

Abstract
Endothelin-1 (ET-1) has been shown to be mitogenic for endothelial and several tumor cells through an autocrine mechanism. In this study we evaluated whether the tumorigenic KS IMM cell line deriving from Kaposi’s sarcoma (KS), a highly angiogenic tumor, is susceptible to ET-1 mitogenic activity. By reverse transcriptase-polymerase chain reaction, we detected ET-1 mRNA expression and both ETA receptor (ETAR) and ETBR mRNA transcripts in the KS IMM cells. High concentrations of ET-1 are released from the KS IMM cells and competition-binding studies demonstrated that these cells also express functional ETAR and ETBR with high affinity for ET-1 and ET-1/ET-3, respectively. Expression of ET-1 and cognate receptors could be detected by immunohistochemical method in vitro, in KS IMM xenograft, and in tissue sections of a human KS lesion. Furthermore ET-1 induces a marked and dose-dependent increase in [3H]thymidine incorporation comparable to that elicited by vascular endothelial growth factor. Addition of both selective ETBR antagonist (BQ 788) and ETAR antagonist (BQ 123), completely blocked ET-1-induced mitogenic response and reduced the basal growth rate of unstimulated cells, suggesting that both receptors mediated the proliferative signal. Such findings demonstrate that ET-1 participates on KS pathogenesis acting as an autocrine growth factor and that ET-1 receptor antagonists may thus be novel candidates for therapeutic intervention.
Kaposi’s sarcoma (KS) is a highly angiogenic tumor characterized by aberrant proliferation of vascular structures with proliferation of endothelial and spindle (tumor) cells and enhanced vascular permeability. 1 Angiogenic molecules produced by KS dictate the progression of the lesion by autocrine and paracrine mechanisms and are likely to play a central role in the development and progression of KS. 2,3 The KS-derived spindle cells induce vascular lesions when inoculated subcutaneously in the nude mouse 4 thus suggesting that these cells secrete factors that induce angiogenesis and enhance vascular permeability. The production in vitro of autocrine growth factors, which include basic fibroblast growth factor, interleukins 1 and 6, and vascular endothelial growth factor (VEGF), by the KS cells, has been previously reported. 5 Endothelin-1 (ET-1), is constitutively produced by endothelial cells and in elevated amounts in many tumors. 6,7 ET-1 acts through two distinct subtypes of G protein-coupled receptors, namely ETA and ETB. ETAR shows selectivity for ET-1 whereas ETBR binds both ET-1 and ET-3. In tumors, such as in ovarian and cervical carcinoma, ET-1 is overexpressed and acts as an autocrine growth factor selectively through ETAR, as demonstrated by the inhibitory effects induced by specific ETAR antagonists. 8-11 The observation that ET-1 is a mitogen for endothelial and tumor cells raises the possibility that ET-1 contributes to the pathogenesis of KS. In this report, we demonstrate that KS IMM, an immortalized KS-derived cell line that retains most of the features of the parental tumor and can induce KS-like sarcomas when injected subcutaneously in nude mice, 12 expresses in vitro and in vivo ETA and ETB receptors, and secretes the potent mitogenic peptide ET-1 that acts as an autocrine growth factor. These findings, together with the inhibitory effect of ET-1 receptor antagonists on cell proliferation, suggest that ET-1 plays an important role in the KS progression and represents a potential important target for therapeutic intervention of tumor growth.
Materials and Methods
Cell Cultures
KS IMM cells were derived from a non-AIDS patient and are immortalized without signs of senescence after more than 120 in vitro passages. 12 KS IMM were grown in Dulbecco’s modified Eagle’s medium and 10% fetal calf serum, supplemented with glutamine, penicillin, and streptomycin. All culture reagents were from GIBCO (Paisley, Scotland). Human umbilical vein endothelial cells were isolated from human umbilical vein (Promocell, Heidelberg, Germany) and maintained in endothelial cell growth medium kit containing 2% fetal calf serum (Promocell).
RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from the KS IMM cells by the guanidium thiocyanate-phenol chloroform extraction method. RT-PCR was performed using a geneAmp RNA PCR kit (Perkin-Elmer Corp., Norway, CT) according to the manufacturer’s instructions. Briefly, 1 μg of RNA was reverse-transcribed using the antisense primer. The primer sets were as follows: 1) ET-1, 5′-TGCTCCTGCTCGTCCCTGATGGATAAAGAG-3′ and 5′-GGTCACATAACGCTCTCTGGAGGGCTT-3′; 2) ETA, 5′-CACTG-GTTGGATGTGTAATC-3′ and 5′-GGAGATCAATGACCA- CATAG-3′; and 3) ETB, 5′-TGAACACGGTTGTGTCCTGC-3′and 5′-ACTGAATAGCCACCAATCTT-3′. 13 Glyceraldehyde-3-phosphate dehydrogenase was used as an internal control. The semiquantitative analysis was done essentially as described by Rieckmann and colleagues. 14 The amplified products were analyzed in a 3% agarose gel and visualized by ethidium bromide. In all experiments, two control reactions, one containing no RNA and another containing RNA but no reverse transcriptase, were included. All 5′ primers covered splice junctions, thus excluding the amplification of genomic DNA.
ET-1 Enzyme-Linked Immunosorbent Assay
ET-1 in the conditioned medium was measured using a ET-1 immunoassay kit (R&D, Minneapolis, MN), following the manufacturer’s instructions. The working range in the enzyme-linked immunosorbent assay for ET-1 assay was 0 to 120 pg/ml. The cross-reactivity of the antiserum for ET-1-related peptides (versus ET-1 = 100%) was: ET-2, 45%; ET-3, 14%; big ET-1, <1%; and sarafotoxin, <2%.
Receptor Binding Assay
KS IMM cells were cultured in 6-well plates until confluent (5 × 10 5 cells/well) and then serum starved for 24 hours. After a wash with assay buffer composed of Hanks’ balanced salt solution, 0.2% bovine serum albumin, and 100 μg/ml bacitracin (Sigma, St. Louis, MO), cells were incubated at 25°C for 60 minutes in 500 μl of assay buffer with increasing concentrations of the radioactive tracer in the presence or absence of an excess of unlabeled ET-1 (1 μmol/L; Peninsula, Belmont, CA). For binding inhibition studies, cells were incubated with tracer (40 pmol/L) and increasing concentrations of ET-1 and ET-3 (Peninsula). At the end of the incubation period, cells were analyzed for bound radioactivity in a γ-spectrometer. ET-1 receptor affinity and the number of binding sites were determined by Scatchard analysis using the LIGAND program.
Thymidine Incorporation Assay
Cells were seeded in 96-well plates to ∼80% confluence (2 × 10 4 cells/well) and incubated in serum-free medium for 24 hours to induce quiescence. VEGF165 (R&D), ET-1, or antagonist (Peninsula) were then added, and after 18 hours, when the cells were confluent, 1 μCi [methyl-3H]thymidine (6.7 Ci/mmol; DuPont, New England Nuclear Research Products, Wilmington, DE) was added to each well. Six hours later the cell-associated radioactivity was determined by liquid scintillation counting.
Cell Proliferation Assay
KS IMM were seeded at 5 × 10 4 cells/well in 12-well plates, cultured for 24 hours, serum-starved for additional 24 hours, and then incubated in 2 ml of serum-free medium in the absence or presence of 1 μmol/L BQ 123 or 1 μmol/L BQ788 or BQ123 + BQ788. At the indicated times, cells were dispersed by trypsinization and counted in a hemocytometer.
KS-Like Lesion Formation by KS IMM Cells in Nude Mice
Healthy 10-week-old athymic nu/nu female mice were obtained from Charles River Laboratories (Como, Italy). KS IMM cells (3 × 106) were inoculated subcutaneously into the lower back of mice. After 15 days from the injection, specimens were taken from the lesional sites, frozen, and ET-1 and ETR expression was analyzed immunohistochemically.
Evaluation of ET-1 and Its Receptors by Immunohistochemistry
Serial frozen sections of a case of KS skin lesions were analyzed by immunohistochemistry. Antibodies (Abs) used were a mouse monoclonal Ab (clone TR.E.T. 48.5; Affinity Bioreagents, Golden, CO) at 1:200 dilution for ET-1 and two rabbit polyclonal antipeptide Abs (a generous gift from Dr. R. Wu-Wong, Abbott, IL) at 1:20 dilution for ETAR and ETBR. For ETAR was an Ab raised against a decapeptide (DNPERYSTNL) of the extracellular NH2-terminal domain of ETAR, for ETBR was an Ab raised against a peptide (CGLSRIWGEERGFPPDRTP) of the NH2-terminal domain of ETBR. Antibody binding was revealed using peroxidase-labeled anti IgG (Vector Laboratories, Burlingame, CA). Reactions were developed using 3-amino-9-ethylcarbazole as the chromogen and sections were counterstained with hematoxylin. To ensure specificity, the primary Ab was preabsorbed 12 hours at 4°C with 50-fold excess of synthetic ET-1 (Peninsula) or substituted with preimmune rabbit serum (negative control for ETAR and ETBR).
