

Summary
Transrepression is widely utilized to negatively regulate gene expression, but the mechanisms by which different nuclear receptors effect gene- and signal-specific transrepression programs remain poorly understood. Here, we report the identification of alternative SUMOylation-dependent mechanisms that enable PPARγ and LXRs to negatively regulate overlapping but distinct subsets of pro-inflammatory genes. Ligand-dependent conjugation of SUMO2/3 to LXRs or SUMO-1 to PPARγ targets them to promoters of TLR target genes, where they prevent the signal-dependent removal of NCoR corepressor complexes required for transcriptional activation. SUMO-1-PPARγ and SUMO-2/3-LXRs inhibit distinct NCoR clearance mechanisms, allowing promoter- and TLR-specific patterns of repression. Mutational analysis and studies of naturally occurring oxysterol ligands indicate that the transactivation and SUMOylation-dependent transrepression activities of LXRs can be independently regulated. These studies define parallel but functionally distinct pathways that are utilized by PPARγ and LXRs to differentially regulate complex programs of gene expression that control immunity and homeostasis.
INTRODUCTION
Members of the nuclear-receptor family of ligand-dependent transcription factors influence immune responses by positively and negatively regulating gene expression in diverse cell types, including macrophages, lymphocytes, and dendritic cells (Glass and Ogawa, 2006; Winoto and Littman, 2002). Recent studies of the glucocorticoid receptor (GR), liver X receptors (LXRs), peroxisome proliferator-activated receptor γ (PPARγ), retinoic acid receptor (RAR), and estrogen receptors (ERs) revealed that each nuclear receptor represses an overlapping, but distinct, pattern of signal-dependent inflammatory gene expression (Galon et al., 2002; Joseph et al., 2003; Ogawa et al., 2005; Welch et al., 2003). While these observations are consistent with the overlapping but distinct effects of specific ligands on inflammatory processes at a biological level, the molecular mechanisms that are utilized to achieve this specificity remain poorly understood.
LXR α and β are members of the nuclear receptor superfamily that play essential roles in cholesterol and fatty acid homeostasis (Janowski et al., 1996; Peet et al., 1998; Schultz et al., 2000). LXRs are required for normal cholesterol efflux from peripheral cells and cholesterol excretion by the liver by positively regulating the expression of genes involved in cholesterol efflux (Costet et al., 2000; Repa et al., 2000) and genes involved in bile acid biosynthesis and transport (Chiang et al., 2001; Lewis and Rader, 2005). The transcriptional activities of LXRs are thought to be regulated in vivo by oxysterols, oxygenated derivatives of cholesterol (Janowski et al., 1999; Janowski et al., 1996).
Recent studies have characterized LXRs as regulators of innate immunity and inflammatory signalling in macrophages (Zelcer and Tontonoz, 2006). LXR agonists enhance macrophage survival during bacterial infection and LXRs are required for normal immunity to Listeria monocytogenes (Castrillo et al., 2003; Joseph et al., 2004; Joseph et al., 2003; Valledor et al., 2004). In addition, LXR activation inhibits the lipopolysaccharide (LPS) induction of a number of pro-inflammatory genes such as inducible nitric oxide synthase (iNOS), cytokines such as interleukin-1β (IL-1β), and chemokines such as monocyte chemoattractant protein-1 (MCP-1) (Joseph et al., 2003). Repression of inflammatory mediators by LXR agonists is abolished in macrophages lacking both LXRs, but not in macrophages containing either LXRα or LXRβ, indicating that both isoforms can mediate anti-inflammatory activity (Joseph et al., 2003). The molecular mechanisms underlying the repression of inflammatory-response genes by LXRs remain to be established.
PPARγ, a nuclear receptor required for fat cell development and normal glucose homeostasis (Spiegelman, 1998; Willson et al., 2001), also exerts broad anti-inflammatory effects in macrophages and other cell types (Delerive et al., 1999; Glass and Ogawa, 2006; Jiang et al., 1998; Ricote et al., 1998), and this activity has been correlated with insulin-sensitizing and anti-atherogenic activities of synthetic PPARγ agonists (Haffner et al., 2002; Li et al., 2000; Staels and Fruchart, 2005). Recent studies of the mechanism by which PPARγ represses activation of the iNOS gene by LPS led to the identification of a SUMOylation-dependent transrepression pathway (Pascual et al., 2005). In this pathway, synthetic PPARγ agonists promote PIAS1-dependent conjugation of SUMO-1 to the PPARγ ligand-binding domain. This in turn targets PPARγ to NCoR corepressor complexes that are normally bound to the iNOS promoter under basal conditions and are required to mediate active repression in the absence of an inflammatory stimulus. The recruitment of SUMO1-PPARγ was found to prevent the signal-dependent ubiquitylation and clearance of the NCoR complex required for full gene activation, resulting in the iNOS gene remaining in a repressed state (Pascual et al., 2005). These findings raised a series of questions, including whether this pathway is used by other nuclear receptors, whether it can be utilized to mediate repression in a gene or signal-specific manner, and whether natural ligands can promote entry of their cognate nuclear receptors into this pathway.
Here, we demonstrate that LXR transrepression of inflammatory target genes also utilizes a SUMOylation and NCoR-dependent pathway, indicating that this mechanism represents a general molecular strategy for transrepression by a subset of nuclear receptors that regulate metabolism and immunity. In contrast to PPARγ, however, LXRs are SUMOylated by SUMO2 and SUMO3 rather than SUMO1, and HDAC4 rather than PIAS1 functions as a required SUMO E3 ligase. As a consequence, transrepression by PPARγ and LXRs can be independently regulated in a signal- and gene-specific manner.
RESULTS
LXR-dependent transrepression requires NCoR
Based on the requirement of NCoR for transrepression by PPARγ, we initially sought to determine whether LXR-mediated repression is also NCoR-dependent. Experiments were performed in primary macrophages transfected with control non-specific or NCoR-specific siRNAs (Fig. S1A), using GW3965 as an LXR-specific agonist. Reduction of NCoR expression significantly reversed the LXR ligand-dependent repression of iNOS expression induced by LPS (Fig. 1A). To confirm this result, iNOS promoter activity was evaluated in RAW264.7 macrophages. As shown in Fig. 1B, knock down of NCoR reversed the LXRα and LXRβ-dependent transrepression of iNOS promoter activity induced by LPS. To determine whether LXRs are recruited to the iNOS promoter and prevents NCoR clearance, chromatin immunoprecipitation (ChIP) experiments were performed examining LXR and NCoR occupancy on the iNOS promoter in primary LPS-stimulated macrophages. LXRs were recruited to the iNOS promoter in a ligand-dependent manner in the presence or absence of LPS and this recruitment was NCoR dependent (Fig. 1C). LXRs were not detected in double LXRα/LXRβ knockout macrophages, indicating that the LXR antibody specifically detected LXRα/LXRβ bound to the iNOS promoter. As expected, NCoR was found to occupy the iNOS promoter under basal conditions and to be dismissed by LPS (Pascual et al., 2005), while treatment with GW3965 prevented NCoR clearance(Fig.1D). These results suggest that LXRα and LXRβ, in addition to PPARγ, repress LPS induction of the iNOS gene by preventing the clearance of NCoR.
Figure 1. LXR transrepression requires NCoR.
