

Abstract
In a previous report we have described the effects of expression of D-type cyclins in epithelial tissues of transgenic mice. To study the involvement of the D-type cyclin partner cyclin-dependent kinase 4 (CDK4) in epithelial growth and differentiation, transgenic mice were generated carrying the CDK4 gene under the control of a keratin 5 promoter. As expected, transgenic mice showed expression of CDK4 in the epidermal basal-cell layer. Epidermal proliferation increased dramatically and basal cell hyperplasia and hypertrophy were observed. The hyperproliferative phenotype of these transgenic mice was independent of D-type cyclin expression because no overexpression of these proteins was detected. CDK4 and CDK2 kinase activities increased in transgenic animals and were associated with elevated binding of p27Kip1 to CDK4. Expression of CDK4 in the epidermis results in an increased spinous layer compared with normal epidermis, and a mild hyperkeratosis in the cornified layer. In addition to epidermal changes, severe dermal fibrosis was observed and part of the subcutaneous adipose tissue was replaced by connective tissue. Also, abnormal expression of keratin 6 associated with the hyperproliferative phenotype was observed in transgenic epidermis. This model provides in vivo evidence for the role of CDK4 as a mediator of proliferation in epithelial cells independent of D-type cyclin expression.
Normal cell growth and differentiation requires precise control of the mechanisms that govern the entry into, passage through, and exit from the cell cycle. Progress through the G1 phase of the mammalian cell cycle is regulated by the ordered synthesis, assembly, and activation of distinct cyclin-dependent kinase (CDK)-cyclin holoenzymes. 1,2 This process is mediated by the D-type cyclins (D1, D2, and D3), whose expression is modulated by growth-stimulatory signals. These cyclins associate with the closely related CDK4 and CDK6 kinases, resulting in their catalytic activation and substrate recognition. A key substrate for G1 cyclin/CDK complexes is the retinoblastoma protein, pRb. Phosphorylation of pRb, a tumor suppressor gene product, has been attributed to cyclin/CDK complexes and implicated in the regulation of proliferation of keratinocytes and other cell types. 1,3,4 The pRb family of proteins, pRb, p107, and p130, negatively regulate the passage of cells from G1 to S phase by sequestering E2F transcription factors and repressing the transcription of critical genes for G1/S transition through binding to histone deacetylase (HDAC). 4-6 Accumulating evidence suggests that the kinase activity of cyclin D-CDK4 is partially responsible for the initial phosphorylation of pRb at specific sites, which allows for subsequent phosphorylation of other sites, presumably by cyclin E-CDK2. 6,7 The activity of CDKs is subject to additional levels of regulation, which include their association with inhibitory molecules such as the INK4 and CIP/KIP family of proteins. 8,9 p16INK4a has a prominent role in regulating cell proliferation by binding and inhibiting CDK4,6. In fact, p16INK4a is a tumor suppressor gene that has been found to be mutated or deleted in many experimental and human tumors. 10 These features suggest that the fundamental role of CDK4/D-type cyclins is to integrate extracellular signals with the cell-cycle machinery. 3 Recently, experiments with CDK4-null and CDK4/cyclin D overexpression in Drosophila have suggested that CDK4 plays a role regulating normal cell growth (mass accumulation) rather than cell-cycle progression. 11,12
Initially, several reports assigned redundant roles to the three members of the D-type cyclin family, but in the last few years, it has become evident that each member plays specific roles and has differential tissue expression. 3 In fact, different tissues are affected in cyclin D1 and cyclin D2 knockout mice. 13-15 Whether these specific roles are dependent on regulatory subunits (cyclins), catalytic subunits (CDKs), or other accessory proteins remain unknown. D-type cyclins have been described as putative oncogenes in different tissues and more recently, CDK-independent functions of D-type cyclins were described. 16-19 On the other hand, less is known about the involvement of CDKs in the tumorigenesis process. The importance of the interaction between CDK4 and p16INK4a became apparent with the identification of a CDK4 mutation in patients with familial melanoma, 20-22 and by reports showing that mutually exclusive mutations of p16INK4a or CDK4 occur in glioblastomas. 23,24