Results
KS IMM Cells Express High mRNA Levels of ET-1 and Its Receptors
Using RT-PCR, we examined the expression of ET-1 and its receptor mRNA in KS IMM cell line. RT-PCR-amplified cDNA fragments for ET-1, ETAR, and ETBR that were detectable in the KS IMM cells are shown in Figure 1A ▶ . The amplified 462-bp ET-1, 367-bp ETAR, and 529-bp ETBR PCR cDNA fragments were of the predicted molecular size. Human umbilical vein endothelial cells also express ET-1 and its receptors mRNA (Figure 1A) ▶ . All samples showed a positive signal for glyceraldehyde-3-phosphate dehydrogenase mRNA, (data not shown), demonstrating that negative results were not because of a degradation of mRNA. The specificity of RT-PCR amplification products for ET-1, ETAR, and ETBR was confirmed by Southern blot (data not shown), revealing the same pattern and intensity of the bands and showing an absence of cross-reactivity.
Figure 1.

A: ET-1, ETAR, and ETBR mRNAs detected by RT-PCR in the KS IMM cells and human umbilical vein endothelial cells. PCR products of 462 bp for ET-1 (lane 1), 367 bp for ETAR (lane 2), and 523 bp for ETBR (lane 3) are shown as visualized by ethidium bromide in the KS IMM and in the human umbilical vein endothelial cells. B: Release of ET-1 from the KS IMM cell line as a function of time. Cells were incubated for 24 hours in serum-free medium before the experiment. The conditioned medium was collected at the indicated times. Each point represents the mean value of data from three independent experiments, each of which was performed in duplicate. Bars, ±SD.
Production of ET-1 by the KS IMM Cells
The levels of ET-1 mRNA in the KS IMM cells are mirrored in their ET-1 secretion. KS IMM cells release high amounts of ET-1 in the culture media as a function of time throughout a 48-hour period, with detectable production as early as 6 hours after the replacement of the culture medium and maximum production at 48 hours (106 ± 12.4 pg/10 6 cell) (Figure 1B) ▶ that persisted for at least 72 hours (data not shown).
Expression of ET Receptors in the KS IMM Cells
KS IMM cells expressed detectable cell surface binding sites with high affinity for 125I-labeled ET-1. Specific binding of 125I-labeled ET-1 to the KS IMM cells occurred rapidly and was time- and temperature-dependent (data not shown). Nonspecific binding, measured in the presence of 1 μmol/L unlabeled peptide, was less than 20% of the total binding. Competition for 125I-labeled ET-1 binding to KS IMM cells by unlabeled ET-1 and ET-3 is shown in Figure 2A ▶ . Whereas the half-maximal inhibitory concentration of ET-1 was 150 pmol/L, that of ET-3 was 350 nmol/L. Therefore, [125I]ET-1 labels a receptor that has high affinity for ET-1 and that can be defined by the following pharmacological profile that defines the ETA receptor: ET-1 > ET-2 ≫ ET-3. Conversely, the competitive inhibition of [125I]ET-3 binding by ETs occurs with the same effectiveness with increasing amounts of unlabeled ET-3 and ET-1, thus also demonstrating the high affinity ET-3 receptor: ETB (Figure 2B) ▶ . Therefore, binding data obtained in the KS IMM cells revealed for ETA receptor a kd of 0.15 nmol/L for ET-1 with estimated binding sites averaging 43,539 sites/cell, and for the ETB receptor a Kd of 0.3 nmol/L for both ET-1 and ET-3, with a receptor capacity of 18,147 sites/cell.
Figure 2.