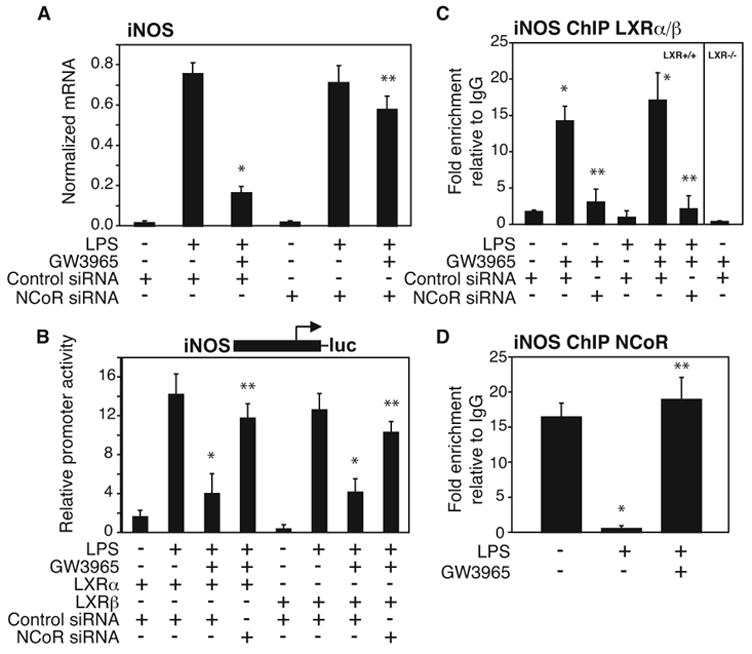
NCoR-specific siRNA abolishes LXR-dependent repression of LPS-induced endogenous iNOS gene expression in primary macrophages (A) and iNOS-luciferase reporter gene activity in RAW264.7 cells (B). After siRNA transfection, macrophages were stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligand (GW3965, 1μM). iNOS expression in peritoneal macrophages was evaluated by QPCR and iNOS promoter activation in RAW264.7 cells was evaluated by luciferase assay. *p < 0.03 vs LPS+control siRNA; **p < 0.02 vs GW3965+LPS control siRNA. C. ChIP assay reveals ligand-dependent and NCoR-dependent LXR recruitment to the iNOS promoter. Primary macrophages were treated with LPS (1μg/ml) with or without the LXR ligand (GW3965, 1μM) for 1h. ChIP assay was performed with antibody against LXR (both isoforms, α and α) or control IgG. Immunoprecipitated DNA was analyzed by QPCR using primers specific for the iNOS-promoter. Chromatin isolated from LXR−/−mice was used as a negative control for specificity of the LXR antibody. * p < 0.05 vs untreated or LPS alone; **p < 0.03 vs GW3965 or GW3965+LPS control siRNA. D. LXR ligand inhibits LPS-stimulated release of NCoR from the iNOS promoter as shown by ChIP assay in peritoneal macrophages, treated as described in panel C. * p < 0.02 vs untreated; **p < 0.02 vs LPS. Results are expressed as the average of three independent experiments. Error bars represent standard deviations.
LXR-dependent transrepression is SUMOylation dependent
The identification of LXR transrepression as NCoR dependent raised the question of whether a SUMOylation-dependent pathway was also involved. The ligation of SUMO to target proteins requires the E2-conjugating enzyme Ubc9 (Hay, 2005). We therefore knocked down Ubc9 expression using a specific siRNA (Fig. S1B) in primary macrophages (Fig. 2A) and in RAW264.7 cells (Fig. 2B) transfected with LXRα and LXRβ expression plasmids. Knockdown of Ubc9 significantly impaired LXR ligand-dependent repression of LPS-induced iNOS expression (Fig. 2A) and iNOS promoter activation (Fig. 2B). ChIP assays were next performed in primary macrophages to determine the role of SUMOylation in ligand-dependent recruitment of LXRα and LXRβ to the iNOS promoter and in the prevention of NCoR clearance. Knockdown of Ubc9 expression prevented ligand-dependent recruitment of LXR to the iNOS promoter (Fig. 2C) and impaired the block in NCoR clearance from the iNOS promoter (Fig. 2D). These results suggest that a SUMOylation-dependent step is critical for the recruitment of LXR to the iNOS promoter to prevent corepressor clearance.
Figure 2. LXR-dependent transrepression is SUMOylation dependent.
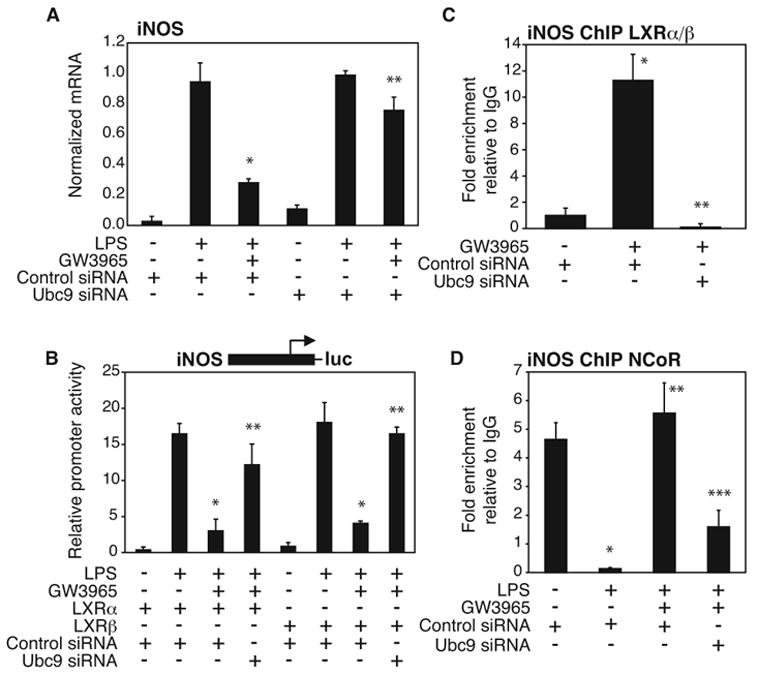
Ubc9-specific siRNA reverses LXR-dependent repression of LPS-induced iNOS expression in primary macrophages (A) and iNOS promoter activation in RAW264.7 cells (B). After siRNA transfection, macrophages were stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligand (GW3965, 1μM). iNOS expression in peritoneal macrophages was evaluated by QPCR and iNOS promoter activation in RAW264.7 was evaluated by luciferase assay. * p < 0.03 vs LPS control siRNA; **p < 0.02 vs GW3965+LPS control siRNA. C,D. siRNA directed against Ubc9 prevents LXR recruitment (C) and abolishes the ability of the LXR agonist to inhibit the NCoR clearance (D) from the iNOS promoter as shown by ChIP. Primary macrophages were treated with LPS (1μg/ml) with or without the LXR ligand (GW3965, 1μM) for 1h. ChIP assay was performed with antibody against LXR or NCoR or control IgG. Immunoprecipitated DNA was analyzed by real time PCR using primers specific for the iNOS-promoter. (C) * p < 0.01 vs untreated + control siRNA; **p < 0.02 vs GW3965 control siRNA. (D) * p < 0.02 vs untreated + control siRNA; **p < 0.02 vs LPS control siRNA, * **p < 0.01 vs GW3965 + LPS control siRNA. Results are expressed as average of 2 independent experiments. Error bars represent standard deviations.
LXRs are modified by SUMO-2/3 dependent on HDAC4 E3 ligase activity
Since a SUMOylation-dependent step is required for LXR transrepression, we tested whether LXR itself was a target of SUMOylation. FLAG-tagged LXRβ was expressed in HeLa cells in the presence or absence of Myc-tagged SUMO-1, SUMO-2 or SUMO-3. High-molecular-weight LXRβ, with an increased size expected for monoSUMOylation, was detected in the presence of SUMO-2 and SUMO-3, but not in the presence of SUMO-1 (Fig. 3A) in a ligand-dependent manner. This high molecular weight species was detected by anti-Myc antibody, and was abolished by knockdown of Ubc9 (Fig. 3B), indicating that it corresponds to SUMOylated LXRβ. LXRα was also SUMOylated by SUMO-2 and SUMO-3 (data not shown). An additional LXR-dependent band indicated by asterisks in Fig. 3, was also observed in some experiments. This band likely represents an unknown posttranslational modification of LXR that is not correlated with SUMOylation.
Figure 3. LXRβ is modified by SUMO-2 and SUMO-3.
