

Abstract
Tissue repair requires an adequate cellular proliferation coordinated with the timely proteolysis of matrix elements. Based on the properties of plasminogen activators in liver cell proliferation and tissue proteolysis, we explored the regulatory role of tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) in liver repair. Using carbon tetrachloride (CCl4) intoxication as a model of acute liver injury, we found that tPA-deficient mice displayed a mild defect in hepatic repair, whereas livers of uPA-deficient mice had a more substantial delay in repair, with injury of centrilobular hepatocytes persisting up to 14 days after CCl4. Notably, functional cooperativity between plasminogen activators was strongly inferred from the profound reparative defect in livers of mice lacking tPA and uPA simultaneously, with persistence of centrilobular injury as far out as 35 days. The defective repair was not because of increased susceptibility of experimental mice to the toxin or to inadequate cellular proliferation. Instead, lack of plasminogen activators led to the accumulation of fibrin and fibronectin within injured areas and poor removal of necrotic cells. These data demonstrate that tPA and uPA play a critical role in hepatic repair via proteolysis of matrix elements and clearance of cellular debris from the field of injury.
Cellular proliferation and reorganization of the extracellular matrix are central features of tissue repair and remodeling of the liver and extra-hepatic systems. After an acute injury, liver cells undergo well-synchronized rounds of proliferation that are terminated when the original liver mass is restored. 1,2 This proliferative response is regulated by growth-related genes, occurs coordinately with glucose homeostasis, and is essential for survival of the organism. 3-6 Concomitant with these processes, remodeling of extracellular matrix components by specialized systems of proteases must occur to clear necrotic debris by cellular scavengers and to form a suitable scaffold for newly formed cells. Although the control of matrix remodeling is not fully understood, transforming growth factor-β1 seems to be an important mediator of matrix production, 7-9 whereas plasminogen plays a key role in the proteolytic clearance of matrix components and necrotic cells in vivo. 10 In plasminogen-deficient mice, the reparative response of the liver after a toxic injury was recently shown to be severely impaired, and these animals are unable to successfully restore lobular architecture throughout prolonged periods. 10
The conversion of the inactive proenzyme plasminogen to the active serine protease, plasmin, by either urokinase-type plasminogen activator (uPA) or tissue-type plasminogen activator (tPA), is critical to the maintenance of hemostasis. Both plasminogen- and plasminogen activator-deficient mice experience widespread microvascular thrombosis, progressive organ damage, and delayed wound healing. 11-15 These adverse pleiotropic effects seem to result from the multisystem accumulation of fibrin, as indicated by the general correction of the spontaneous phenotypic abnormalities observed in plasminogen-deficient mice when a loss of fibrinogen is genetically superimposed. 16 However, a number of observations support a broader role for the plasminogen activation system outside fibrinolysis, 17-19 particularly in the hepatic microenvironment. 20-25 Perhaps the most persuasive of these observations is that plasminogen-deficient mice are unable to reorganize hepatic matrix and/or remove necrotic hepatocytes from sites of acute liver injury, regardless of the presence or absence of circulating fibrinogen. 10 Interestingly, poor removal of necrotic cells has also been reported in the heart of mice lacking uPA; necrotic cardiomyocytes within ischemic heart tissue persist as mummified ghosts, and are not readily replaced by fibrotic scar tissue in uPA-deficient mice. 26 Together, these studies suggest that plasminogen- and uPA-mediated clearance of necrotic cells may be central to tissue repair.
In the current studies, we explored the role of uPA and its functional homologue tPA in liver cell proliferation, matrix remodeling, and clearance of necrotic cells in vivo. Using a well-established model of acute liver injury in mice genetically engineered to lack tPA (tPAo), uPA (uPAo), or both (tPAo/uPAo), 12 we report that although cellular proliferation is not affected by the lack of plasminogen activators, repair is severely impaired. Mice lacking each plasminogen activator individually have a mild (tPAo mice) to moderate (uPAo mice) impairment in repair, whereas the combined loss of uPA and tPA results in severe defect in lobular reorganization with matrix accumulation and poor clearance of necrotic hepatocytes.
Materials and Methods
Gene-Targeted Mice
Mice with a targeted disruption of the genes coding for tPA (tPAo) or uPA (uPAo) were of a mixed 129/C57BL/6 genetic background. 12 To generate mice with simultaneous deficiency of tPA and uPA (tPAo/uPAo), we bred double heterozygous mice, and offspring were genotyped by polymerase chain reaction using specific primers that identify endogenous and targeted alleles for the tPA and uPA genes using ear biopsy DNA as template. 12,27 All experimental challenges were performed in 2- to 6-month-old littermates. All mice were housed together, fed laboratory chow ad libitum, and were kept in the same room under supervision by the same investigator throughout the study period. Animal protocols were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital Research Foundation (Cincinnati, OH).