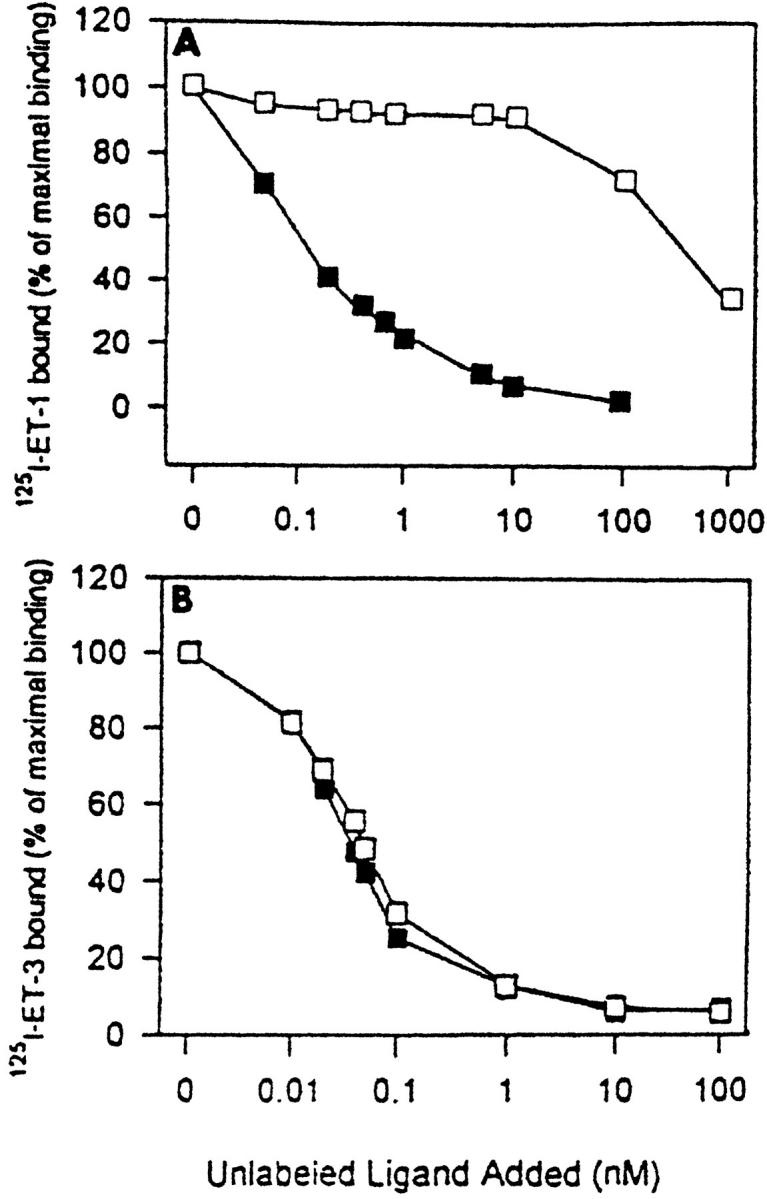
Specific displacement curves of [125I]-ET-1 and [125I]ET-3 binding by unlabeled ET-1 and ET-3 in the KS IMM cells. A: Inhibition of [125I]-ET-1 binding by increasing concentrations of ET-1 (filled square) and ET-3 (open square). B: The competitive inhibition of [125I]-ET-3 binding by increasing concentrations of ET-1 (filled square) and ET-3 (open square). Binding is expressed as a percentage of bound radioligand in the absence of unlabeled peptide. The data are the mean of triplicate determinations and are representative of three independent experiments.
Proliferative Effects of ET-1 on the KS IMM Cells
When the KS IMM were incubated in serum-free medium with increasing concentrations (0.1 nmol/L to 100 nmol/L) of ET-1 for 24 hours, [3H]thymidine incorporation was markedly stimulated in a dose-dependent manner with a maximal effect at 100 nmol/L (70% increase over control, P < 0.0001). In the same experiment, 100 nmol/L of selective ETB agonists, ET-3, and sarafotoxin 6c (S6c) induced mitogenic effects similar to that induced by ET-1, indicating that both ET-1 and ET-3 are capable of stimulating proliferative responses in the KS IMM cells (Figure 3A) ▶ . Because previous studies have shown that VEGF is able to induce mitogenic effect on the KS-derived cells, 15 we compared the mitogenic effects of ET-1 and VEGF on the KS IMM cells. In this study, ET-1 (100 nmol/L) was equipotent with VEGF (10 ng/ml) in stimulating mitogenesis. Moreover, because ET-1 stimulated mitogenic responses in the KS IMM cell line, we evaluated which receptor subtype mediated the ET-1 mitogenic signaling by co-incubating the cells with ET-1 and ETA receptor antagonist BQ 123 and with the ETB receptor antagonist BQ 788. In this experiment, the stimulatory action of 100 nmol/L ET-1 on [3H]thymidine incorporation by the KS IMM cells was partially inhibited in the presence of 1 μmol/L BQ 123 or BQ 788 (Figure 3B) ▶ . In the same experiment, exposure of the cells to BQ 788 in combination with BQ 123, so as to inhibit both ETA and ETB receptors, completely abolished the ability of the KS IMM cells to respond to ET-1. The KS IMM cell proliferation induced by VEGF was not inhibited by both the addition of BQ 788 and BQ 123, indicating that the inhibitory effect induced by these antagonists was not because of cytotoxicity (data not shown). Taken together, these data indicate that mitogenic signaling by ET-1 is mediated by both ETA and ETB receptor subtype.