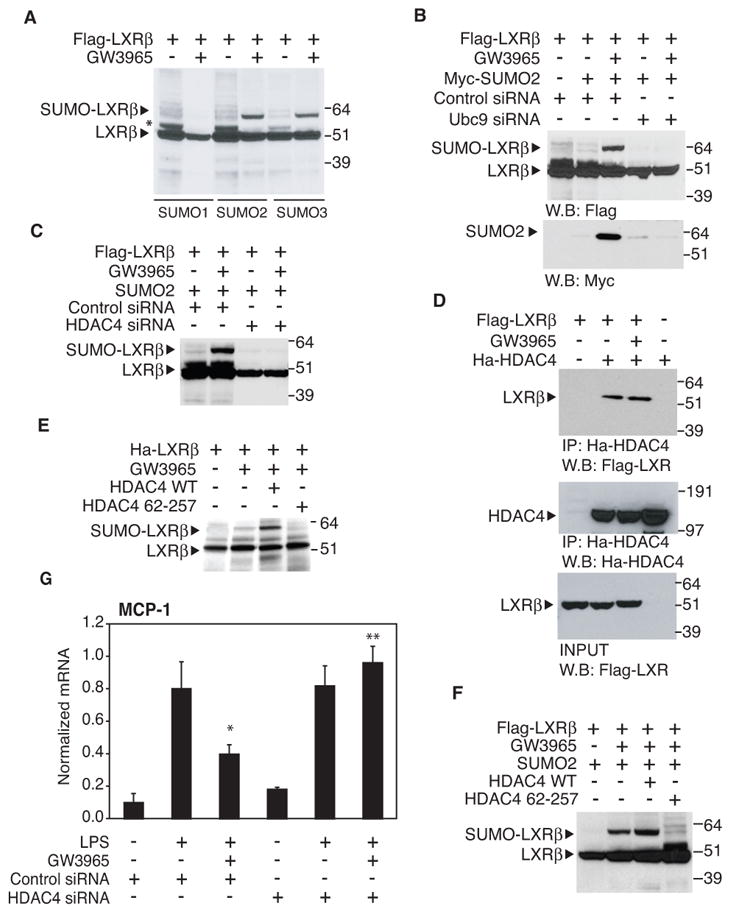
A. LXRβ is SUMOylated by SUMO-2 and SUMO-3. HeLa cells were transfected with FLAG-LXRβ, Ubc9 and with SUMO-1, SUMO-2 or SUMO-3 expression vector (as indicated) and treated with GW3965 for 1h (1μM). Whole cell lysates were immunoblotted for FLAG-tag. B. Knock down of Ubc9 by specific siRNA blocks the SUMO-2 conjugation of LXRβ. HeLa cells were transfected with siControl, siUbc9 (where indicated), FLAG-LXRβ and MYC-SUMO-2 expression vectors. Whole cell lysated were immunoprecipitated with FLAG-antibody and immunoblotted for FLAG-tag and SUMO2- MYC-tag. C. An siRNA directed against HDAC4 blocks LXRβSUMOylation. HeLa cells were transfected with siControl, siHDAC4 (where indicated), FLAG-LXRβ and SUMO-2 expression vectors and treated with GW3965 for 1h (1μM). Whole cell lysates were immunoblotted for FLAG-tag. D. LXRβ and HDAC4 physically interact. HeLa cells were transfected with FLAG-LXRβand Ha-HDAC4 and treated with GW3965 for 1h as indicated. Whole cell lysate was immunoprecipitated with HA antibody (middle panel) and LXRβ was detected with FLAG antibody (upper panel). In the lower panel the INPUT shows an equal expression of LXRβin each sample. Cells transfected with only LXRβ or HDAC4 (first and last lane) were used as negative control for the IP. E. LXRβ is SUMOylated by HDAC4 in vitro. The in vitro SUMOylation assay was performed as described in Experimental Procedures, adding to the reaction HDAC4 or HDAC4 mutant (67-257) as indicated. The proteins were immunoblotted for HA-tag. F. HDAC4 modulates LXRβ SUMOylation in vivo. HeLa cells were transfected with FLAG-LXRβ, Ubc9, SUMO-2 and HDAC4 or HDAC4 mutant (67-257) expression vector (as indicated) and treated with GW3965 for 1h (1μM). Whole cell lysates were immunoblotted for FLAG-tag. G. Knock down of HDAC4 blocks LXR transrepression of MCP1. Primary macrophages were transfected with control or HDAC4 specific siRNA for 48h and then stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligand (GW3965, 1μM). MCP1 expression was evaluated by real time PCR. Results are expressed as average of two independent experiments. Error bars represent standard deviations. * p < 0.02 vs LPS+control siRNA; **p < 0.03 vs GW3965+LPS+ control siRNA.
Several SUMO E3 ligases have been identified that promote transfer of SUMO from the E2 to specific substrates. To date, three types of SUMO E3 ligases have been well characterized: the protein inhibitor of activated signal transducer and activator of transcription (PIAS) family, the RanBP2/Nup358 protein and the human Polycomb member Pc2 (Melchior et al., 2003). Although SUMO-PPARγ mediated transrepression is PIAS1 dependent (Pascual et al., 2005), the systematic knockdown of all PIAS E3 ligases, RanBP2/Nup358, and PC2 did not reverse LXR ligand-dependent repression in primary macrophages (data not shown). Recently, HDAC4 and related members of class II deacetylases, such as HDAC5, have been described as SUMO E3 ligases for MEF2 transcription factor (Gregoire and Yang, 2005; Zhao et al., 2005). Knocking down HDAC4 expression (Fig. S1C) resulted in a complete inhibition of LXRβ SUMOylation (Fig. 3C). Consistent with these findings, a physical interaction between LXRβ and HDAC4 was revealed by co-immunoprecipitation assays (Fig. 3D). To determine whether HDAC4 directly promotes LXRs SUMOylation, we performed an in vitro SUMOyation assay. Ligand-dependent LXRβ SUMOylation was significantly increased by the addition of in vitro-translated HDAC4 protein in the presence of ligand (Fig. 3E), but not in the absence of ligand (data not shown). In contrast, an HDAC4 mutant (67-257), that does not have the coiled-coil domain required for the interaction with Ubc9 (Zhao et al., 2005), abolished LXRβ SUMOylation. Similar effects were observed in HeLa cells over-expressing WT HDAC4 or the HDAC4 mutant (67-257) (Fig. 3F). In concert, these findings strongly suggest that HDAC4 functions as a SUMO E3 ligase for LXRs in vivo.
We next evaluated whether HDAC4 was required for LXRs-mediated transrepression in primary macrophages. Unexpectedly, knockdown of HDAC4 prevented LPS-activation of iNOS, precluding an assessment of its role in transrepression of this specific target (data not shown). Several recent reports suggest that activation of inflammatory response genes by IRFs is blocked by HDAC inhibitors (Chang et al., 2004; Nusinzon and Horvath, 2003; Sakamoto et al., 2004). As the iNOS gene is also a target of these factors, we speculate that HDAC4 is one of the key HDACs that are required for activation of this cohort of genes.
To identify alternative LPS-target genes that would be suitable for determining the role of HDAC4 in LXR-dependent transrepression, microarray experiments were performed to identify LPS-inducible, LXR-sensitive target genes that retained LPS-induction in the presence of the HDAC inhibitor TSA (Table S1). MCP-1 was chosen as one of the most representative chemokine genes meeting these criteria. To determine whether the MCP-1 gene was an appropriate model, we performed transrepression experiments in primary macrophages and confirmed that MCP-1 transrepression was NCoR and Ubc9-dependent (Fig. S2). ChIP assays confirmed that LXRs were recruited to the MCP-1 promoter in an NCoR and Ubc9-dependent manner and prevented NCoR clearance (Fig. S3A and B). Knockdown of HDAC4 (Fig. S1C) had no effect on induction of MCP-1, as predicted by the microarray data, but reversed LXR transrepression (Fig. 3G). Taken together, these results demonstrate that HDAC4 interacts with LXR and regulates LXR transrepression activity by promoting its SUMOylation. Furthermore, these findings indicate that, although both LXR and PPARγ transrepression are SUMOylation-dependent, LXR is SUMOylated in a ligand-dependent manner in vivo by SUMO-2 and SUMO-3 while PPARγ is modified by SUMO-1 (Floyd and Stephens, 2004; Ohshima et al., 2004; Pascual et al., 2005; Yamashita et al., 2004).