Liver Injury
Gene-targeted mice and control littermates were injected intraperitoneally with 0.5 ml of carbon tetrachloride (CCl4) (Aldrich Chemical Inc., Milwaukee, WI) per kg of body weight as a 25% solution in corn oil. 28,29 Mice were examined daily and sacrificed 2 to 35 days after CCl4 treatment, as previously described. 10 In brief, mice were weighed, anesthetized, and blood samples were collected from the inferior vena cava. The liver was removed, blot dried, and weighed; samples were obtained from lobes, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for histological analysis. The remainder of the liver was immediately frozen in liquid nitrogen. Biochemical markers of liver function and injury were determined in plasma within 4 hours of collection by an automated enzymatic assay using the Vistros Chemistry Systems 950 (Johnson & Johnson, Rochester, NY). 29
Hepatocyte Proliferation
The proliferative response after CCl4 injection was measured by the incorporation of bromodeoxyuridine (BrdU), which was administered intraperitoneally to all mice 2 hours before sacrifice. 10 BrdU-labeled hepatocytes were identified on 4-μm sections of paraffin-embedded liver samples according to instructions provided in the Cell Proliferation kit (Amersham, Life Science, Arlington Heights, IL). 29 For each liver sample, hepatocyte labeling index (% of hepatocytes incorporating BrdU) was calculated by counting BrdU-labeled and -unlabeled hepatocytes in 10 high-power fields (∼100 hepatocyte nuclei/field) by an investigator unaware of animal genotype. Hepatocyte proliferation was then expressed as the mean (±SD) for all mice in each group, and statistical significance was determined using unpaired t-test, with a significance level of P < 0.05.
Plasminogen Activator Zymography
Liver extracts were prepared from frozen liver samples homogenized in phosphate-buffered saline containing 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, and 10% protease inhibitor cocktail (Sigma Chemical Co., St. Louis, MO). Homogenates were centrifuged at 12,000 × g, for 15 minutes, at 4°C, and protein concentration was determined in the supernatant using the Bradford method-based Bio-Rad assay (Bio-Rad Lab., Inc., Hercules, CA). One hundred μg of homogenate protein were analyzed by zymography using polyacrylamide gels cast with casein and plasminogen as described previously. 11,30
Staining of Extracellular Matrix Substrates
Immunohistochemical detection of fibrinogen and fibronectin (two components of the provisional matrix of wound fields) was performed in liver sections using a rabbit anti-fibrinogen antiserum, 10 rabbit anti-fibronectin IgG (Sigma Chemical Co.), and the Vectastain ABC-AP detection system (Vector Laboratories, Burlingame, CA); omission of primary antibodies was used in at least one section for each experiment as a negative control. 15 Fast Red TR/naphthol AS-MX (Sigma Chemical Co.) was used to detect alkaline phosphatase activity in situ. 10
Results
Deficiency of Plasminogen Activators Leads to Abnormal Liver Repair
CCl4 is a specific hepatotoxin that destroys a subset of hepatocytes proximal to the central vein that enzymatically convert CCl4 to the free radical CCl3 and other highly-reactive species. 31 To explore the role of plasminogen activators in liver repair after an acute toxic injury, mice with single and combined deficits in tPA and uPA were challenged with a single dose of CCl4. Control (tPA+/uPA+) and plasminogen activator-deficient animals displayed a similar susceptibility to CCl4-induced liver damage. Plasma alanine aminotransferase (ALT, an indicator of hepatocyte injury) levels increased >80-fold within 2 days of the initial challenge and returned to near baseline by 7 days, regardless of the genotype (Figure 1) ▶ . Therefore, deficiency of plasminogen activators did not seem to affect either the initial extent of liver damage or the short-lived toxicity of CCl4. Consistent with the ALT data, visual inspection of livers 2 days after CCl4 revealed a similar diffuse pale/lacy appearance in mice of all four genotypes (Figure 2) ▶ . The gross appearance of the livers of mice expressing both plasminogen activators (tPA+/uPA+) or expressing solely uPA (ie, tPAo) normalized by 7 days. In contrast, the diffuse pale/lacy appearance persisted in the livers of both uPAo and tPAo/uPAo mice, with only marginal improvement in the former at 14 days. To further define the long-term outcome of hepatic repair, we further carried the study to 35 days. At this time, the livers of tPA+/uPA+, tPAo, and uPAo mice invariably displayed a normal appearance, whereas the livers of tPAo/uPAo mice remained grossly abnormal, and similar to the appearance that had been seen 2 days after CCl4 (data not shown).
Figure 1.