Figure 3.

A: Stimulatory actions of ET-1 and VEGF on [3H]thymidine incorporation. Quiescent KS IMM cells (2 × 104) were treated with different concentrations (0.1 nmol/L up to 100 nmol/L) of ET-1 or with ETB agonists, ET-3 (100 nmol/L) and S6c (100 nmol/L), or with VEGF (10 ng/ml) for 24 hours before measuring [3H]thymidine incorporation as described in Materials and Methods. a, P < 0.0001 compared to control; b, P < 0.05 compared to control. B: Effects of ET-1 receptor antagonists on ET-1-stimulated incorporation of [3H]thymidine. Quiescent KS IMM cells were treated with ETAR antagonist, BQ 123 (1 μmol/L), or ETBR antagonist, BQ 788 (1 μmol/L), or with a combination of both antagonists in the absence or presence of 100 nmol/L of ET-1. Bars, mean ± SD of data from eight independent experiments each of which was performed in sextuplicate. a, P ≤ 0.0001 compared to control; b, P ≤ 0.005 compared to ET-1; c, P ≤ 0.0001 compared to ET-1; d, P ≤ 0.0001 compared to BQ 123 + ET-1 and to BQ 788 + ET-1. C: Effects of ET-1 receptor antagonists on the spontaneous growth rate of the KS IMM cells. To evaluate the autocrine action of ET-1 on cell growth, the KS IMM cells were seeded at 5 × 10 4 cells/well in 12-well plates and cultured in serum-free medium in the absence (filled diamond) or presence of 1 μmol/L BQ 123 (filled circle) or 1 μmol/L BQ788 (filled triangle) or BQ123 +BQ788 (filled square), and cell numbers were measured at the indicated times in a hemocytometer. Bars, ± SD. *, P < 0.02; **, P < 0.004 compared to control.
In addition to defining the receptor-mediated action on thymidine incorporation by the KS IMM cells, we also determined the extent to which such growth responses were manifested as an increase in cell proliferation (Figure 3C) ▶ . To determine whether endogenous ET-1 produced by the KS IMM cells could drive KS proliferation, cells were incubated, under serum-free conditions, for up to 72 hours in the absence or presence of ET-1 receptor antagonists. Addition of 1 μmol/L of BQ123 and 1 μmol/L of BQ788 significantly inhibited the spontaneous growth rate of the KS IMM cells in response to endogenous ET-1, demonstrating that ET-1 acts as an autocrine regulator of the KS cell proliferation.
Expression of ET-1 and Its Receptors in Vitro and in Vivo
In all nude mice that were subcutaneously inoculated with 3 × 10 6 KS IMM cells, a lesion developed at the site of inoculation within 5 to 7 days. After 15 days from the injection, specimens were taken from the lesional sites. Histologically, the neoplastic lesion consisted of round and spindle cells, with vascular structures and capillaries and some infiltrated inflammatory cells. We analyzed the presence of ET-1, ETA, and ETB receptors by immunohistochemistry (Figure 4) ▶ . A strong cytoplasmic staining for ET-1, ETA, and ETB receptors was observed in the cultured KS IMM cells (Figure 4; A, B, and C ▶ ) and a similar pattern of expression of ET-1 and its cognate receptors was detected in the KS IMM-derived lesions in nude mice (Figure 4; D, E, and F ▶ ) demonstrating that in vitro and in vivo KS-derived cells co-expressed ETA and ETB receptors. To test whether the expression of ET-1 and its receptor could have a role a pathophysiological function in KS, we extend our observations to human specimens of primary KS (Figure 4; G, H, and I ▶ ). In human KS lesions, spindle cells were found to express ET-1 protein. Both tumor cells and vessels surrounding and invading the tumor express ETAR and ETBR, suggesting that ET-1 could have a bifunctional role, both acting as an autocrine growth factor on the KS spindle cells and favoring the vascularization of the lesions. Incubation with preimmune rabbit serum did not give a detectable staining (not shown).