In previous work, we have demonstrated that the kinetics of CDK4-cyclin D1 complex formation followed a pattern similar to cyclin D1 expression 25 in mouse keratinocytes induced by phorbol esters and in chemically induced mouse skin tumors. 26 These results and the fact that the CDK4 protein level remains constant showed that CDK4 is not the rate-limiting factor in this model. In concordance with these data, cyclin D1, cyclin D2, and cyclin D3 transgenic mice showed a hyperproliferative epidermis and increased CDK4 and CDK6 kinase activities. 27-29 To study the involvement of CDK4 in keratinocyte proliferation and differentiation, we generated transgenic mice overexpressing CDK4 in the epidermis. For this purpose we used a keratin 5 promoter that was used previously in our laboratory for the generation of cyclin D1, cyclin D2, and cyclin D3 transgenic mice. Here, we described the phenotypic consequences of overexpression of CDK4 in vivo. The transgenic mice developed severe epidermal hyperplasia and hypertrophy in basal and suprabasal cell layers. In addition to epidermal changes, transgenic mice developed dermal fibrosis with marked atrophy of the panniculum adiposum (subcutaneous adipose tissue). Protein expression in epidermal tissue of the transgenic mice showed that none of the D-type cyclins were overexpressed. Therefore, forced expression of CDK4 resulted in a hyperproliferative phenotype independent of D-type cyclin regulation. Increased binding of p27Kip1 to CDK4 in transgenic mice was associated with an elevated kinase activity of CDK2. Taken together, these results show that CDK4 overexpression has dramatic consequences in mouse skin compared with D-type cyclin transgenic mice. The strong effect in the dermal tissue suggests that an indirect mechanism, more likely through growth factor secretion, is associated with CDK4 deregulation.
Materials and Methods
Generation of Transgenic Mice
An EcoRV/XbaI fragment containing the human cyclin-dependent kinase 4 cDNA was excised from the plasmid pBluescript II and inserted into the polylinker of the vector pBK5 that contained the 5.2-kb bovine keratin 5 (K5) regulatory sequences, β-globin intron 2 and the 3′ polyadenylation sequences. This construct was designated as pK5-CDK4 (Figure 1) ▶ . The transgene was excised from the plasmid vector by digestion with BssHII, separated by low-melting-point agarose electrophoresis, and purified using a Geneclean II Kit (BIO101; Geneclean, Vista, CA). This transgene was microinjected into the FVB mouse strain by the Transgenic Mouse Development Facility at Science Park, M. D. Anderson Cancer Center. Transgenic mice were crossed for two generations with SSIN strain to generate 75% SSIN background mice. All of the experiments were performed in this background.
Figure 1.

pK5-CDK4 construct and PCR screening. A: Diagram of the K5CDK4 construct. B: PCR amplification of DNA extracted from mouse tails. β-globin sequence was amplified resulting in a 450-bp product.
Transgene-Specific Polymerase Chain Reaction (PCR)
Genomic DNA was extracted from mouse tail clips and used for PCR detection of the transgene. We used an upstream primer (TTCAGGGTGTTGTTTAGAATGG) and a downstream primer (CAATAAGAATATTTCCACGCCA) specific for the β-globin intron 2 sequence. With this process, we screened all of the transgenic mouse lines. The DNA amplification renders a 450-bp PCR product. PCR was performed by denaturation at 95°C for 1 minute, followed by 32 cycles of amplification as follows: denaturation at 95°C for 30 seconds, annealing at 55°C for 40 seconds, and extension at 72°C for 45 seconds, with a final extension at 72°C for 10 minutes.
CDK4 Immunohistochemical Stains
Immunohistochemical staining of formalin-fixed paraffin-embedded tissues was performed with polyclonal mouse anti-CDK4 (C-22; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Epithelial cell proliferation was measured by intraperitoneal injection of 60 μg/g of 5-bromodeoxyuridine (BrdU) 30 minutes before the mice were killed. BrdU incorporation was detected by immunohistochemical staining of paraffin-embedded sections using mouse anti-BrdU monoclonal antibody (Becton Dickinson immunocytometry system; Becton Dickinson, San Jose, CA). The reaction was visualized with a biotin-conjugated anti-mouse antibody (Vector Laboratories, Inc. Burlingame, CA) and avidin-biotin-peroxidase kit (Vectastain Elite, Vector Laboratories, Inc.) with diaminobenzidine as the chromogen.
