80°C. For the EMSA, 10 μg of nuclear proteins were incubated in binding buffer (Promega, Madison, WI) with 3.5 pmol of double-stranded DNA oligonucleotide containing a NF-κB consensus binding sequence (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) which was labeled with [γ-32P]-ATP using T4 polynucleotide kinase (Promega). Binding reactions were completed by incubating reaction solution for 30 minutes at room temperature. Protein-DNA complexes were separated from the unbound DNA probe by electrophoresis through 5% native polyacrylamide gels containing 0.5× Tris-Borate-EDTA. The gel was dried and exposed to BiomaxMR film (Kodak, Rochester, NY). Specificity of binding was verified by competition with a 40-fold molar excess of unlabeled double-stranded consensus oligonucleotide. Two micrograms of the antibody for the p65 subunit of NF-κB (Santa Cruz Biotechnology, Santa Cruz, CA) was used for the supershift assay.
Immunohistochemistry for NF-κB and the Neoepitope of Caspases 3/7
Antisera for the p65 subunit of NF-κB and the neoepitope of caspases 3/7 were obtained from Santa Cruz Biotechnology and from Anu Srinivasan at IDUN Pharmaceuticals (La Jolla, CA), respectively. The caspase antisera recognizes a common neoepitope shared by activated caspase 3 and 7. 10 Liver tissue was fixed and sectioned as previously described by us. 11 The liver specimens were incubated with blocking buffer (PBS containing 0.5% bovine serum albumin, 0.3% Triton X-100, and 0.2% horse serum or 0.2% goat serum; horse serum was used for the NF-κB immunohistochemistry, goat serum for the caspase immunohistochemistry). The primary antibody was used at a 1:50 dilution in PBS containing 0.5% bovine serum albumin plus 0.2% horse serum for NF-κB; and at a 1:100 dilution in PBS containing 0.5% bovine serum albumin plus 0.2% goat serum for caspases 3/7. The primary antibodies were incubated with the specimens for 1 hour at 25°C, followed by washing with PBS. The immunoreactivity was visualized using the avidin and biotinylated horseradish peroxidase macromolecular complex and diaminobenzidine tetrahydrochloride system (Vector Laboratories, Burlingame, CA). Specimens were counterstained with eosin using standard techniques. Hematoxylin and eosin staining was also performed for histological examination.
Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate Nick-End Labeling (TUNEL) Assay
The TUNEL assay was performed as we have previously described in detail using the fluorescein fluorescence-based In Situ Cell Death Detection Kit (Boehringer Mannheim, Indianapolis, IN). Specimens were observed by fluorescent microscope Eclipse TE200 (Nikon, Melville, NY). Hepatocyte nuclei were counted in a minimum of five low power fields (20 × 10) per specimen. TUNEL-positive nuclei were expressed as a percentage of all hepatocyte nuclei in the low power field as previously described. 11
Determination of Serum Alanine Aminotransferase (ALT)
Serum ALT was measured using the Sigma (St. Louis, MO) diagnostics kit No. 505.
Measurement of Liver Caspase 3/7-Like Protease Activity
Caspase 3/7-like activity was measured in liver cytosol using the fluorogenic substrate DEVD-AMC as we have previously described in detail. 11
Statistical Analysis
All data are expressed as mean ± SD from at least 3 separate experiments. Differences between groups were compared using an analysis of variance for repeated measures and a post hoc Bonferroni test to compare for multiple comparisons. All statistical analyses were performed using Instat Software (GraphPAD, San Diego, CA).
Results
Is NF-κB Activated in Hepatocytes of BDL Mice?