Figure 4.
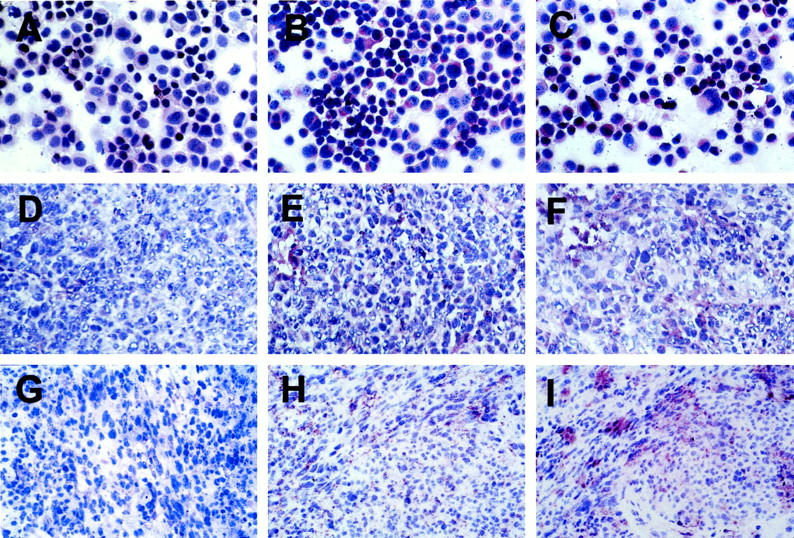
Expression of ET-1, ETAR, and ETBR in the KS IMM cultured cells, in nude mice lesions induced by the KS IMM cells, and in human specimens of primary KS. Immunohistochemical staining of ET-1 (A), ETAR (B), and ETBR (C) in the cultured KS IMM cells. Tissue specimens of in vivo lesions induced after 15 days by KS IMM injection were stained by ET-1 mAb (D), ETAR Ab (E), and ETBR Ab (F). Expression of ET-1 (G), ETA (H), and ETB (I) receptors in a section of human primary KS. Original magnification, ×400; ABC-peroxidase, counterstained with hematoxylin.
Discussion
An abnormal expression of different angiogenic factors has been demonstrated to be involved in the pathogenesis of KS. 3,15,16 This report indicates that ET-1 is an autocrine growth factor for KS through its receptors, which could be targeted for therapy. The KS cell line produces abundant amounts of ET-1 protein. Elevated expression of ET-1 has been reported in many tumors 8,9,17,18 and using an ET-1-specific assay we have, for the first time, demonstrated that ET-1 is produced by the KS IMM cells and accumulates in culture media to biologically active levels. RT-PCR revealed that the KS cell line that expresses ET-1 co-express mRNA encoding ETAR and ETBR mRNA. Competitive displacement experiments demonstrated that both ETAR and ETBR subtypes are the functional receptor present in the KS cell line producing ET-1. We further characterized by immunohistochemistry the expression of the ET receptors showing that in vitro and in vivo the KS IMM cells and human primary tumor of KS express ET-1, ETAR, and ETBR protein.
ET-1 elicits biological responses by binding to two separate subtypes, ETAR and ETBR, that are differentially expressed in a wide variety of tissues. 6 The signal transduction of the mitogenic response in endothelial cells seems to occur exclusively through ETBR. 19 Based on the phenotype of the KS cells that share markers with endothelial cells, we hypothesized that the KS IMM may also express ETBR. Because ETAR is the predominant receptor expressed in various tumors but not in their normal counterparts, 17,18 expression of both ETAR and ETBR in the KS cell line suggests that this cell line has a mixed tumor-endothelial phenotype. 20
In the present study, addition of endothelin caused a consistent and dose-dependent increase in DNA synthesis in the KS IMM cells. High concentrations of ET-1 and VEGF stimulated similar increases in thymidine incorporation, demonstrating that ET-1 is as effective as a typical angiogenic factor such as VEGF, which stimulates growth response in the KS through an autocrine mechanism. 15 The demonstration of receptors in cells that produce as well as require ET-1, provides evidence for an ET-1 autocrine loop in KS. In the KS IMM cells, mitogenic effects induced by ET-1 were partially inhibited by BQ 123 or BQ 788, whereas addition of the two antagonists completely inhibited ET-1 stimulated growth, suggesting the involvement of both receptor types in transducing the proliferative signal and that ET-1 is involved in the growth control of KS. Furthermore this study suggests that ET-1 exerts a significant autocrine action in the growth control of KS, as demonstrated by the decrease of spontaneous cell proliferation in the presence of ET-1 receptor antagonists.