Western Blotting Analysis
The dorsal sides of the mice were shaved. After they were sacrificed, the dorsal skins were treated with a depilatory agent for 1 minute and then washed off. The epidermal tissue was scraped off with a razor blade, placed into homogenization buffer (50 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 2.5 mmol/L EGTA, 1 mmol/L ethylenediaminetetraacetic acid, 0.1% Tween-20, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethyl sulfonyl fluoride, 0.2 U/ml aprotinin, 10 mmol/L β-glycerophosphate, 0.1 mmol/L sodium vanadate, and 1 mmol/L NaF) and homogenized using a manual homogenizer. The epidermal homogenate was centrifuged at 11,000 × g to collect the supernatant, which was used directly for Western blotting analysis or stored at −80°C. The protein concentration was measured with the Bio-Rad protein assay system (Bio-Rad Laboratories, Richmond, CA). Protein lysates (25 μg from each sample) were electrophoresed through 12% acrylamide gels and electrophoretically transferred onto nitrocellulose membranes. After being blocked with 5% nonfat powdered milk in Dulbecco’s phosphate-buffered saline (Sigma Chemical Co.), the membranes were incubated with 1 μg/ml specific antibodies. The following antibodies were used: polyclonal antibodies against CDK4 (C22), CDK2 (M2), CDK6 (C21), p107 (C18), p130 (C20) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and mouse monoclonal antibody against pRb (14001A) (BD Pharmingen, San Diego, CA). Horseradish peroxidase-conjugated secondary antibody (Amersham Corp., Arlington Heights, IL), followed by enhanced chemiluminescence (ECL detection kit; Amersham Corp.) were used for immunoblotting detection. Bio-image analysis was used to quantitate the expression levels of those proteins.
To study p27Kip1/CDK complex formations and kinase activities, we used polyclonal antibodies against CDK4, CDK2, and p27Kip1 conjugated with protein A-Sepharose beads (Life Technologies Inc., Grand Island, NY) to immunoprecipitate fresh protein lysates for 1 hour at 4°C with constant rotation. After washing three times with extraction buffer, Western blot analysis was performed as described above. To study the kinase activities of CDK4 and CDK2, protein lysates were obtained as described above, but the homogenate was frozen on powdered dry ice, thawed in ice water, incubated on ice for 15 minutes, and centrifuged at 10,000 × g for 10 minutes at 4°C. The supernatant was collected and used for a kinase assay. Five hundred μg of protein lysate were immunoprecipitated with antibodies against CDK4 or CDK2. Thirty μl of precoated antibody beads were incubated with the lysate for 1 hour at 4°C. The beads were washed twice with Nonidet P-40 buffer (Tris, pH 7.5, 150 mmol/L NaCl, Nonidet P-40 0.5%, 50 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethyl sulfonyl fluoride) and twice with kinase buffer (50 mmol/L HEPES, pH 7.0, 10 mmol/L MgCl2, 5 mmol/L MnCl2). Then, 30 μl of kinase buffer, 0.5 μg of pRb substrate (Santa Cruz Biotechnology, Inc.); 5 μCi 32P-ATP (6000 Ci/mmol), 1 mmol/L dithiothreitol, 5 μmol/L ATP) was added to the bead pellet and incubated for 30 minutes at 30°C. Sodium dodecyl sulfate sample buffer was added, and each sample was boiled for 5 minutes and electrophoresed through an 8% acrylamide gel.