NF-κB activation in the BDL mouse liver was assessed by EMSA, and immunohistochemistry was performed to determine whether NF-κB was activated in hepatocytes. The EMSA revealed that nuclear protein extracts from the livers of BDL mice contained activated NF-κB (Figure 1A) ▶ . However, activated NF-κB was not observed in sham-operated mice. Binding specificity was verified by loss of binding in the presence of a 40-fold molar excess of unlabeled oligonucleotide and supershift of the band by p65 antisera. Immunohistochemistry demonstrated that in hepatocytes of sham-operated mice, the immunoreactivity for the p65 subunit of NF-κB was excluded from the nucleus and localized exclusively to the cytoplasm. In contrast, immunoreactivity for NF-κB was predominantly detected in hepatocyte nuclei of BDL mice (Figure 1B) ▶ . Translocation of NF-κB to the nuclei of other liver cell types was not observed (data not shown). Thus, NF-κB is translocated to the nucleus from cytosol in hepatocytes of the BDL mouse, consistent with hepatocyte NF-κB activation in the BDL animals.
Figure 1.
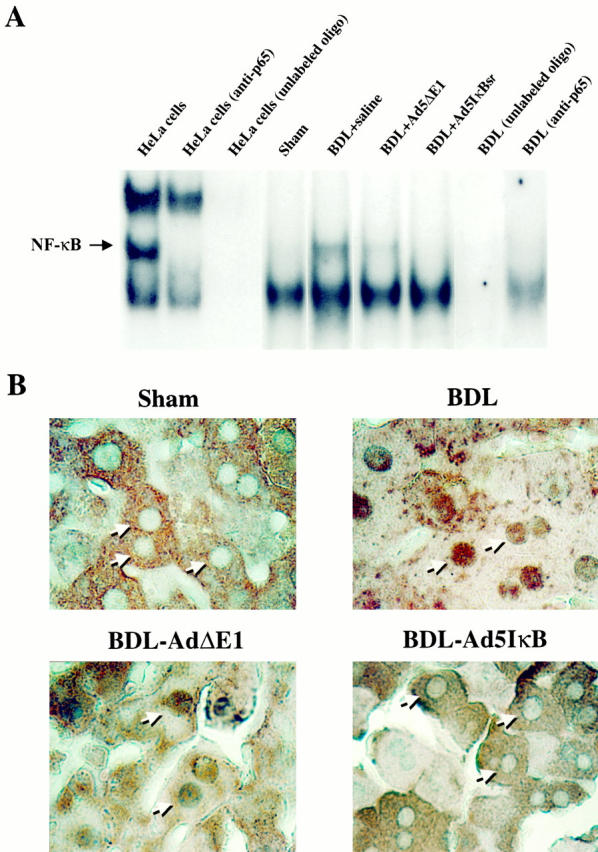
NF-κB is activated in the hepatocytes of BDL mice. A: An EMSA was performed using nuclear protein extracts isolated from whole liver and from commercially available HeLa cell nuclear extracts used as a positive control (Promega). The NF-κB binding complex was positively identified in HeLa cells by demonstrating loss of the complex with a p65 antisera and competitive inhibition by consensus unlabeled oligonucleotide This positive control served to help identify the complex in the mouse liver nuclear extracts. Binding of NF-κB to its consensus DNA binding motif was observed with nuclear protein extracts obtained from BDL animals treated with saline or the empty virus (Ad5ΔE1). However, no binding of NF-κB to the radiolabeled DNA oligonucleotide binding motif was observed with nuclear protein extracts obtained from BDL mice treated with the Ad5IκB superrepressor. Confirmation of the complex as NF-κB was obtained by demonstrating disruption of the complex with anti-p65 antisera and competitive inhibition with unlabeled oligonucleotide. B: Immunohistochemistry for NF-κB (p65) was performed on mouse liver tissue (original magnification, ×400). Immunoreactivity for NF-κB was identified in the nuclei of hepatocytes in BDL animals but was restricted to the cytoplasm of control mice. However, nuclear immunoreactivity for the p65 subunit of NF-κB was not observed in BDL mice treated with the Ad5IκB superrepressor. Collectively, the data demonstrate NF-κB activation in hepatocytes of BDL mice, but not controls.