Increased levels of alanine aminotransferase after CCl4. Levels of alanine aminotransferase (ALT) in the serum of mice lacking plasminogen activators and control (tPA+/uPA+) littermates demonstrate a nearly 80-fold increase 2 days after a single dose of CCl4. Thereafter, values return to baseline levels at 7 to 14 days in a similar manner in mice of all genotypes (mean ± SD, n = 7 for each group at 2 days, and n = 3 to 4 at 0, 7, and 14 days; 0 day = no treatment; no statistical significance among groups within each time point).
Figure 2.

Delayed restoration of normal liver appearance after toxic injury in mice lacking plasminogen activators. Visual inspection of livers from mice lacking plasminogen activators and control littermates shows an indistinguishable pale/lacy appearance in all livers 2 days after CCl4. Although livers of tPA+/uPA+ and tPAo mice restore normal appearance at 7 days, uPAo livers appear mildly abnormal at 14 days, and tPAo/uPAo livers display a pronounced pale/lacy appearance at both time points.
To determine the microscopic basis for the abnormal appearance after CCl4 in mice lacking plasminogen activators, we performed histological analyses of paraffin-embedded liver sections. Two days after injury, all livers (n = 7 for each genotype) had widespread liquefaction necrosis in centrilobular hepatocytes regardless of the genotype, compromising 30 to 50% of the entire lobule (Figure 3) ▶ . There was minimal inflammation and intact cellular components in the unaffected areas of the liver lobule; venular, sinusoidal endothelial, and nonparenchymal cells were well preserved. Systematic analysis of liver samples at 7 to 35 days (n = 3 to 4 for each genotype) showed that the centrilobular injury of tPA+/uPA+ mice resolved at 7 days, whereas tPAo livers had minimal residual necrotic hepatocytes with inflammatory infiltrate in the centrilobular region in three of four mice. At 14 and 35 days, liver sections of tPA+/uPA+ mice and tPAo littermates were normal and indistinguishable. Normalization of centrilobular injury was delayed in uPAo mice, with near resolution seen 14 to 35 days after the initial challenge (Figure 3) ▶ . These data suggest that in the absence of tPA, uPA can independently promote the reparative response after CCl4 injury and nearly normalize hepatic histology within 7 days. In contrast, tPA cannot support efficient repair in the absence of uPA, as demonstrated by a persistent centrilobular injury at 14 days in uPAo livers. Nevertheless, tPA remains an important physiological adjunct in view of the profound delay in normalization of lobular appearance of livers lacking both tPA and uPA; these livers display persistent centrilobular injury as far out as 35 days after a single dose of CCl4 (Figure 3) ▶ .
Figure 3.
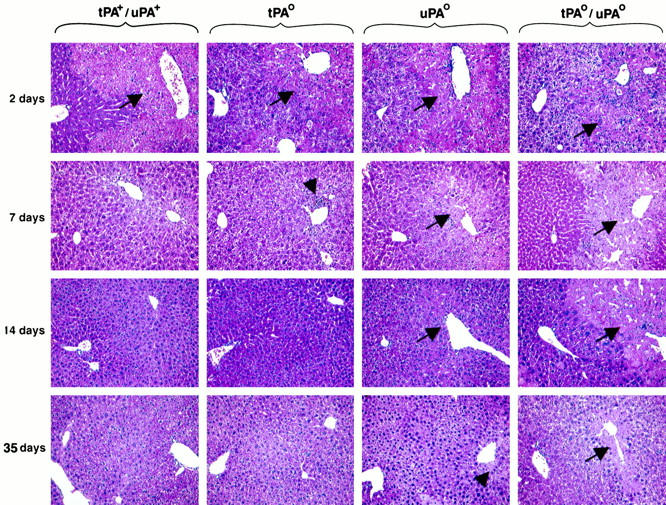
Variable degree of abnormal repair of centrilobular injury in mice lacking plasminogen activators. H&E staining of liver sections after CCl4 shows a similar type and degree of centrilobular injury in mice of all genotypes (arrows). The lesion is completely resolved by 7 days in control mice, whereas there is focal area of necrosis and inflammation in tPAo livers (arrowhead). In the absence of uPA, centrilobular injury is still present at 14 days, but only in a few centrilobular areas at 35 days (arrowhead). In contrast, the centrilobular injury of tPAo/uPAo livers persists through 14 days, with minimal evidence of repair at 35 days. Original magnification, ×200.