Results
Generation of Transgenic Mice Expressing CDK4
To determine the function of cyclin-dependent kinase 4 (CDK4) in the epidermis, we generated transgenic mice that overexpressed CDK4 under the regulation of the keratin 5 promoter. The K5CDK4 construct was made by subcloning the human CDK4 cDNA into a vector containing a 5.2-kb fragment of the bovine K5 promoter, the rabbit β-globin intron 2, and the SV40 polyadenylation signal (Figure 1A) ▶ . The K5 promoter fragment was previously shown to direct transgene expression to the basal cell compartment of stratified squamous epithelia. 30 All of the transgenic mice were generated in the genetic background FVB. Transgenic mice were identified by PCR analysis of tail DNA using primers specific for the β-globin sequence (Figure 1B) ▶ . Based on those results, five integration-positive mice were selected as founders and crossed with SSIN inbred mice. A second screening to verify transgene expression was performed by immunohistochemical staining for CDK4. Because CDK4 was poorly detected in normal epidermis by immunohistochemistry (Figure 2F) ▶ , positive CDK4 staining was used as an indication of transgene expression (Figure 2E) ▶ . These data are consistent with the expected activity of the K5 promoter. We selected two transgenic lines with high CDK4 expression (2303 and 2305), which were backcrossed with SSIN inbred mice to produce animals with 75% SSIN background that were used in all of the experiments presented here.
Figure 2.
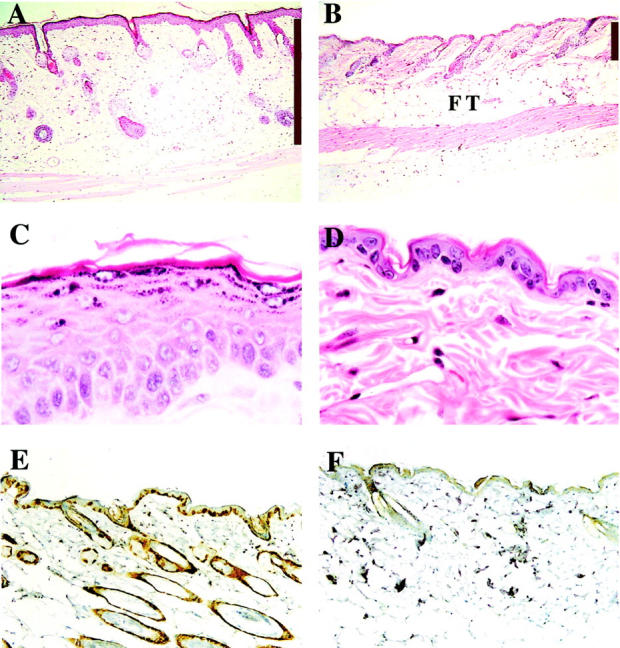
Skin phenotype of K5CDK4 transgenic mice. Representative paraffin-sections of skin from K5CDK4 transgenic (A and C) and normal sibling (B and D) mice were stained with H&E. Expression of CDK4 in transgenic (E) and wild-type skin (F) was determined with specific antibody. Antibody binding was detected by secondary antibody conjugated with horseradish peroxidase, and the reaction was developed with diaminobenzidine. Original magnifications: ×4 (A and B), ×40 (C and D). The bars indicate the thickness of the dermis (A and B). FT, fat tissue.
CDK4 Overexpression Results in Epidermal Hyperplasia, Hypertrophy, and Dermal Fibrosis
The newborn CDK4 transgenic mice did not demonstrate any obvious developmental abnormalities and there were no differences in size compared to wild-type littermates. To determine whether the expression of transgene influenced the rate of proliferation and/or the architecture of mouse skin, we analyzed the interfollicular epidermis of transgenic and normal sibling mice. Immunohistochemical staining for CDK4 expression in paraffin-embedded sections showed that both transgenic lines expressed high levels of CDK4 protein compared with normal sibling littermates (Figure 