Mutation of LXR SUMO acceptor sites abolishes transrepression but not transactivation activity
Two lysine residues in the human LXRβ primary amino acid sequence, the N-terminal Lys-31 and the C-terminal Lys-448, conform with high probability to the proposed consensus motif for SUMO-conjugation. In addition, a lower probability site was identified at Lys-410. To determine whether these lysine residues are targets for SUMOylation, mutants with lysine to arginine substitutions of K31, K410 and K448 were generated and assayed for SUMOylation. SUMOylation of LXRβ was reduced when the cells were transfected with mutants K410 and K448, and completely abolished with K410/448 double mutation, while mutation of K31 had no effect (Fig. 4A).
Figure 4. Mutation of LXRβ SUMO acceptor sites abolishes transrepression but not transactivation activity.
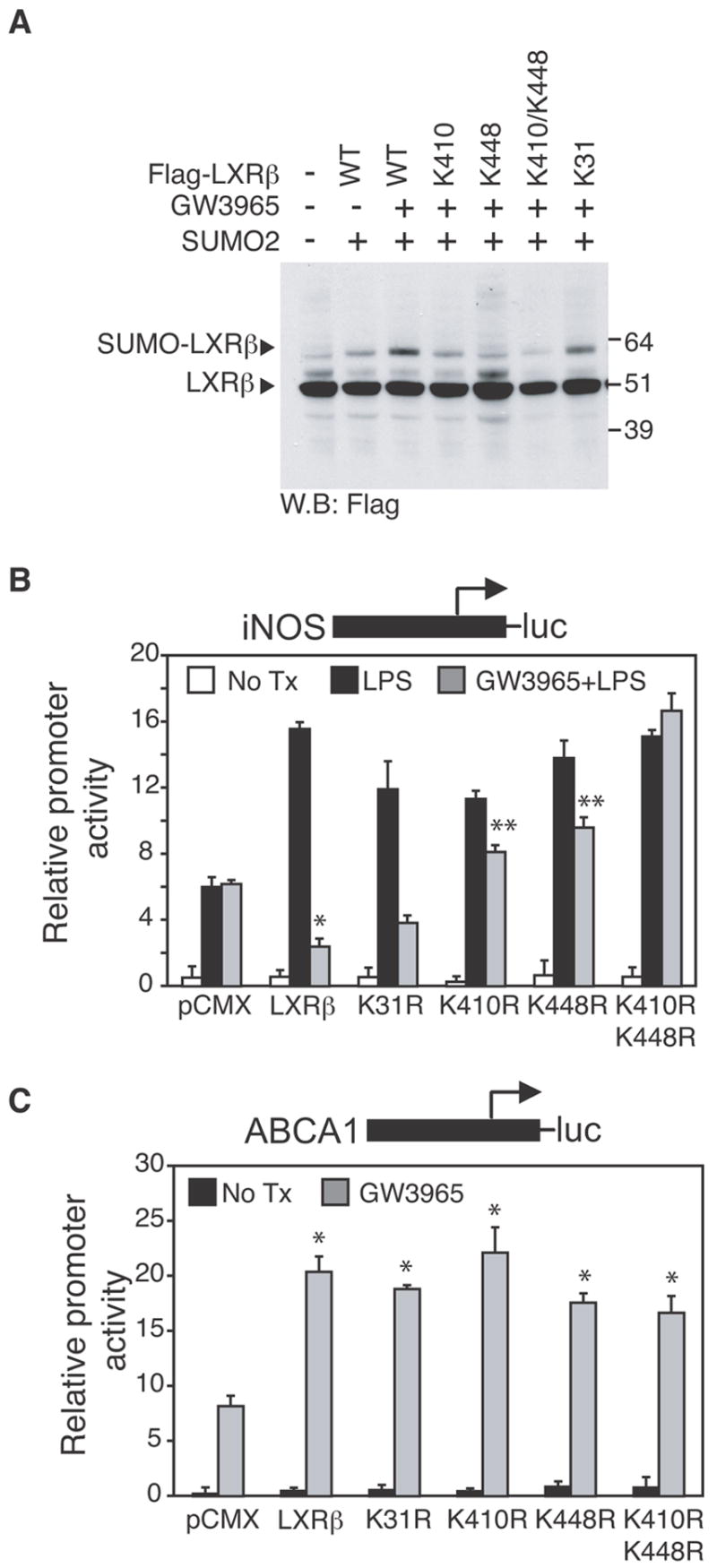
A. SUMOylation of LXRβ occurs at K410 and K448, but not at K31, as demonstrated by SUMOylation assay (performed as described in Fig 3A) in HeLa cells transfected with WT FLAG-LXRβ or FLAG-mutants K31, K410, K448 or double mutants K410/K448 as indicated. B,C. LXRβ K410 and K448 are required for transrepression but not for transactivation. RAW264.7 cells were transfected with iNOS-luc (B) or ABCA1-luc (C) reporter plasmids and with an expression vector for LXRβ wild type or mutated in K31, K410, K448 or K410/K448 as indicated. 24h after transfection, cells were treated with DMSO (white bars) or with LPS (1μg/ml) for 12h in the absence (black bars) or presence of LXR ligand (GW3965, 1μM, grey bars). Results are expressed as average of three independent experiments. Error bars represent standard deviations. (B) * p < 0.01 vs LPS; **p < 0.05 vs GW3965+LPS+ LXRβ wild type. (C) * p < 0.03 vs pCMX.
To determine the functional significance of LXR SUMOylation, the ability of the LXRβ mutants to transrepress the iNOS promoter and transactivate the ABCA1 promoter were tested in RAW264.7 cells. Individual mutations of K410 and K448 partially impaired transrepression activity, while the K410/448 double mutant fully abolished transrepression (Fig. 4B). Similar results were observed for the MCP1 promoter (Fig. S3C). In contrast, these same lysine mutations did not impair ligand-dependent transactivation of the ABCA1 promoter. Parallel studies of LXRα demonstrated ligand-dependent SUMOylation of lysine residues 328 and 434 (corresponding to residue 448 of LXRβ). Mutation of these residues to arginine impaired LXRα transrepression but not transactivation (data not shown). In concert, these experiments indicate that specific lysine residues in the ligand-binding domains of LXRα and LXRβ are SUMOylated by SUMO-2 and SUMO-3 and that their SUMOylation is required for transrepression.
Endogenous LXRs ligands promote entry into the transrepression pathway
A number of naturally occurring oxysterols are potent transcriptional activators of LXRs (Janowski et al., 1999; Janowski et al., 1996). In macrophages, endogenous oxysterols are thought to be produced in proportion to intracellular cholesterol levels and induce the expression of a number of genes involved in LXR-dependent cholesterol efflux, such as ABCA1 (Repa et al., 2000). To investigate the relative abilities of naturally occurring LXR ligands to both positively and negatively regulate gene expression their effects on ABCA1 gene expression and LPS-dependent induction of the endogenous iNOS gene were evaluated in primary macrophages. These experiments revealed that 22(R)-hydroxycholesterol (22R), 24(S), 25 epoxycholesterol (EC) and 24-hydroxycholesterol (24HC) each repressed iNOS activation and induced ABCA1 expression. This anti-inflammatory effect was LXR-dependent as it was not observed in LXR−/− macrophages (Fig. 5B). In contrast, 25-hydroxycholesterol (25HC) and 27-hydroxycholesterol (27HC) were able to activate ABCA1, but did not repress iNOS activation even at significantly higher concentrations than allow activation of the ABCA1 gene (Fig. 5C), indicating that activation and transrepression activities can be chemically separated. Furthermore, the transrepression activities of 22(R)-hydroxycholesterol (22R), 24(S), 25epoxycholesterol (EC) and 24-hydroxycholesterol (24HC) are impaired by knocking down Ubc9, suggesting that a subset of the naturally occurring oxysterol ligands of LXRs can promote their entry into the SUMOylation-dependent transrepression pathway.
Figure 5. Endogenous LXRs ligands promote entry into transrepression pathway.