Lack of Plasminogen Activators Does Not Impair Hepatocyte Proliferation
Plasminogen activators have been previously linked to hepatocyte proliferation, 20-25 and one potential mechanism underlying the defective repair of tPAo/uPAo livers is impaired proliferative response. Using BrdU incorporation by hepatocytes as an indicator of proliferation, we found that mice lacking plasminogen activators mounted a proliferative response that was similar to control animals in regards to localization of BrdU-labeled cells within the hepatic lobule and degree of proliferation (Figure 4) ▶ . BrdU-labeled hepatocytes were uniformly dispersed along noninjured areas of the liver lobule in an indistinguishable manner in both targeted and nontargeted littermates (Figure 4A) ▶ . Analysis of the number of BrdU-labeled hepatocytes 2 days after CCl4 showed a >10-fold increase greater than baseline levels in all livers regardless of the genotype. Although the increase tended to be more pronounced in the livers of tPAo/uPAo mice, this finding did not reach statistical significance (Figure 4B) ▶ . Interestingly, the number of BrdU-labeled hepatocytes returned to near baseline levels in nontargeted littermates as well as in tPAo/uPAo mice despite the persistence of pronounced centrilobular tissue damage at 7 and 14 days. This apparent dissociation between proliferative response and hepatic repair probably results from the restoration of normal cellular mass in mice lacking plasminogen activators leading to normalization of hepatic function. The maintenance of plasma albumin at normal levels at both time points in control and plasminogen activator-deficient mice is consistent with a prompt restoration of liver function (data not shown). Interestingly, the coexistence of an adequate proliferative response and the inability to clear necrotic cells in uPAo and tPAo/uPAo livers resulted in an increase in liver mass in these mice to 47 to 49% greater than control and tPAo littermates at 14 days (Figure 5) ▶ .
Figure 4.
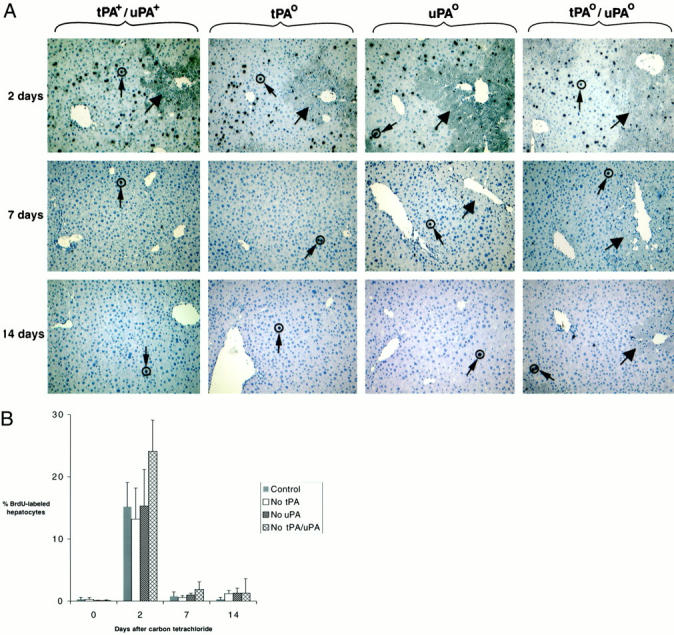
Adequate proliferation of hepatocytes after CCl4. A: BrdU-labeled hepatocytes (small arrows) are easily detected throughout unaffected lobular regions of all livers 2 days after CCl4. Thereafter, labeled cells are inconspicuous in all genotypes in a similar manner, despite the persistent centrilobular injury (large arrows) in uPAo and tPAo/uPAo livers. Original magnification, ×200. B: Quantification of BrdU-labeled hepatocytes demonstrates a similar increase in proliferating hepatocytes at 2 days to more than 10-fold greater than pretreatment levels (day 0), followed by a return to low proliferation at 7 and 14 days concomitantly in livers of all genotypes (control = tPA+/uPA+).
Figure 5.

Increased liver mass in mice lacking plasminogen activators after CCl4. Expression of liver weight as a percentage of body weight shows an increase in liver mass in all mice 2 days after CCl4, followed by a gradual decrease in control and tPAo littermates between 7 to 14 days. In contrast, this decrease is not observed in mice lacking uPA or both tPA/uPA, which display a greater liver mass at 14 days (mean ± SD; P < 0.01 for uPAo and tPAo/uPAo versus tPAo and tPA+/uPA+ at 14 days; control = tPA+/uPA+).
Functional Overlap between Plasminogen Activators during Liver Repair
Increased uPA activity and plasminogen activation in liver remnants after partial hepatectomy have been suggested to play a regulatory role in liver regeneration. 20,22 Therefore, we used zymography to define the hepatic activity of plasminogen activators after a toxic injury and to explore whether the loss of an individual plasminogen activator species is accompanied by a compensatory increase in the level of the remaining plasminogen activator in injured livers. In tPA+/uPA+ mice, liver injury resulted in a rapid and substantial increase in the activity of hepatic plasminogen activators, primarily uPA (Figure 6) ▶ . The activity of plasminogen activators progressively returned to basal levels throughout the 14-day study period, seemingly in parallel with the microscopic resolution of centrilobular damage. A similar pattern of transiently increased activity of hepatic uPA was seen in tPAo mice after CCl4 administration, although the decrease in uPA activity appeared delayed. Interestingly, the transient change in hepatic uPA activity coincided with the timely resolution of the centrilobular injury of tPAo livers, suggesting that uPA-mediated proteolysis efficiently directs hepatic repair. In uPAo mice, hepatic tPA activity was markedly increased at 7 and 14 days; however, based on the slow normalization of hepatic architecture in these mice, the elevated tPA activity was not sufficient to direct timely liver repair. Taken together, these data point to a coordinate regulation of plasminogen activator expression within an injured liver tissue that serves to promote repair. When this combined proteolytic response is completely abolished by the simultaneous inactivation of the tPA and uPA genes the reparative response to the injury is profoundly impaired.