2, E and F) ▶ . This result correlated well with the CDK4 protein levels determined by Western blot (see Figure 6 ▶ ) and showed that expression of CDK4 by the keratin 5 promoter was occurring in the epidermal basal cell layer. Paraffin-embedded hematoxylin and eosin-stained sections were analyzed to determine modifications in the skin architecture of transgenic mice. The epidermis of wild-type mice consists of one to two nucleated cell layers (basal and suprabasal cell layers) and the hair follicles are regularly distributed (Figure 2, B and D) ▶ . Both transgenic lines show an increased proliferation rate and hyperplasia in areas of the interfollicular epidermis (Figure 2 ▶ and Figure 3 ▶ ). No obvious modifications in the morphology or in the pattern of distribution of hair follicles were observed in transgenic mice compared with wild-type littermates. The proliferation status of keratinocytes was determined by BrdU incorporation in DNA of cells in S phase. In the transgenic mice as well as in the wild-type littermates, proliferation of the epidermis was restricted to the basal layer (see Figure 4, A and B ▶ ). However, the number of proliferating cells was fourfold higher in the epidermis of transgenic mice (n = 7) compared with control mice (n = 9), demonstrating that overexpression of CDK4 stimulates proliferation of basal keratinocytes (Figure 4C) ▶ . The epidermis of transgenic mice was hyperplastic, presenting an increased number of nucleated cells (Figure 2 and 3) ▶ ▶ . The average thickness of transgenic epidermis was 21.36 μm compared to 10.60 μm in normal littermates (Figure 3B) ▶ . The spinous and granular layers were greatly increased as compared with the normal epidermis. A mild keratosis (accumulation of keratinized cells in the epidermal surface) with areas of parakeratosis were also observed in transgenic epidermis (Figure 2C) ▶ . In addition to epidermal changes, we observed dermal fibrosis with severe hypoplasia of the subcutaneous adipose tissue (panniculum adiposum) (Figure 2A) ▶ . The average thickness of the dermal tissue increased 2.8-fold in transgenic compared with normal sibling mice (Figure 2, A and B) ▶ . These data show that forced expression of cdk4 in epidermis results in an essential increase of both epidermis and dermis.
Figure 6.
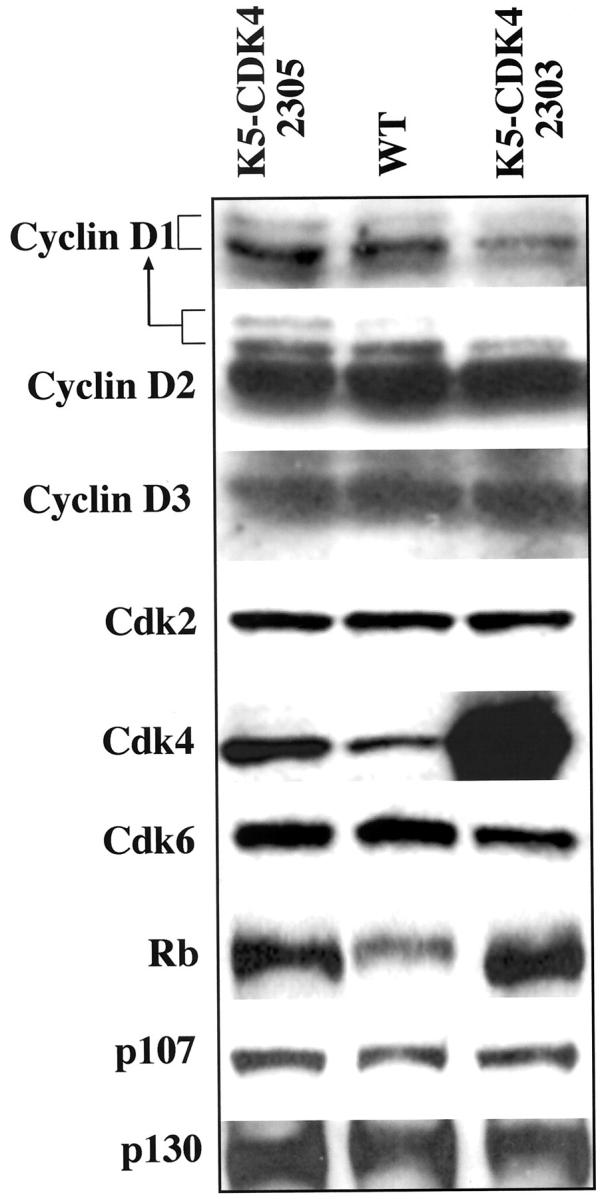
Western blot analysis of D-type cyclins, CDKs, and pRb family proteins in epidermis of transgenic and wild-type mice. Proteins were separated by SDS-PAGE and blotted onto a nitrocellulose membrane. Primary antibodies against CDK4, CDK6, CDK2, p107, p130, pRb, and each of the D-type cyclins were used for immunoblot analysis. The level of CDK4 protein was quantified with a densitometer.
Figure 3.