Is NF-κB Activation Inhibited by the IκB Superrepressor in BDL Mice?
We sought to determine the role of NF-κB in cholestasis by transfecting hepatocytes with the Ad5IκB superrepressor to inhibit NF-κB activation. This mutated IκB cannot be phosphorylated and thus remains tightly bound to NF-κB, preventing its activation and translocation to the nucleus despite appropriate activation stimuli. Although inhibition of NF-κB by the Ad5IκBsr has been demonstrated in normal liver, its effectiveness in the BDL mouse liver has not established. The EMSA demonstrated activated NF-κB in liver nuclear protein extracts isolated from BDL animals treated with Ad5ΔE1 (a control for virus infection) but not in nuclear protein extracts from Ad5IκBsr-treated BDL animals (Figure 1A) ▶ . Immunohistochemistry for NF-κB also showed translocation of NF-κB to the nucleus in the Ad5ΔE1-treated BDL mice, but not in BDL animals treated with the Ad5IκBsr (Figure 1B) ▶ . These results directly demonstrate that Ad5IκBsr effectively blocks NF-κB nuclear translocation in hepatocytes of the BDL mice.
Is TNF-α Responsible for the Activation of NF-κB in BDL Mice?
TNF-α is increased in the serum of BDL mice and is known to activate NF-κB in hepatocytes by binding to the TNF receptor 1 (TNF-R1). 12,13 To determine whether hepatocyte NF-κB activation during cholestasis is potentially mediated by TNF-α, we performed EMSA and immunohistochemistry for NF-κB in BDL TNF-R1 knockout mice. Both the EMSA and immunohistochemistry demonstrated NF-κB translocation to the nucleus in hepatocytes of the TNF-R1 knockout mice (Figure 2, A and B) ▶ . Moreover, hepatocyte apoptosis in the BDL TNF-R1 knockout mouse was similar to that observed in the BDL-wild-type animal (8.3 ± 3.4 vs. 7.2 ± 1.0, P = n.s.). These data suggest that both hepatocyte NF-κB activation and apoptosis in BDL mice is independent of the TNF-R1 signaling pathway and further support our hypothesis that bile acids may mediate the hepatocyte activation of this transcription factor.
Figure 2.

NF-κB is activated in the hepatocytes of BDL mice from TNF-R1-deficient animals. A: An EMSA was performed using nuclear protein extracts isolated from BDL TNF-R1 knockout mice. Binding of NF-κB to its radiolabeled consensus DNA oligonucleotide binding motif was observed in nuclear protein extracts from BDL TNF-R1 knockout mouse. Specificity of binding was confirmed by demonstrating competition with an excess of unlabeled oligonucleotide. The identity of NF-κB was confirmed as the DNA binding protein by showing a supershift with antisera for the p65 subunit of NF-κB. B: Immunohistochemistry for NF-κB on liver tissue from TNF-R1 knockout BDL mice (original magnification, ×400). Immunoreactivity for NF-κB was observed in nuclei of hepatocytes in the BDL TNF-R1 knockout mouse. Collectively, the data suggest TNF-R1 is not necessary for the activation of NF-κB in acutely BDL mice.
Does Inhibition of NF-κB Result in Caspase Activation and Apoptosis of Hepatocytes in the BDL Mice?