Figure 6.

Activity of plasminogen activators after CCl4. Representative zymography demonstrating plasmin-mediated casein lysis by tPA and uPA in liver extracts of gene-targeted mice and control littermates. Injection of CCl4 results in an increase in activity of plasminogen activators in the first 7 days, with no detectable activity at 14 days. Activities of uPA (7 days) and tPA (14 days) are greater than control littermates in liver extracts from mice lacking the functional homologue.
Loss of Plasminogen Activators Leads to Impaired Clearance of the Provisional Fibrin-Rich Matrix in Diseased Livers
At 7 and 14 days after injury, the most prominent histological feature of tPAo/uPAo mice was the presence of necrotic hepatocytes and accumulation of eosinophilic material in the centrilobular region. This finding suggested that the absence of plasminogen activators resulted in defective proteolysis of cellular debris and/or extracellular matrix that occurs during normal liver repair. Because fibrin and fibronectin are important components of the provisional matrix within hepatic and extra-hepatic wound fields, 15,32 we explored whether impaired clearance of these matrix elements is associated with the defect in liver repair observed in tPAo/uPAo mice. Immunohistochemical analysis of liver sections 2 days after CCl4 showed that fibrin (Figure 7) ▶ and fibronectin (not shown) deposition was a prominent feature of damaged centrilobular zones of all mice, regardless of the genotype. In livers of tPA+/uPA+ mice, clearance of both substrates occurred concomitantly with resolution of the lobular injury, which is consistent with the effective proteolytic clearance of provisional matrix during repair. In contrast, impaired fibrin/fibronectin clearance was noted in all areas of defective repair in livers of mice lacking plasminogen activators. In tPAo mice, there was minimal residual fibrin/fibronectin in centrilobular areas beyond day 7, suggesting that induction of proteolysis by its functional counterpart, uPA, can efficiently direct clearance of both substrates in the liver. The primary importance of uPA in fibrin/fibronectin clearance is further supported by the persistent accumulation of fibrin/fibronectin in diseased centrilobular areas 7 to 14 days after CCl4 in uPAo livers. However, the even more severe impairment in fibrin/fibronectin clearance observed in tPAo/uPAo livers at both 7 and 14 days clearly points to a strong functional cooperation between both plasminogen activators in clearance of fibrin-related matrices during liver repair.
Figure 7.
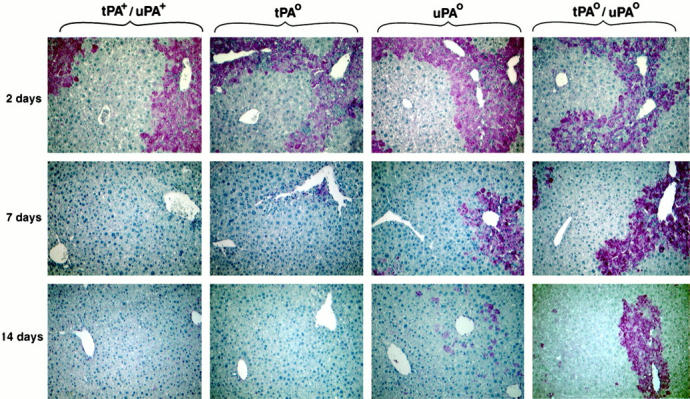
Fibrin accumulation in livers of mice lacking plasminogen activators. Immunohistochemical staining detects fibrin deposition in injured centrilobular areas in livers of all mice 2 days after CCl4. Acquisition of normal histology is associated with clearance of fibrin in tPA+/uPA+ and tPAo livers, whereas fibrin deposition is a dominant feature in areas of defective repair in uPAo and tPAo/uPAo livers.
Discussion
These studies show that plasminogen activators play a critical role in the reparative reorganization of the liver lobule after an acute toxic injury. However the relative importance of the two known plasminogen activators is not identical, with loss of tPA resulting in a mild defect in tissue repair and the loss of uPA resulting in a substantial delay in the resolution of the centrilobular injury. Consistent with the concept of a functionally overlapping role of tPA and uPA in the control of hepatic repair, resolution of the centrilobular injury is markedly delayed in mice lacking both tPA and uPA. The defective repair is not because of an increased susceptibility of experimental mice to toxic injury or to impaired hepatocellular proliferation. Rather, tPAo/uPAo mice display a pronounced impairment in the clearance of necrotic debris and the fibrin-rich matrix found at the site of injury. As a result, liver size increases out of proportion to body weight, and injured livers display an abnormal, lacy appearance far beyond the time required for control animals to restore normal liver morphology.