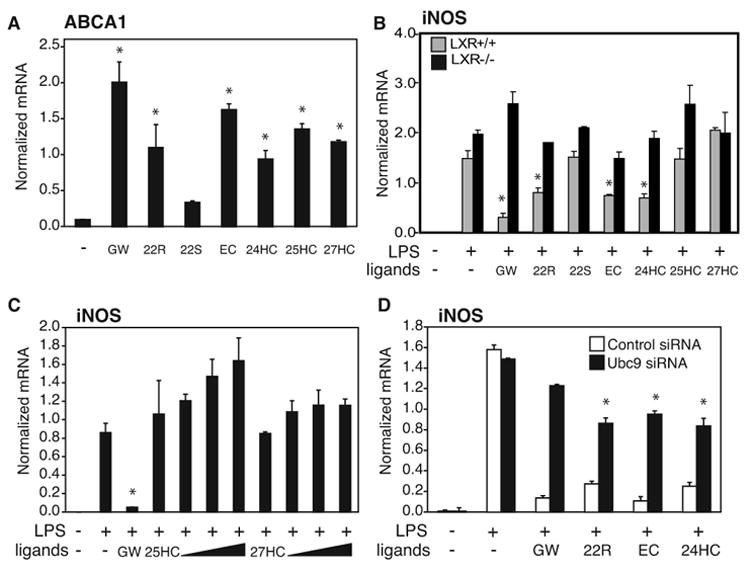
A,B. Natural LXRs ligands induce ABCA1 expression and repress iNOS activation. Primary macrophages from WT (panel A and B, grey bars) and LXR−/− mice (panel B, black bars) were treated with GW3965 (1μM) or 5μM oxysterols 22R, 22S, 24(S),25EC, 24HC, 25HC and 27HC (A) for 18 hours. In the B panel cells were pretreated with the indicated ligands and then treated with LPS (1μg/ml) for 6h. Expression of ABCA1 (A) and iNOS (B) was evaluated by QPCR. (A) * p < 0.03 vs untreated; (B) * p < 0.01 vs LPS. C. Dose-response experiment of 25HC and 27HC. Primary macrophages were pretreated with GW3965(1μM), 25HC and 27HC at increasing concentrations (1μM, 5 μM, 10 μM, 50 μM) and then treated with LPS (1μg/ml) for 6h. Expression of iNOS was evaluated by QPCR. * p < 0.01 vs LPS. D. Natural LXRs ligands require Ubc9 to transrepress iNOS expression. Primary macrophages were transfected with Ubc9 siRNA (black bars) or non specific siRNA (white bars) for 48h and then stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligands as indicated (5μM). iNOS expression in peritoneal macrophages was evaluated by QPCR. * p < 0.05 vs control siRNA. Results are expressed as average of two independent experiments. Error bars represent standard deviations.
Gene-specific utilization of parallel NCoR and SUMOylation-dependent pathways by LXRs and PPARγ
The finding that a SUMOylation/NCoR-dependent pathway parallel to that initially identified to mediate transrepression of inflammatory responses by PPARγ is utilized by LXRs to repress iNOS activation raised the questions of the extent to which these parallel pathways are utilized to repress other LPS target genes and whether they can be utilized in a gene-specific manner. Previous expression profiling studies demonstrated that PPARγ and LXR agonists repressed overlapping but distinct sets of genes induced in primary macrophages by LPS or the TLR3-activator poly I:C (Ogawa et al., 2005)(The complete data set is available at www.lipidmaps.org). These relationships are illustrated in Fig. 6A, filtered for genes induced at least 5-fold for each TLR agonist and exhibiting at least 50% repression with respect to activation in the absence of PPARγ or LXR agonist. We initially evaluated several endogenous LPS-inducible target genes that were found to be transrepressed by both nuclear receptors in primary macrophages. The majority of the LPS-inducible genes meeting these criteria, exemplified by MIP-1β, MCP-1, VCAM-1 and IL-15, were found to be transrepressed by PPARγ and LXRs by an NCoR and SUMOylation-dependent mechanism (Fig. S2). These findings indicate that the NCoR and SUMOylation-dependent pathways are likely to be widely utilized by LXRs and PPARγ to transrepress proinflammatory target genes.
Figure 6. Gene-specific utilization of NCoR and SUMOylation-dependent pathways in LXRs and PPARγ transrepression.
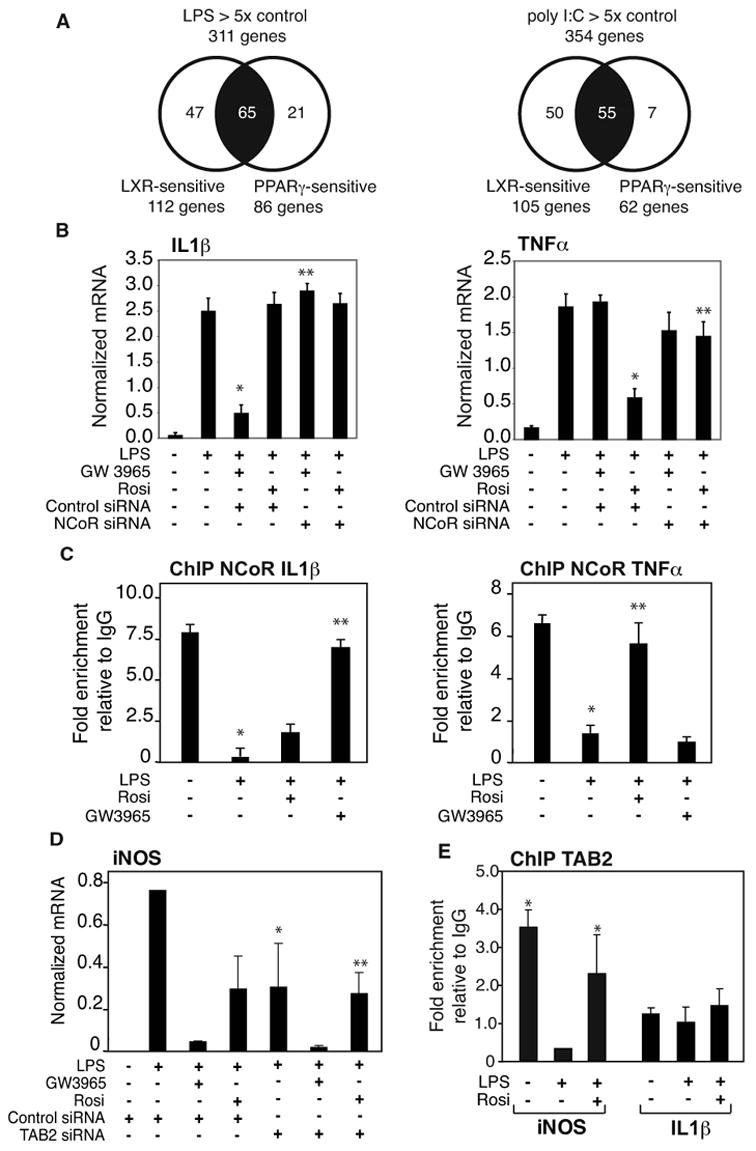
A. Relationship of genes subject to transrepression by LXR or PPARγ agonists in primary macrophages stimulated with either LPS or poly I:C as determined by expression profiling assays. Genes were filtered based on at least 5-fold induction by LPS and poly I:C, and at least 50% repression by each PPARγ or LXR agonist. Data is derived from (Ogawa et al., 2005). B. Receptor-specific repression of IL1β and TNFα. Primary macrophages were transfected with siRNA control or NCoR specific siRNA for 48h and then stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligand (GW3965, 1γM) or PPARγ ligand (Rosiglitazone, 1μM). IL1β and TNFα expression was evaluated by QPCR. * p < 0.01 vs LPS; ** p < 0.01 vs GW3965 (left panel) or Rosi (right panel) +LPS control siRNA. C. Differential inhibition of NCoR clearance from IL1β and TNFα promoters by PPARγ and LXR ligands. Primary macrophages were treated with LPS (1μg/ml) with or without PPARγ (Rosiglitazone, 1μM) or LXR ligand (GW3965, 1μM) for 1h. ChIP assays were performed with antibody against NCoR and or control IgG. * p < 0.01 vs untreated; ** p < 0.08 vs LPS. D. PPARγ transrepression is TAB2-dependent. Primary macrophages were transfected with siRNA control or TAB2 specific siRNA for 48h and then stimulated with LPS (1μg/ml) for 6h in the absence or presence of LXR ligand (GW3965, 1μM) or PPARγ ligand (Rosiglitazone, 1μM). iNOS expression was evaluated by QPCR. * p < 0.01 vs LPS; ** p < 0.01 vs Rosi +LPS control siRNA. E. Tab2 is present in the basal state on the iNOS promoter, but not on the IL1β promoter. ChIP assays were performed with antibody against Tab2 or control IgG. * p<0.01 vs control IgG. Results are expressed as the average of 2 independent experiments. Errors bars represent standard deviations.