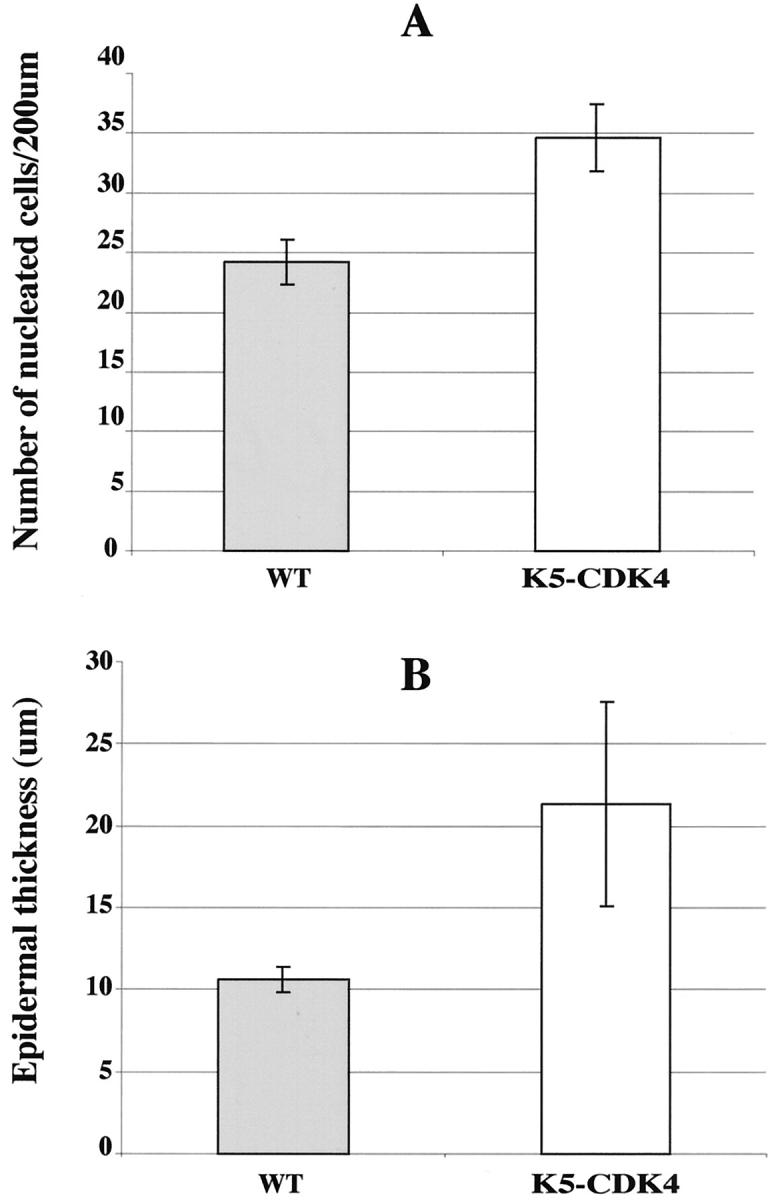
Quantification of epidermal thickness and hyperplasia in transgenic and normal sibling skin. A: The bars indicate the number of nucleated cells in 200 μm of interfollicular epidermis. B: The bars indicate the thickness of whole mouse skin in μm. Shaded bars, transgenic animals; open bars, normal sibling.
Figure 4.