The reparative response of the liver after hepatocellular necrosis requires proliferation of relatively sedentary hepatocytes, matrix synthesis, and reorganization of the lobule. In support for the role of uPA as a facilitator of cellular proliferation during liver regeneration, 20-25 we found a robust induction of hepatic uPA activity in nontargeted mice after CCl4, which correlates with a previous report of increased uPA activity in the regenerating rat liver. 22 Despite these data, deficiency of either or both plasminogen activators in vivo seems to have no untoward effect on hepatocyte proliferation after an acute toxic injury. The number of hepatocytes entering the cell cycle and the distribution of proliferating cells along the noninjured portion of the liver lobule were unaffected by the absence of uPA and/or tPA. However, these findings do not conflict with or argue against the reported mitogenic role for uPA during liver regeneration. 24 Rather, our results highlight important differences among the molecular pathways regulating the hepatic response to a physical and toxic injury. 33-35 The finding of normal hepatocyte proliferation in tPAo/uPAo mice after a toxic injury suggests the existence of tPA/uPA-independent molecular pathways that effectively direct the restoration of liver mass. Nevertheless, despite the appropriate proliferative response, the ability of hepatocytes to populate diseased centrilobular areas is severely impaired because of a defect in the removal of components of provisional matrix and clearance of necrotic cells.
The striking similarities between the reparative defects observed in our studies of tPAo/uPAo mice and previous studies in plasminogen-deficient mice 10 strongly imply that the failure to restore normal liver architecture is mechanistically because of a failure in local plasmin-mediated proteolysis, rather than some independent functional property of plasminogen activator (eg, uPAR-mediated signaling events) 36-39 or plasminogen (eg, angiostatin-mediated events). 40 Two critical questions that remain to be resolved are: 1) What target(s) of plasmin-mediated proteolysis is critical to the clearance of necrotic debris and reorganization of damaged tissue? and 2) Is there a general role of plasmin in re-organizing necrotic tissue in organ systems other than the liver? Interestingly, loss of plasmin-mediated proteolysis is known to be a major impediment to repair of other tissues (eg, skin incisional wounds 15 and corneal scrape wounds 41,42 ), and these defects can be effectively corrected by the genetic elimination of fibrin(ogen). 16 Therefore, fibrin must be considered as a prime suspect in the search for plasmin targets related to hepatic repair after toxin injury. Indeed, fibrin is a persistent feature in the damaged centrilobular zones in tPAo/uPAo mice, and an inability to infiltrate, organize, and clear fibrin may be sufficient to impede hepatic repair. Nevertheless, it is highly unlikely that fibrin is the sole target of the plasminogen activation system that is relevant to hepatic repair. Notably, unlike repair in the skin, the absence of fibrin(ogen) does not restore normal liver repair in plasminogen-deficient mice after toxic injury. 10 Therefore, our current working hypothesis is that a failure of plasmin-mediated proteolysis of nonfibrin substrates (eg, necrotic cells and associated extracellular matrices) may impede repair as effectively as a failure of plasmin-mediated fibrin clearance.
Proteolysis within the field of injury is critical to tissue repair through removal of matrix components and clearance of necrotic cells. In wound environments where tissue necrosis is limited (eg, skin incisional wounds), the most important physiological target of plasmin may be fibrin-rich matrices. 16 In contrast, when cellular necrosis is the main feature of an injured tissue (eg, liver), fibrin-unrelated matrix proteolysis and removal of necrotic cells must occur to ensure effective repair. 10 Based on the findings in the liver, one might anticipate that the absence of circulating fibrinogen is unlikely to restore the normal reparative pattern of cardiac 26,43,44 and hepatic tissues in mice with defects in the plasminogen activation system. Together, these data point to a potential defect in nonfibrin targets that may relate to tissue repair, perhaps through plasmin-mediated activation of transforming growth factor or metalloproteinases. 26,43,44 Alternately, effective removal of necrotic cells may be hampered by a defect in recruitment and/or activation of inflammatory cells within the field of injury. In this regard, using the same model of acute CCl4 injury in plasminogen-deficient mice, we found no obvious impairment in recruitment of inflammatory cells to damaged centrilobular regions after the toxic injury; however, mononuclear phagocytes failed to ingest abundant cellular debris from neighboring necrotic hepatocytes or within distorted sinusoidal spaces (Jorge A. Bezerra, unpublished work). Therefore, the plasminogen system may be critical to mononuclear phagocyte-mediated proteolytic clearance of necrotic cells during tissue repair.