We next evaluated subsets of LPS-inducible genes that were transrepressed only by the LXR agonist (represented in Fig. 6B by IL1β) or only by the PPARγ agonist (represented in Fig. 6B by TNFα). Unexpectedly, although IL1β and TNFα were differentially transrepressed by LXRs and PPARγ ligands, both cases involved an NCoR-dependent transrepression mechanism (Fig. 6B). This result was confirmed by ChIP experiments, showing that the LXR agonist, but not the PPARγ agonist, was able to inhibit the clearance of NCoR from IL1β promoter (Fig. 6C, left panel). Conversely, NCoR was cleared from the TNFα promoter in the presence of the LXR agonist, but not following treatment with the PPARγ agonist (Fig. 6C, right panel). We therefore performed ChIP experiments in primary macrophages to determine whether the SUMOylated receptors were differentially recruited to these promoters. These assays indicated that PPARγ and LXRs were both recruited to the iNOS, IL1β and TNFα promoters in a ligand-dependent manner (Fig. S4A), suggesting that the SUMOylated receptors are interrupting distinct signalling inputs that are responsible for NCoR clearance. This finding is supported by the observation that saturating concentrations of PPARγ and LXRs agonists exerted additive repressive effects on the iNOS promoter, which is sensitive to transrepression by both receptors, suggesting that they operate through independent mechanisms (Fig. S4 B).
To explore the roles of specific signalling adapter proteins in LPS activation and transrepression, we investigated Tab2, which plays a cytoplasmic role in TLR4-dependent signal transduction and is also a component of a subset of NCoR complexes (Baek et al., 2002; Jiang et al., 2004; Takaesu et al., 2000; Zhu et al., 2006). Knockdown of Tab2 in macrophages (Fig. S1D) reduces the fold activation of iNOS in response to LPS by about 50%. Intriguingly, the residual activation of iNOS remained fully sensitive to repression by LXRs agonist, but was now completely resistant to repression by the PPARγ agonist (Fig. 6D). Furthermore, ChIP experiments demonstrated that Tab2 was present on the iNOS promoter under basal conditions, was cleared in response to LPS treatment, but was retained in the presence of LPS and PPARγ agonist. In contrast, Tab2 was not enriched above background (IgG control) levels on the IL1β promoter under any condition. In concert, these results are consistent with Tab2 playing a promoter-specific role in mediating PPARγ-dependent, but not LXR-dependent, transrepression.
Signal-specific utilization of parallel NCoR and SUMOylation-dependent pathways by LXRs and PPARγ
Recent studies demonstrated that the ability of a particular nuclear receptor to repress specific subsets of inflammatory response genes could be modulated in a signal specific manner, e.g., a cohort of genes that was sensitive to GR-mediated repression when induced by LPS were GR-resistant when activated by poly I:C (Ogawa et al., 2005). A similar relationship was observed for genes subject to transrepression by LXR and PPARγ agonists, illustrated in Figure 7A. To investigate whether genes that exhibit signal-specific repression by LXRs and PPARγ utilize an NCoR-dependent mechanism, we evaluated the IL1β gene. While this gene is strongly repressed by LXR agonists in an NCoR-dependent manner when activated by LPS, it is almost completely resistant when activated by poly I:C (Fig. 7B). As a second example, we evaluated repression of the iNOS gene when activated by the TLR1/2 agonist Pam3CSK (Omueti et al., 2005).
Figure 7. Signal-specific regulation of NCoR and SUMOylation-dependent transrepression.
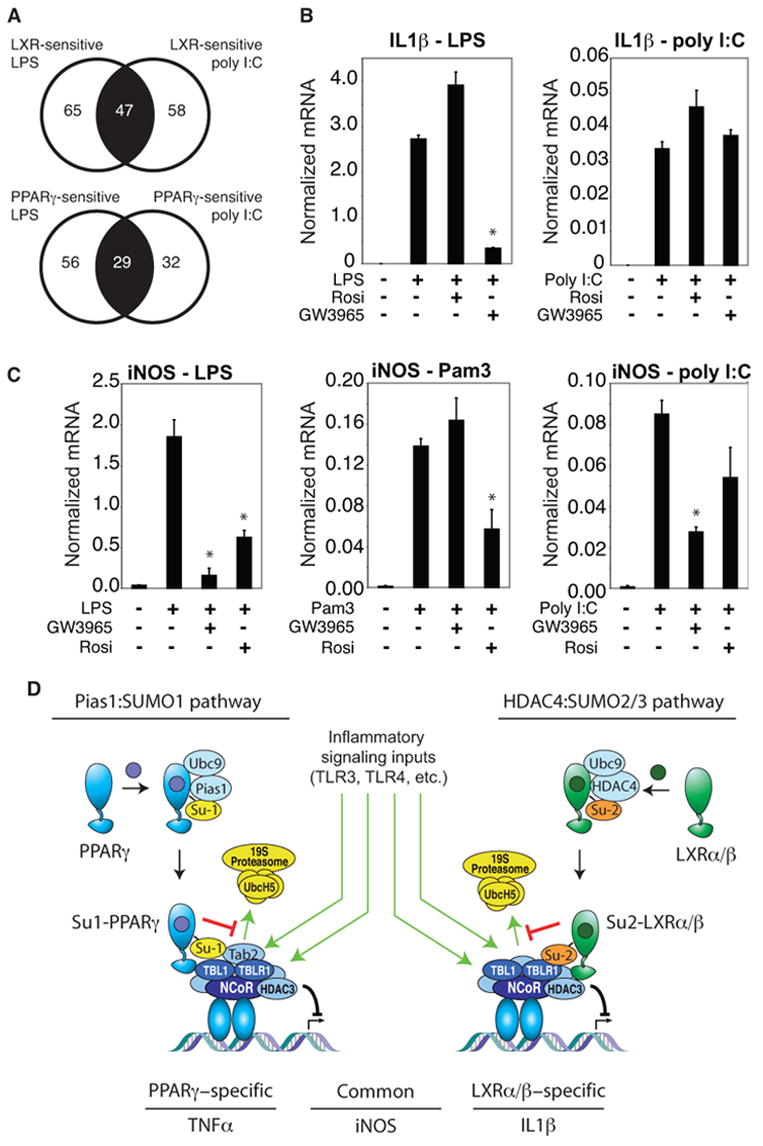
A. Signal-dependence of repression by PPARγ and LXR agonists. Genes in the central sector of each diagram were repressed when induced by either LPS or poly I:C. Genes in the left sector were only repressed when induced by LPS, while genes in the right sector were only repressed when induced by poly I:C. B. The IL-1β gene is induced by both LPS and poly I:C, but is only sensitive to LXR repression when induced by LPS. Primary macrophages were treated with TLR4 specific agonist (LPS,1μg/ml), or TLR3 specific agonist (polyI:C, 50 ng/ml) for 6h with or without PPARγ (Rosiglitazone, 1μM) or LXR ligand (GW3965, 1μM). IL-1β expression was evaluated by QPCR. * p < 0.02 vs LPS. C. Differential transrepression of the iNOS gene by LXR and PPARγ ligands. Primary macrophages were treated with the LPS (1 μg/ml), TLR2-specific agonist (PAM3, 300 ng/ml) or poly I:C (50 ng/ml) for 6h with or without PPARγ (Rosiglitazone, 1μM) or LXR ligand (GW3965, 1μM). iNOS expression was evaluated by real time PCR. * p < 0.02 vs LPS (left), Pam 3 (middle), poly I:C (right). Results are expressed as the average of 2 independent experiments performed in duplicate. Error bars represent standard deviations. D. Model depicting parallel transrepression pathways utilized by PPARγ and LXRs. See text for discussion.