It has been reported that NF-κB inhibits apoptosis by inducing the transcription of anti-apoptotic proteins. 14 Inhibition of NF-κB would, therefore, be predicted to increase hepatocyte apoptosis in the BDL animal. To examine the role of NF-κB in modulation of apoptosis in the BDL mouse liver, we next performed the TUNEL assay to quantitated apoptosis in the liver tissue from BDL and control mice (Figure 3) ▶ . Consistent with our previously published observations, 11 the TUNEL assay demonstrated increased apoptosis in BDL mice as compared to control mice (Figure 3) ▶ . Moreover, apoptosis was significantly increased in BDL mice treated with the Ad5IκBsr (Figure 3) ▶ . To confirm the occurrence of apoptosis, immunohistochemistry for the activated forms of the effector caspases 3/7 was performed (Figure 4) ▶ . Effector caspases are proteases that participate in the apoptosis program, and their activation is biochemical evidence in support of apoptosis. 15 These proteases, synthesized as zymogens, reveal neoepitopes when activated by proteolytic cleavate. Immunohistochemistry for the common neoepitope of active caspases 3/7 demonstrated immunoreactivity in livers of BDL mice, but not in sham-operated mice. Although BDL mice treated with the control virus Ad5ΔE1 demonstrated caspase 3/7 activation, caspase 3/7 activation was increased in BDL mice treated with the Ad5IκB superrepressor (Figure 4) ▶ . Indeed, caspase 3/7 activity in liver homogenates was 2.5-fold greater in BDL mice treated with the Ad5IκBsr compared to control BDL mice, as assessed using the fluorogenic substrate DEVD-AMC (data not shown). Taken together, these data suggest that inhibition of NF-κB in the BDL mouse increases hepatocyte caspase 3/7 activation.
Figure 3.

Inhibition of NF-κB increases TUNEL-positive hepatocytes in the BDL mouse. The TUNEL assay was performed as described in Experimental Protocol on fixed liver tissues. TUNEL-positive cells were increased in IκB superrepressor-treated BDL animals as compared to no-operated, sham-operated, or saline- or empty (Ad5ΔE1) virus-treated BDL mice.
Figure 4.
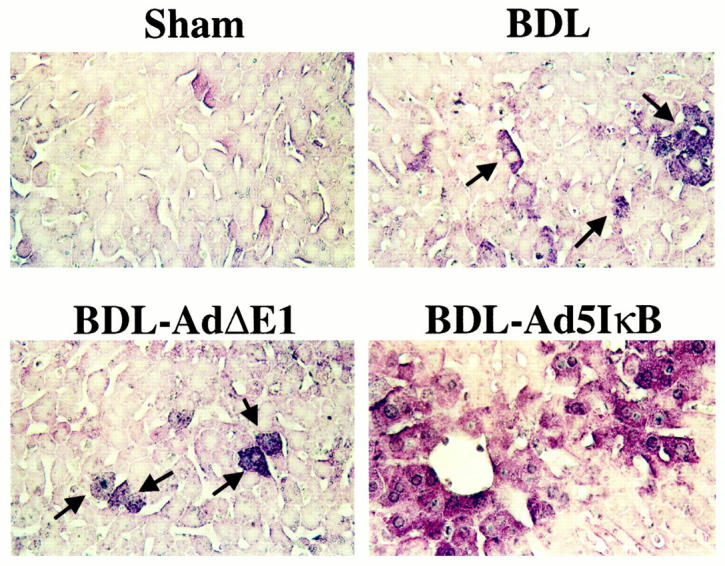
Caspases are activated in the hepatocytes of BDL mice. Immunohistochemistry was performed on mouse liver tissue using the antisera against the neoepitopes of active caspases 3/7 (original magnification, ×250). Immunohistochemistry showed that BDL induced caspase 3/7 activation in hepatocytes and treatment of BDL mice with the Ad5IκB superrepressor treatment enhances caspase 3/7 activation.
Is the Increase in Apoptosis Caused by Inhibiting NF-κB Activation Associated with an Exacerbation of Liver Injury?
Liver injury was examined by measuring serum ALT concentrations and assessing liver histology. Although serum ALT values were increased above control values in BDL mice and BDL treated with the Ad5ΔE1 (Figure 5A) ▶ , the ALT values in these mice remained below 103 IU/L. In contrast, serum ALT values were 475 ± 78 IU/L in BDL mice receiving the Ad5IκBsr, P < 0.01 (Figure 5A) ▶ . As has been previously described by others, 16 histological studies showed liver damage characterized by bile infarcts in BDL animals (Figure 5B) ▶ . Consistent with the ALT values, these infarcts were larger and more numerous in BDL animals treated with the Ad5IκB but not in BDL mice receiving the empty virus Ad5ΔE1 (Figure 5B) ▶ . Collectively, these data demonstrate that inhibition of NF-κB in the BDL mouse is associated with increased liver injury.