In conclusion, these studies reveal a key regulatory role of tPA and uPA in the reparative response of the liver to an acute injury. The shared features of defective repair in mice lacking plasminogen activators and plasminogen strongly imply that plasmin serves a central proteolytic role in tissue repair. Future studies will determine whether tPA and uPA act solely by generation of plasmin-mediated proteolysis, or whether one or both proteases also act independently of plasmin generation to support tissue repair. Finally, it remains to be established what nonfibrin targets of the plasminogen activation system are important in the reorganization of necrotic tissue.
Acknowledgments
We thank Alicia Emley for assistance with illustrations, and Dr. William Balistreri for insightful review of the manuscript.
Footnotes
Address reprint requests to Jorge A. Bezerra, Division of Gastroenterology and Nutrition, Children’s Hospital Medical Center, 3333 Burnet Ave., Cincinnati OH 45229-3039. E-mail: jorge.bezerra@chmcc.org.
Supported in part by the Hoffman-LaRoche, Ltd., Grant ROTRF 477924785 (to J. A. B.) and National Institutes of Health grants DK 55710 (to J. A. B.) and HL 47826 (to J. L. D.).
References
- 1.Higgins GM, Anderson RM: Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol 1931, 12:126-202 [Google Scholar]
- 2.Grisham JW: A morphologic study of deoxyribonucleic acid synthesis and cell proliferation in regenerating rat liver; autoradiography with thymidine-H3. Cancer Res 1962, 22:842-849 [PubMed] [Google Scholar]
- 3.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R: Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274:1379-1383 [DOI] [PubMed] [Google Scholar]
- 4.Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R: CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest 1998, 102:996-1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rai RM, Lee FY, Rosen A, Yang SQ, Lin HZ, Koteish A, Liew FY, Zaragoza C, Lowenstein C, Diehl AM: Impaired liver regeneration in inducible nitric oxide synthase deficient mice. Proc Natl Acad Sci USA 1998, 95:13829-13834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamada Y, Kirillova I, Peschon JJ, Fausto N: Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 1997, 94:1441-1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman SL: Cytokines and fibrogenesis. Semin Liver Dis 1999, 19:129-140 [DOI] [PubMed] [Google Scholar]
- 8.Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA: The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999, 30:77-87 [DOI] [PubMed] [Google Scholar]
- 9.Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H: Blockade of type beta transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci USA 1999, 96:2345-2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, Degen JL: Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci USA 1999, 96:15143-15148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugge TH, Flick MJ, Daugherty CC, Degen JL: Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 1995, 9:794-807 [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC: Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994, 368:419-424 [DOI] [PubMed] [Google Scholar]
- 13.Drew AF, Kaufman AH, Kombrinck KW, Danton MJ, Daugherty CC, Degen JL, Bugge TH: Ligneous conjunctivitis in plasminogen-deficient mice. Blood 1998, 91:1616-1624 [PubMed] [Google Scholar]
- 14.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D: Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 1995, 92:2585-2593 [DOI] [PubMed] [Google Scholar]
- 15.Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K: Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 1996, 2:287-292 [DOI] [PubMed] [Google Scholar]
- 16.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL: Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 1996, 87:709-719 [DOI] [PubMed] [Google Scholar]
- 17.Chen ZL, Strickland S: Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997, 91:917-925 [DOI] [PubMed] [Google Scholar]
- 18.Brunner G, Gabrilove J, Rifkin DB, Wilson EL: Phospholipase C release of basic fibroblast growth factor from human bone marrow cultures as a biologically active complex with a phosphatidylinositol-anchored heparan sulfate proteoglycan. J Cell Biol 1991, 114:1275-1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Odekon LE, Blasi F, Rifkin DB: Requirement for receptor-bound urokinase in plasmin-dependent cellular conversion of latent TGF-beta to TGF-beta. J Cell Physiol 1994, 158:398-407 [DOI] [PubMed] [Google Scholar]
- 20.Kim TH, Mars WM, Stolz DB, Petersen BE, Michalopoulos GK: Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997, 26:896-904 [DOI] [PubMed] [Google Scholar]