We further characterized the expression of ET-1 and its receptors in human sections of KS lesions by immunohistochemistry. The intense in vivo immunostaining signals for ETAR and ETBR were localized in tumor cells and on some vessels adjacent to nests of tumor cells. These results raise the possibility that ET-1 may act as a growth factor by autocrine mechanism on receptor-expressing spindle (tumor) cells and also through a paracrine mechanism involving angiogenic effects on vascular elements. It has been shown that immunohistochemically detectable ET-1 expression in human gliomas correlates with the degree of tumor vascularity. 21
Recently, we demonstrated that ET-1 induces angiogenic responses including proliferation, migration, invasion, and protease production and morphogenesis in cultured endothelial cells predominantly through ETBR, and that by acting in concert with VEGF, ET-1 stimulates angiogenesis in vivo. 22 Moreover, in tumors such as ovarian carcinoma, in which ET-1 acts as an autocrine growth factor selectively via ETAR 9 and where the presence of ET-1 correlates with neovascularization and with VEGF expression, ET-1 and VEGF play coordinated roles during neovascularization and malignant ascites formation. 23 These observations together with the results reported in this work strongly suggest that ET-1 acting as growth factor could promote KS lesions by regulating functions of both KS spindle cells and associated endothelial cells.
In addition to the VEGF family, basic fibroblast growth factor, placental growth factor, the identification of ET-1 as an autocrine growth factor expands our knowledge of the biology of KS. We therefore hypothesize that the complex process of angiogenesis as well as of tumor progression require the cooperation of multiple factors and the overexpression of some key members, as ET-1 and VEGF, is able to trigger both processes. New therapeutic strategies using specific antagonists for ET receptors 24 provide an additional approach for the treatment of malignancies characterized by active angiogenesis, as KS, and of other angiogenesis-dependent diseases.
Footnotes
Address reprint requests to Anna Bagnato, Laboratory of Molecular Pathology and Ultrastructure, Regina Elena Cancer Institute, Via delle Messi d’Oro 156, 00158 Rome, Italy. E-mail: bagnato@ifo.it.
Supported by grants from the Associazione Italiana Ricerca sul Cancro.
Dr. Laura Rosanò and Dr. Debora Salani are recipients of fellowships from Fondazione Italiana Ricerca sul Cancro.
References
- 1.Ruszczak Z, Mayer DA, Silva AM, Orfanos CE: Kaposi’s sarcoma in AIDS. Multicentric angioneoplasia in early skin lesions. Am J Dermatopathol 1987, 9:388-398 [PubMed] [Google Scholar]
- 2.Salahuddin SZ, Nakamura S, Bebergold P, Kaplan MM, Markham PD, Larsson L, Gallo RC: Angiogenic properties of Kaposi’s sarcoma-derived cells after long-term culture in vitro. Science 1988, 242:430-433 [DOI] [PubMed] [Google Scholar]
- 3.Ensoli B, Nakamura S, Salahuddin SZ, Biberfeld P, Larsson L, Beaver B, Wong-Staal F, Gallo RC: AIDS-Kaposi’s sarcoma-derived cells express cytokines with autocrine and paracrine growth effects. Science 1989, 243:223-226 [DOI] [PubMed] [Google Scholar]
- 4.Lunardi-Iskandar Y, Gill P, Lam VH, Zeman RA, Michaels F, Mann DL, Reitz MS, Kaplan M, Berneman ZN, Carter D: Isolation and characterization of an immortal neoplastic cell line (KS Y-1) from AIDS-associated Kaposi’s sarcoma. J Natl Cancer Inst 1995, 87:974-981 [DOI] [PubMed] [Google Scholar]
- 5.Ensoli B, Barillari G, Gallo RC: Cytokines and growth factors in the pathogenesis of AIDS-associated Kaposi’s sarcoma. Immunol Rev 1992, 127:147-155 [DOI] [PubMed] [Google Scholar]
- 6.Levin ER: Endothelins. N Engl J Med 1995, 333:356-363 [DOI] [PubMed] [Google Scholar]
- 7.Bagnato A, Catt KJ: Endothelins as autocrine regulators of tumor cell growth. Trends Endocrinol Metab 1998, 9:378-383 [DOI] [PubMed] [Google Scholar]