Figure 5.

Inhibition of NF-κB potentiates liver injury in the BDL mouse. A: Serum was collected from mice as described in Experimental Protocol. Serum ALT values were increased in BDL mice treated with the Ad5IκB superrepressor as compared to no-operated, sham-operated, or saline- or empty (Ad5ΔE1) virus-treated BDL mice. B: Liver histology was examined to assess liver injury. Bile infarcts were not observed in controls or sham-operated animals. Although bile infarcts were observed in BDL mice and the BDL mice treated with empty virus (Ad5ΔE1), the number and magnitude of bile infarct were increased in BDL mice transfected with the Ad5IκB superrepressor. Taken together, these data suggest activated NF-κB in the BDL mouse functions to reduce liver injury.
Discussion
The principal findings reported here relate to the role of NF-κB in cholestatic liver diseases. Using the 3-day BDL mouse as a model of obstructive cholestasis, the results demonstrate that (i) NF-κB is activated in the hepatocytes, (ii) NF-κB was activated in TNF-R1-deficient mice, (iii) the Ad5IκBsr inhibited NF-κB activation, and (iv) the Ad5IκBsr potentiated hepatocyte apoptosis and liver injury. These data directly demonstrate that NF-κB is activated in obstructive cholestasis and functions to reduce liver injury. The rate and progression of human cholestatic liver diseases may depend on the activation status of NF-κB within hepatocytes.
Using both EMSA and immunohistochemistry, we were able to demonstrate NF-κB in hepatocytes of BDL mice. NF-κB can be activated by a wide variety of stimuli including cytokines, oxidant free radicals, ionizing radiation, and UV light. 17,18 Although serum levels of TNF-α are increased in obstructive cholestasis and binding of this cytokine to the TNF-R1 receptor is known to activate NF-κB in hepatocytes, 19 we observed NF-κB activation in hepatocytes of BDL TNF-R1-deficient mice. These data suggest the TNF-α and TNF-R1 system is not involved in NF-κB activation during cholestasis. We have recently demonstrated NF-κB activation by selected bile acids via a PI3-kinase-dependent mechanism. 8 Because tissue bile acid concentrations are increased within the liver during cholestasis, 2 it is likely that bile acids contribute to the activation of NF-κB in the BDL mouse model of cholestasis. However, we cannot exclude other mechanisms such as oxidant stress, which has also been demonstrated in the cholestatic liver. 20
We demonstrated an increase in hepatocyte apoptosis as assessed by the TUNEL assay when NF-κB activation was inhibited by the Ad5IκBsr. We confirmed the presence of apoptosis by also demonstrating caspase activation, proteases essential for the execution of the apoptosis cell death program. 21 NF-κB has been shown to either prevent or potentiate hepatocyte apoptosis, depending on the model. For example, NF-κB prevents TNF-α- and Fas death receptor-mediated hepatocyte apoptosis, but contributes to hepatocyte apoptosis by hydrogen peroxide. 22 We have previously reported that hepatocyte apoptosis occurs in BDL mice by a Fas death receptor-dependent process using lpr Fas-deficient mice. 11 Thus, our previous observations in conjunction with the work of others demonstrating inhibition of Fas-mediated hepatocyte apoptosis by NF-κB are consistent and implicate Fas as the mediator of hepatocyte apoptosis in cholestasis. 11,23
Ad5IκBsr-treated BDL animals had increased rates of apoptosis, significantly increased serum higher ALT values, and more histological liver injury than saline- or empty virus (Ad5ΔE1)-treated animals. Because NF-κB functions to inhibit apoptosis, these observations imply a link between hepatocyte apoptosis and liver injury. Indeed, we previously observed a relationship between hepatocyte apoptosis, serum ALT values, and animal survival when comparing outcomes in BDL Fas-deficient and wild-type animals. 11 Although apoptosis is not thought to elicit an inflammatory response or contribute to tissue destruction, this concept is derived primarily from developmental studies. Inhibition of apoptosis with caspase inhibitors has been shown to block liver inflammation after administration of galactosamine and endotoxin. 24 Rapid and extensive induction of hepatocyte apoptosis with Fas-agonistic antisera is associated with fulminant hepatic failure. 25 Therefore, hepatocyte apoptosis in inflammatory conditions and disease states likely contributes to the potentiation of liver injury. 26 In this respect, NF-κB activation in cholestasis likely functions not only to reduce hepatocyte apoptosis, but also to reduce overall liver injury.