Although repressed by both LXR and PPARγ agonists when induced by LPS, the ability of the LXR agonist to transrepress iNOS was selectively abolished when macrophages were stimulated by the TLR1/2 agonist (Fig. 7C). We therefore evaluated whether the ability of LXRs agonist to transrepress inflammatory responses is correlated with their ability to block NCoR clearance induced by different TLR agonists. ChIP experiments revealed that LXR agonists inhibited NCoR clearance from the iNOS promoter in response to LPS treatment, but failed to inhibit NCoR clearance when cells were treated with the TLR1/2 agonist Pam3CSK (Fig. S5A). In addition, poly I:C was found to promote clearance of NCoR from the IL1β promoter even when the cells were treated with LXRs ligand, consistent with the failure of GW3965 to transrepress IL1βactivation in response to this signal (Fig. S5B). In concert, these results indicate that the parallel SUMOylation-dependent transrepression pathways are themselves subject to regulation, and can be over-ridden by specific signals in a gene specific manner.
DISCUSSION
A general SUMO/NCoR transrepression mechanism
Many nuclear receptors can act in trans to repress inflammatory responses, with recent studies indicating that ligand-dependent repression of inflammatory target genes occurs in a gene and signal-specific manner (Ogawa et al., 2005). Although a large number of transrepression mechanisms have been suggested from the analysis of artificial and transiently transfected target genes, relatively few mechanisms have been established for endogenous target genes in primary cells. The present studies demonstrate that ligand-dependent transrepression of a number of endogenous inflammatory response genes in primary macrophages by LXRs is NCoR- and SUMOylation-dependent. Moreover, ChIP analysis revealed that LXRs are recruited to the iNOS promoter in a SUMOylation-dependent manner and inhibit corepressor clearance induced by LPS. These observations extend the NCoR and SUMOylation-dependent pathway, initially identified for PPARγ (Pascual et al., 2005), to two additional members of the nuclear receptor family, LXRα and LXRβ. Thus, SUMOylation-dependent targeting of a nuclear receptor to corepressor complexes to prevent their signal-dependent clearance is likely to represent a general molecular strategy for transrepression of proinflammatory target genes, expanding upon previously described mechanisms by which SUMO proteins function to negatively regulate gene expression (Gill, 2005).
Parallel SUMO-dependent transrepression pathways
Although these studies indicate that LXRs and PPARγ both utilize a SUMOylation-dependent mechanism that converges on NCoR corepressor complexes, they also provide evidence for distinct and parallel pathways that are used in a receptor-specific and gene-specific manner (Fig. 7). These conclusions are based on the findings that PPARγ and LXRs are conjugated to different SUMO proteins by different E3 ligases and differentially regulate proinflammatory target genes such as IL1β and TNFα. While PIAS1 has been described as the E3 ligase involved in PPARγ SUMOylation (Ohshima et al., 2004; Pascual et al., 2005), the present studies strongly suggest that SUMOylation of LXRs requires HDAC4 E3 ligase activity. In the case of PPARγ, a ligand-induced allosteric change in the position of the lysine residue that is the point of SUMO1 conjugation was proposed to be the basis for ligand-dependent SUMOylation (Pascual et al., 2005). In the case of LXRs, crystal structures of apo receptors are not available, so it is not yet possible to assess ligand-dependent changes in the lysine residues that represent the essential SUMO acceptor sites. However, association of LXRβ with HDAC4 does not appear to be ligand-dependent, suggesting that ligands alter the specific configurations of the lysine residues that serve as SUMO acceptor sites to provide the mechanism for ligand-dependence.
Since the conjugated forms of SUMO-2 and SUMO-3 form a distinct subfamily and are only 50% identical in sequence to SUMO-1 (Hay, 2005), we propose that LXRs and PPARγ are functionally differentiated based on their differential SUMOylation and represent distinct, parallel transrepression pathways that can be utilized in a gene and signal-specific manner (Figure 7D). Gene-specific utilization of these parallel pathways is exemplified by the selective transrepression of IL1β by LXRs and TNFα by PPARγ, while signal-specific utilization is exemplified by the differential sensitivities of the IL1βand iNOS genes to LXR ligands when activated by TLR1/2, TLR3 and TLR4 agonists.
These findings thus raise a number of intriguing possibilities regarding the molecular targets of SUMOylated PPARγ and LXRs within the NCoR complex and how the docking of these proteins prevents NCoR clearance. ChIP experiments have now localized NCoR complexes on nearly all LPS-responsive genes we have examined. Furthermore, it appears that SUMOylated PPARγ and LXRs localize to these complexes prior to an activating signal. In all cases, whether or not a gene is sensitive or resistant to PPARγ or LXR agonists is strictly correlated with whether or not they prevent NCoR clearance. Previous studies suggest that signal-dependent clearance of NCoR complexes from target promoters involves the activation of the ubiquitin E3 ligase activities of the Tbl1 and TblR1 proteins that are core components of these complexes (Ogawa et al., 2004; Perissi et al., 2004), leading to the recruitment of the Ubc5/19S proteosome machinery required for ubiquitylation of NCoR complexes and their subsequent dismissal. SUMOylation of PPARγ and its interaction with the NCoR complex has been shown to block signal-dependent recruitment of UbcH5 to the iNOS promoter (Pascual et al., 2005), but whether this is due to inhibition of Tbl1/TblR1 phosphorylation, or some distal step remains to be defined. The finding that Tab2 is required for transrepression of the iNOS gene by PPARγ, but not LXRs, suggests that the SUMOylated receptors inhibit distinct signalling inputs that are involved in activating the ubiquitinligase activities of Tbl1/TblR1. Biochemical approaches have recently identified components of corepressor complexes as interacting targets of SUMO2 (Rosendorff et al., 2006) and it will be of interest to utilize SUMOylated LXRs and PPARγ as molecular probes to further elucidate the specific events that allow NCoR corepressor complexes to be cleared from inflammatory response genes in a signal-specific manner.
Coordinate and independent regulation of transactivation and transrepression
In macrophages, LXRs play critical roles in maintaining normal cholesterol homeostasis by positively regulating the expression of genes involved in cholesterol storage and efflux. The observation that synthetic LXR agonists are also capable of potently repressing inflammatory response genes raises the question of whether the production of endogenous oxysterols might also exert anti-inflammatory effects by binding to LXRs. Consistent with this possibility, we find that 22R, 24(S), 25EC, and 24HC can both induce ABCA1 expression and repress iNOS activation in primary macrophages. Intriguingly, 25HC and 27HC can activate ABCA1, but do not repress iNOS activation. These findings suggest that the transactivation and transrepression activities of LXRs may be independently regulated based on the specific ligands that are produced under different contexts. Intriguingly, the oxysterols that induce transrepression are not found at any significant levels in cultured macrophages, while the oxysterols that are made at high levels by cultured macrophages do not induce transrepression (Fu et al., 2001). It is possible that enzymes necessary for producing natural ligands that activate this pathway are expressed in specific subsets of macrophages in vivo. Alternatively, this pathway may operate as a paracrine system, e.g., Kuppfer cells in liver may be exposed to hepatocyte-derived oxysterols that activate the transrepression pathway.
The ability of many ligand-dependent nuclear receptors to inhibit inflammatory gene expression is also of considerable interest from the perspective of the development of new approaches to modulate inflammatory diseases. This goal is often complicated by the fact that potent nuclear receptor agonists are associated with unwanted side effects: e.g., PPARγ agonists promote weight gain and edema formation in diabetic patients (Staels and Fruchart, 2005; Yki-Jarvinen, 2005), while LXR agonists have been shown to cause marked hypertriglyceridemia in animals, at least in part due to activation of SREBP1c in liver (Schultz et al., 2000). The present studies suggest that it should be possible to develop LXR ligands that separate transrepression from transactivation activities. Ligands that specifically promote entry of LXR into the SUMO-dependent transrepression pathway may be of therapeutic utility in diseases in which inflammation plays a significant pathogenic role.