- 8.Bagnato A, Tecce R, Moretti C, Di Castro V, Spergel DJ, Catt KJ: Autocrine actions of endothelin-1 as a growth factor in human ovarian carcinoma cells. Clin Cancer Res 1995, 1:1059-1066 [PubMed] [Google Scholar]
- 9.Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra MR, Venuti A, Natali PG: Expression of endothelin-1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res 1999, 59:1-8 [PubMed] [Google Scholar]
- 10.Bagnato A, Tecce R, Di Castro V, Catt KJ: Activation of mitogenic signaling by endothelin-1 in ovarian carcinoma cells. Cancer Res 1997, 57:1306-1311 [PubMed] [Google Scholar]
- 11.Venuti A, Salani D, Manni V, Poggiali F, Bagnato A: Expression of endothelin-1 and endothelin A receptor in HPV associated cervical carcinoma: new potential target for anticancer therapy. FASEB J 2000, 14:2277-2283 [DOI] [PubMed] [Google Scholar]
- 12.Albini A, Paglieri I, Orengo G, Carbone S, Aluigi MG, De Marchi R, Matteucci C, Mantovani A, Carozzi F, Donini S, Bonelli R: The β-core fragment of human chorionic gonadotropin (hCG) inhibits growth of Kaposi’s sarcoma-derived cells and a new immortalized Kaposi’s sarcoma cell line. AIDS 1997, 11:713-721 [DOI] [PubMed] [Google Scholar]
- 13.Pekonen F, Nyman T, Ammala M, Rutanen EM: Decreased expression of messenger RNAs encoding endothelin receptors and neutral endopeptidase 24.11 in endometrial cancer. Br J Cancer 1992, 71:59-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rieckmann P, Albrecht M, Ehrenreich H, Weber T, Michel U: Semiquantitative analysis of cytokine gene expression in blood and cerebrospinal fluid cells by reverse transcriptase polymerase chain reaction. Res Exp Med 1995, 195:17-29 [DOI] [PubMed] [Google Scholar]
- 15.Masood R, Cai J, Zheng T, Smith DL, Naidu Y, Gill PS: Vascular endothelial growth factor/vascular permeability factor is an autocrine growth factor for AIDS-Kaposi sarcoma. Proc Natl Sci Acad Sci USA 1997, 94:979-984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ensoli B, Sturzl M: Kaposi’s sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev 1998, 9:63-83 [DOI] [PubMed] [Google Scholar]
- 17.Nelson JB, Hendican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, Simons JW: Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med 1996, 1:944-949 [DOI] [PubMed] [Google Scholar]
- 18.Nelson JB, Chan-Tack K, Hendican SP, Magnuson SM, Opgenorth TJ, Bova GS, Simons JW: Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res 1996, 56:663-668 [PubMed] [Google Scholar]
- 19.Morbidelli L, Orlando C, Maggi CA, Ledda F, Ziche M: Proliferation and migration of endothelial cells is promoted by endothelins via activation of ETB receptors. Am J Physiol 1995, 269:4686-4695 [DOI] [PubMed] [Google Scholar]
- 20.Uccini S, Ruco L, Monardo F, Stoppacciaro A, Dejana E, La Parola I, Cerimele D, Baroni C: Co-expression of endothelial cells and macrophage antigens in Kaposi’s sarcoma cells. J Pathol 1994, 173:23-31 [DOI] [PubMed] [Google Scholar]
- 21.Stiles JD, Ostrow PT, Balos LL, Greenberg SJ, Plunkett R, Grand W, Heffner RR: Correlation of endothelin-1 and transforming growth factor β1 with malignancy and vascularity in human gliomas. J Neuropathol Exp Neurol 1997, 56:435-439 [DOI] [PubMed] [Google Scholar]
- 22.Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A: Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol 2000, 157:1703-1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salani D, Di Castro V, Nicotra MR, Rosanò L, Tecce R, Venuti A, Natali PG, Bagnato A: Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol 2000, 157:1537-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lahav R, Heffner G, Patterson PH: An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci USA 1999, 96:11496-11500 [DOI] [PMC free article] [PubMed] [Google Scholar]