In conclusion, our working hypothesis is that, during cholestasis, there is retention and accumulation of bile acids within hepatocytes. The resulting increased hepatocellular concentrations of glycine conjugates of hydrophobic mono- or dihydroxy bile salts present a strong pro-apoptotic stimulus to the hepatocyte via activation of Fas. 27 However, taurine conjugates of these bile acids may induce NF-κB activation functioning as an adaptive mechanism to attenuate hepatocyte apoptosis and liver injury. The ability of potentially toxic bile acids to activate NF-κB may represent a pathophysiological adaptation that helps permit survival and continued function of hepatocytes and the liver despite cholestatic dysfunction. The rate and progression of human cholestatic liver diseases, therefore, may be influenced by the status of hepatocyte NF-κB activation. Therapeutic maneuvers designed to potentiate NF-κB activation in the cholestatic liver disease (eg, oral administration of NF-κB-activating bile acids) may help slow the progression of human cholestatic liver diseases such as primary sclerosing cholangitis and primary biliary cirrhosis.
Acknowledgments
The superb secretarial assistance of Sara Erickson is gratefully acknowledged.
Footnotes
Address reprint requests to Gregory J. Gores, M.D., Professor of Medicine, Mayo Medical School, Clinic, and Foundation, 200 First Street S.W., Rochester, MN 55905. E-mail: gores.gregory@mayo.edu.
Supported by grants from the National Institutes of Health DK 41876 (to G.J.G.) and the Deutsche Forschungsgemeinschaft ( to C.R.) and by the Mayo Foundation, Rochester, MN.
References
- 1.Trauner M, Meier PJ, Boyer JL: Molecular pathogenesis of cholestasis. N Engl J Med 1998, 339:1217-1227 [DOI] [PubMed] [Google Scholar]
- 2.Greim H, Trulzsch D, Czygan P, Rudick J, Hutterer F, Schaffner F, Popper H: Mechanism of cholestasis. 6. Bile acids in human livers with or without biliary obstruction. Gastroenterology 1972, 63:846-850 [PubMed] [Google Scholar]
- 3.Kaplan M: Primary biliary cirrhosis: a first step in prolonging survival. N Engl J Med 1994, 330:1386-1387 [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues C, Fan G, Ma X, Kren B, Steer C: A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane pertubation. JClin Invest 1998, 101:2790-2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, Tanner MS, Kagalwalla AF, Nemeth A, Pawlowska J, Baker A, Mieli-Vergani G, Freimer NB, Gardiner RM, Thompson RJ: A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998, 20:233-238 [DOI] [PubMed] [Google Scholar]
- 6.Patel T, Roberts LR, Jones BA, Gores GJ: Dysregulation of apoptosis as a mechanism of liver disease: an overview. Semin Liver Dis 1998, 18:105-114 [DOI] [PubMed] [Google Scholar]
- 7.Patel T, Bronk SF, Gores GJ: Increases of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J Clin Invest 1994, 94:2183-2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rust C, Karnitz LM, Paya CV, Moscat J, Simari RD, Gores GJ: The Bile Acid Taurochenodeoxycholate Activates a Phosphatidylinositol 3-Kinase-dependent Survival Signaling Cascade. J Biol Chem 2000, 275:20210-20216 [DOI] [PubMed] [Google Scholar]
- 9.Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA: NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J Clin Invest 1998, 101:802-811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ: In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ 1998, 5:1004-1016 [DOI] [PubMed] [Google Scholar]