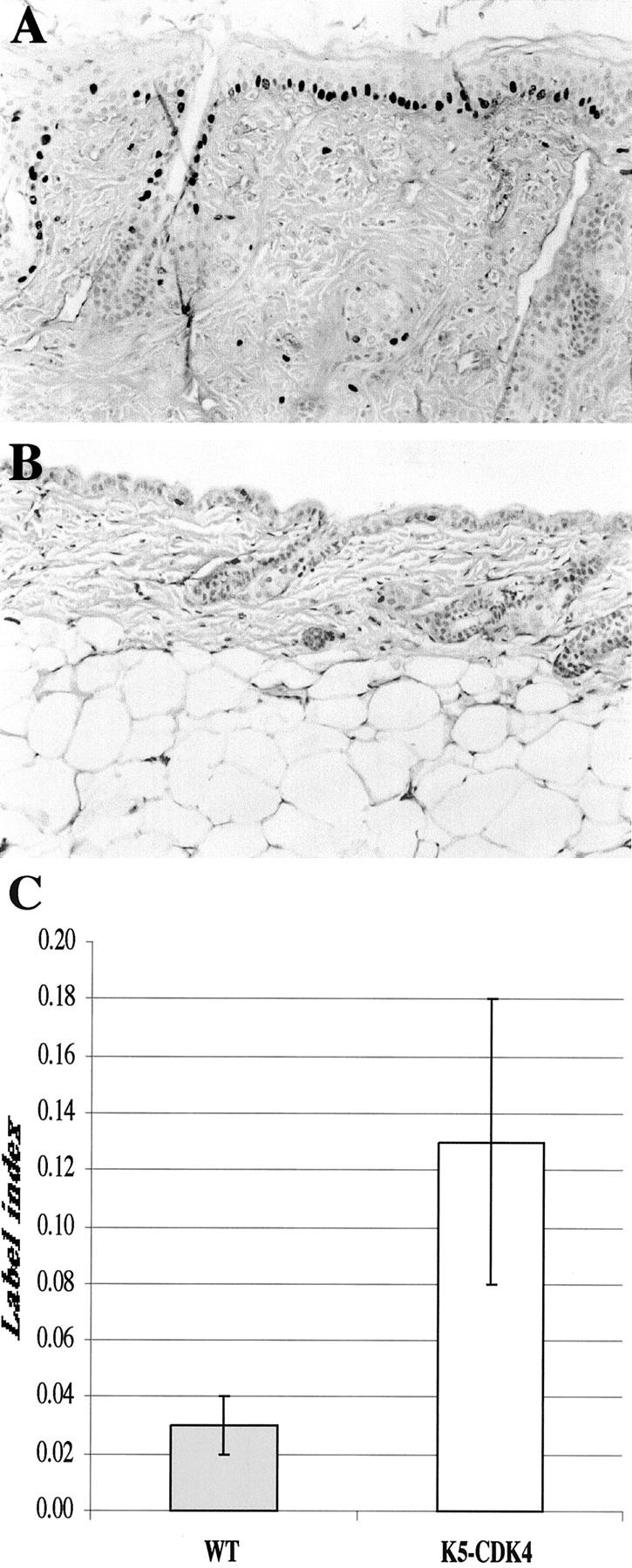
Epidermal proliferation in transgenic and normal sibling mice. BrdU incorporation in transgenic (A) and normal (B) skin was detected in paraffin sections with mouse anti-BrdU antibody and a secondary antibody conjugated with horseradish peroxidase. C: The bars indicate the label index or the percentage of BrdU incorporation in basal cell layer from interfollicular epithelia.
An unexpected result was the increase in the size of the nucleus and cells in the interfollicular epidermis and hair follicles of transgenic animals as compared to their normal siblings (Figure 2, C and D) ▶ . The average diameter of the transgenic nucleus in the basal and suprabasal cell layers was 10.5 μm whereas the wild-type animals had a diameter of 4.5 μm. Thus, the transgenic K5CDK4 nuclear size increased 2.3-fold compared to normal sibling mice. The more relevant difference between lines 2303 and 2305 is the extent of the hyperplasia, which was more severe in line 2305. A third transgenic line (2301) that was discontinued, showed the same phenotype as that of line 2305, indicating that the phenotype observed was independent of insertion site of the transgene.
Keratin Expression in Transgenic Mice
During epidermal differentiation there are sequential changes in the expression of the keratins. Keratin 5 and keratin 14 are the major products of basal epidermal cells, the proliferative compartment of the epidermis. 31 One of the earliest changes associated with the commitment to differentiation and migration into the spinous layer is the induction of the differentiation-specific keratin pair, K1/K10, 31-33 and decreased expression of the keratin pair, K5/K14. In addition, mouse skin tumors and hyperproliferative epidermis express keratins 6 and 16. 34-36 To study whether CDK4 expression affected the normal pattern of keratin expression, the expression of K5, K1, and K6 was analyzed by immunohistochemistry. The expression of K5 was observed in the basal and suprabasal layers in both wild-type and transgenic mice (Figure 5, A and B) ▶ . Keratin 1 was restricted to terminally differentiated cells as is seen in wild-type adult mice (Figure 5, C and D) ▶ . As expected, the wild-type mice showed no expression of keratin 6 (Figure 5E) ▶ , but interestingly the transgenic mice showed a very high level of expression of this keratin in the interfollicular epidermis (Figure 5F) ▶ . This result indicated that CDK4 overexpression in vivo induces cell proliferation and the expression of a marker associated with hyperproliferative changes and tumorigenesis.