EXPERIMENTAL PROCEDURES
Reagents and plasmids
TLR4 agonist (LPS), TLR3 agonist (poly I:C) were obtained from Sigma, TLR1/2 agonist (Pam3CysSK4) from ECM Microcollections, GW3965 and Rosiglitazone were provided by GlaxoSmithKline. 22(R)-hydroxycholesterol, 22(S)-hydroxycholesterol, 25-hydroxycholesterol were purchased from Sigma, 24(S),25epoxycholesterol and 24-hydroxycholesterol from Biomol, 27-hydroxycholesterol from Research Plus, Inc. Point mutations in Flag-hLXRα and Flag-hLXRβ were made using the QuickChange side-directed mutagenesis kit (Stratagene). The reporter plasmid iNOS-luc, Ubc9 and SUMO-1 expression vectors have been previously described (Pascual et al., 2005), the reporter plasmid ABCA1-luc was kindly donated by Dr. B. Wagner, the reporter plasmid MCP1-luc was kindly donated by Dr. R. Natarajan and the HDAC4 mutant (67-257) was kindly donated by Dr.T.Yao.
Cell culture and transient transfection
Thioglycollate-elicited macrophages were prepared as previously described (Pascual et al., 2005) from LXRα β+/+ and LXRα β−/− mice (Wagner et al., 2003) or C57BL/6 mice (Charles River). For RNAi experiments in primary macrophages, cells were transfected with control or smart-pool siRNAs (100 nM, Dharmacon) directed against NCoR, Ubc9, HDAC4 and TAB2 using lipofectamine 2000 (Invitrogen). Cells were used for experiments after 48 h incubation and target gene knockdown was validated by QPCR (Supplemental Figure S1). RAW 264.7 were transiently transfected with iNOS, ABCA1 or MCP1 promoters directing luciferase expression using Superfect reagent (Qiagen). A β-galactosidase expression vector was cotrasfected as an internal control. For siRNA experiments in RAW264.7 macrophages, cells were transfected with siRNAs (40 nM) using Superfect reagent for 48 h.
ChIP assays
ChIP assays were performed as previously described (Pascual et al., 2005). Anti-LXR (Calbiochem), anti- PPARβ (ActiveMotif) and anti-NCoR (Affinity Bioreagents) antibodies were used. Rabbit preimmune serum was used as a control for non-specific binding. 150-bp regions of the iNOS, MCP1, IL1β and TNFα proximal promoters were amplified by real time PCR. Primer sequences are available upon request.
RNA isolation and QPCR
Total RNA (isolated by RNeasy kit, Qiagen) was prepared from primary macrophages. One μg of total RNA was used for cDNA synthesis, and 1 μl of cDNA was used for real time PCR using inflammatory-gene-specific primers. Primer sequences are available upon request. PCR (SYBERgreen) analysis was performed on an Applied Biosystems 7300 Real-time PCR system. Values are normalized with GAPDH content.
Co-IP assay
HeLa cells were transfected with Flag-LXRβ and Ha-HDAC4 and lysed in low salt lysis buffer. Ha-HDAC4 was immunoprecipitated using anti-HA beads (Covance) and Flag-LXRβ was detected using anti-FLAG antibody (Sigma).
In vitro SUMOylation assay
In vitro SUMOylation reactions were carried out in 20 μl of reaction buffer by adding recombinant SUMO, SAE-1, SAE-2, Ubc9 and in vitro translated HDAC4 or HDAC4 mutant (67-257) to the in vitro translated LXRβ. Reactions were incubated for 60 min at 37°C supplemented with 2mM ATP and analyzed by SDS–PAGE and immunoblotted using anti-HA (Covance) antibody.
In vivo SUMOylation assay
Whole protein extract was prepared in presence of NEM (N-Ethylmaleimide, Calbiochem) from HeLa cells transfected with FLAG-tagged wild-type LXRα and LXRβ, or lysine mutants and Myc-tagged SUMO-1, SUMO-2 or SUMO-3, Ubc9 and HDAC4 or HDAC4mutant (67-257) expression vectors. Extracts were resolved by SDS–PAGE and immunoblotted using anti-FLAG (Sigma) or anti-Myc (Santa Cruz) antibodies.
Supplementary Material
Acknowledgments
We thank B. Wagner (X-ceptor Therapeutics) for the ABCA1-luc plasmid, R. Natarajan (City of Hope, Duarte) for the MCP1-luc plasmid, T. Yao (Duke University, Durham) for the HDAC4 mutant (67-257) plasmid, A. Gamliel (University of California, San Diego) for the SUMO-2 and SUMO-3 plasmids. We thank the DERC/BIOGEM core facility for assistance with microarray experiments and A. Howarth for assistance with manuscript preparation. These studies were supported by an American Heart Association Postdoctoral Fellowship to S.G and NIH grants CA52599, HL56989, DK063491 and GM 069338 to CKG. MGR is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:9578–9583. doi: 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY, Kimmel R, Stroup D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha) Gene. 2001;262:257–265. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]
- Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X Receptor/Retinoid X receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- Delerive P, Martin-Nizard F, Chinetti G, Trottein F, Fruchart JC, Najib J, Duriez P, Staels B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ Res. 1999;85:394–402. doi: 10.1161/01.res.85.5.394. [DOI] [PubMed] [Google Scholar]
- Floyd ZE, Stephens JM. Control of Peroxisome Proliferator-Activated Receptor {gamma}2 Stability and Activity by SUMOylation. Obes Res. 2004;12:921–928. doi: 10.1038/oby.2004.112. [DOI] [PubMed] [Google Scholar]
- Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem. 2001;276:38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. Faseb J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- Gill G. Something about SUMO inhibits transcription. Curr Opin Genet Dev. 2005;15:536–541. doi: 10.1016/j.gde.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner SM, Greenberg AS, Weston WM, Chen H, Williams K, Freed MI. Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation. 2002;106:679–684. doi: 10.1161/01.cir.0000025403.20953.23. [DOI] [PubMed] [Google Scholar]
- Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPARγ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci U S A. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O'Connell RM, Cheng G, Saez E, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- Li A, Brown K, Silvestre M, Willson T, Palinski W, Glass C. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J CIin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior F, Schergaut M, Pichler A. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci. 2003;28:612–618. doi: 10.1016/j.tibs.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Nusinzon I, Horvath CM. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci U S A. 2003;100:14742–14747. doi: 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors mediating counter-regulation of inflammatory responses. Cell. 2005 doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci USA. 2004;101:14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Koga H, Shimotohno K. Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. J Biol Chem. 2004;279:29551–29557. doi: 10.1074/jbc.M403866200. [DOI] [PubMed] [Google Scholar]
- Omueti KO, Beyer JM, Johnson CM, Lyle EA, Tapping RI. Domain exchange between human toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J Biol Chem. 2005;280:36616–36625. doi: 10.1074/jbc.M504320200. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet DJ, Janowski BA, Mangelsdorf DJ. The LXRs: a new class of oxysterol receptors. Curr Opin Genet Dev. 1998;8:571–575. doi: 10.1016/s0959-437x(98)80013-0. [DOI] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, Shi Y, Shi Y, Gill G. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc Natl Acad Sci U S A. 2006;103:5308–5313. doi: 10.1073/pnas.0601066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto S, Potla R, Larner AC. Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J Biol Chem. 2004;279:40362–40367. doi: 10.1074/jbc.M406400200. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM. PPARγ: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54:2460–2470. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci USA. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, et al. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARγ and PPARδ negatively regulate specific subsets of lipopolysaccharide and IFNγ target genes in macrophages. Proc Natl Acad Sci USA. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- Winoto A, Littman DR. Nuclear hormone receptors in T lymphocytes. Cell . 2002;109(Suppl):S57–66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Yamaguchi T, Shimizu M, Nakata N, Hirose F, Osumi T. The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes Cells. 2004;9:1017–1029. doi: 10.1111/j.1365-2443.2004.00786.x. [DOI] [PubMed] [Google Scholar]
- Yki-Jarvinen H. The PROactive study: some answers, many questions. Lancet. 2005;366:1241–1242. doi: 10.1016/S0140-6736(05)67504-6. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sternsdorf T, Bolger TA, Evans RM, Yao TP. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25:8456–8464. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.