- 11.Miyoshi H, Rust C, Roberts P, Burghart L, Gores G: Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999, 117:669-677 [DOI] [PubMed] [Google Scholar]
- 12.Beierle EA, Vauthey JN, Moldawer LL, Copeland EM, 3rd: hepatic tumor necrosis factor-alpha production and distant organ dysfunction in a murine model of obstructive jaundice. Am J Surg 1996, 171:202-206 [DOI] [PubMed] [Google Scholar]
- 13.Bemelmans MH, Gouma DJ, Greve JW, Buurman WA: Cytokines tumor necrosis factor and interleukin-6 in experimental biliary obstruction in mice. Hepatology 1992, 15:1132-1136 [DOI] [PubMed] [Google Scholar]
- 14.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS, Jr: NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281:1680-1683 [DOI] [PubMed] [Google Scholar]
- 15.Cohen GM: Caspases: the executioners of apoptosis. Biochem J 1997, 326(Pt 1):1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Sakamoto T, Ezure T, Yokomuro S, Murase N, Michalopoulos G, Demetris AJ: Interleukin-6, hepatocyte growth factor, and their receptors in biliary epithelial cells during a type I ductular reaction in mice: interactions between the periductal inflammatory and stromal cells and the biliary epithelium. Hepatology 1998, 28:1260-1268 [DOI] [PubMed] [Google Scholar]
- 17.Mercurio F, Manning AM: Multiple signals converging on NF-kappaB. Curr Opin Cell Biol 1999, 11:226-232 [DOI] [PubMed] [Google Scholar]
- 18.Baeuerle PA, Baichwal VR: NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv Immunol 1997, 65:111-137 [PubMed] [Google Scholar]
- 19.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP: Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol 1999, 17:331-367 [DOI] [PubMed] [Google Scholar]
- 20.Singh S, Shackleton G, Ah-Sing E, Chakraborty J, Bailey ME: Antioxidant defenses in the bile duct-ligated rat. Gastroenterology 1992, 103:1625-1629 [DOI] [PubMed] [Google Scholar]
- 21.Thornberry NA, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 22.Jones BE, Lo CR, Liu H, Pradhan Z, Garcia L, Srinivasan A, Valentino KL, Czaja MJ: Role of caspases and NF-kappaB signaling in hydrogen peroxide- and superoxide-induced hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol 2000, 278:G693-G699 [DOI] [PubMed] [Google Scholar]
- 23.Hatano E, Bradham CA, Stark A, Iimuro Y, Lemasters JJ, Brenner DA: The mitochondrial permeability transition augments Fas-induced apoptosis in mouse hepatocytes. J Biol Chem 2000, 275:11814-11823 [DOI] [PubMed] [Google Scholar]
- 24.Jaeschke H, Fisher MA, Lawson JA, Simmons CA, Farhood A, Jones DA: Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J Immunol 1998, 160:3480-3486 [PubMed] [Google Scholar]
- 25.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S: Lethal effect of the anti-Fas antibody in mice [published erratum appears in Nature 1993, 365: 568]. Nature 1993, 364:806-809 [DOI] [PubMed] [Google Scholar]
- 26.Maher JJ, Gores GJ: Apoptosis: silent killer or neutron bomb? [editorial; comment]. Hepatology 1998, 28:865-867 [DOI] [PubMed] [Google Scholar]
- 27.Faubion W, Guicciardi M, Miyoshi H, Bronk S, Roberts P, Svingen P, Kaufmann S, Gores G: Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest 1999, 103:137-145 [DOI] [PMC free article] [PubMed] [Google Scholar]