- 21.Liu ML, Mars WM, Zarnegar R, Michalopoulos GK: Collagenase pretreatment and the mitogenic effects of hepatocyte growth factor and transforming growth factor-alpha in adult rat liver. Hepatology 1994, 19:1521-1527 [PubMed] [Google Scholar]
- 22.Mars WM, Liu ML, Kitson RP, Goldfarb RH, Gabauer MK, Michalopoulos GK: Immediate early detection of urokinase receptor after partial hepatectomy and its implications for initiation of liver regeneration. Hepatology 1995, 21:1695-1701 [PubMed] [Google Scholar]
- 23.Mars WM, Kim TH, Stolz DB, Liu ML, Michalopoulos GK: Presence of urokinase in serum-free primary rat hepatocyte cultures and its role in activating hepatocyte growth factor. Cancer Res 1996, 56:2837-2843 [PubMed] [Google Scholar]
- 24.Roselli HT, Su M, Washington K, Kerins DM, Vaughan DE, Russell WE: Liver regeneration is transiently impaired in urokinase-deficient mice. Am J Physiol 1998, 275:G1472-G1479 [DOI] [PubMed] [Google Scholar]
- 25.Michalopoulos GK, DeFrances MC: Liver regeneration. Science 1997, 276:60-66 [DOI] [PubMed] [Google Scholar]
- 26.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJAP, Carmeliet P: Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 1999, 5:1135-1142 [DOI] [PubMed] [Google Scholar]
- 27.Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Romer J, Dano K, Carmeliet P, Collen D, Degen JL: Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci USA 1996, 93:5899-5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulsen JE: The time-course of mouse liver regeneration after carbon tetrachloride injury is influenced by circadian rhythms. Chronobiol Int 1990, 7:271-275 [PubMed] [Google Scholar]
- 29.Yazigi NA, Carrick TL, Bucuvalas JC, Schmidt CS, Balistreri WF, Bezerra JA: Expansion of transplanted hepatocytes during liver regeneration. Transplantation 1997, 64:816-820 [DOI] [PubMed] [Google Scholar]
- 30.Heckel JL, Sandgren EP, Degen JL, Palmiter RD, Brinster RL: Neonatal bleeding in transgenic mice expressing urokinase-type plasminogen activator. Cell 1990, 62:447-456 [DOI] [PubMed] [Google Scholar]
- 31.Recknagel RO, Glende EA, Jr, Dolak JA, Waller RL: Mechanisms of carbon tetrachloride toxicity. Pharmacol Ther 1989, 43:139-154 [DOI] [PubMed] [Google Scholar]
- 32.Neubauer K, Knittel T, Armbrust T, Ramadori G: Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology 1995, 108:1124-1135 [DOI] [PubMed] [Google Scholar]
- 33.Czaja MJ, Flanders KC, Biempica L, Klein C, Zern MA, Weiner FR: Expression of tumor necrosis factor-α and transforming growth factor-β1 in acute rat liver injury. Growth Factors 1989, 1:219-226 [DOI] [PubMed] [Google Scholar]
- 34.Fausto N, Laird AD, Webber EM: Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J 1995, 9:1527-1536 [DOI] [PubMed] [Google Scholar]
- 35.Schmiedeberg P, Biempica L, Czaja MJ: Timing of protooncogene expression varies in toxin-induced liver regeneration. J Cell Physiol 1993, 154:294-300 [DOI] [PubMed] [Google Scholar]
- 36.Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F: The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem 1987, 262:4437-4440 [PubMed] [Google Scholar]
- 37.Behrendt N, Ploug M, Patthy L, Houen G, Blasi F, Dano K: The ligand-binding domain of the cell surface receptor for urokinase-type plasminogen activator. J Biol Chem 1991, 266:7842-7847 [PubMed] [Google Scholar]
- 38.Gyetko MR, Todd RF, III, Wilkinson CC, Sitrin RG: The urokinase receptor is required for human monocyte chemotaxis in vitro. J Clin Invest 1994, 93:1380-1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA: Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem 1994, 269:32380-32388 [PubMed] [Google Scholar]
- 40.Claesson-Welsh L, Welsh M, Ito N, Anand-Apte B, Soker S, Zetter B, O’Reilly M, Folkman J: Angiostatin induces endothelial cell apoptosis and activation of focal adhesion kinase independently of the integrin-binding motif RGD. Proc Natl Acad Sci USA 1998, 95:5579-5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kao WW, Kao CW, Kaufman AH, Kombrinck KW, Converse RL, Good WV, Bugge TH, Degen JL: Healing of corneal epithelial defects in plasminogen- and fibrinogen-deficient mice. Invest Ophthalmol Vis Sci 1998, 39:502-508 [PubMed] [Google Scholar]
- 42.Drew AF, Schiman HL, Kombrinck KW, Bugge TH, Degen JL, Kaufman AH: Persistent corneal haze after excimer laser photokeratectomy in plasminogen-deficient mice. Invest Ophthalmol Vis Sci 2000, 41:67-72 [PubMed] [Google Scholar]
- 43.Martin P: Wound healing—aiming for perfect skin regeneration. Science 1997, 276:75-81 [DOI] [PubMed] [Google Scholar]
- 44.Kojima S, Hayashi S, Shimokado K, Suzuki Y, Shimada J, Crippa MP, Friedman SL: Transcriptional activation of urokinase by the Kruppel-like factor Zf9/COPEB activates latent TGF-beta1 in vascular endothelial cells. Blood 2000, 95:1309-1316 [PubMed] [Google Scholar]