Figure 5.
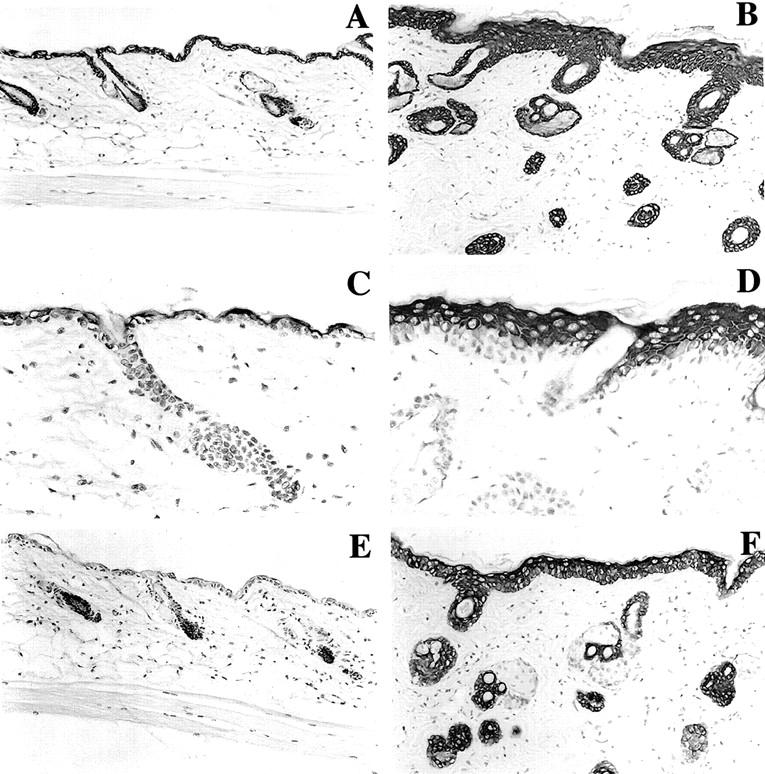
Keratin expression in transgenic and normal sibling mice. Keratins 1, 5, and 6 were determined on representative paraffin sections of skin from K5CDK4 transgenics (B, D, and F) and normal sibling (A, C, and E). Specific antibody binding was detected by secondary antibody conjugated with horseradish peroxidase, and the reaction was developed with diaminobenzidine. Keratin 5 (A, B), Keratin 1 (C, D) and Keratin 6 (E, F).
Expression of CDKs, Cyclins, and the pRb Family of Proteins
To quantify the level of CDK4 protein expression, we isolated the epidermis of transgenic and normal sibling mice. Protein lysates were used for immunoblotting analysis as was previously described. 25 The CDK4 transgene is expressed at high levels in both K5CDK4 lines compared to their normal siblings. CDK4 protein levels were elevated 7.5-fold in 2303 and 2.5-fold in 2305 transgenic lines (Figure 6) ▶ . Although line 2303 showed a higher level of CDK4 expression (Figure 6) ▶ , similar hyperproliferative changes were observed in both transgenic lines. As regulatory subunits of CDKs, D-type cyclins are rate-limiting controllers of G1 phase progression in mammalian cells. 2,3 CDK4 and CDK6 are the major catalytic partners of D-type cyclins, assembling them into holoenzymes. 37,38 To test whether the hyperproliferative phenotype of transgenic mice was because of an increased activity of CDK4 caused by elevated expression of the regulatory subunit, we analyzed the D-type cyclin protein levels. No differences in the protein levels of cyclin D1, cyclin D2, or cyclin D3 were observed between transgenic and wild-type animals (Figure 6) ▶ .
Because CDK4 and CDK6 have common functional and biochemical properties as pRb kinases, 39 we examined whether the CDK6 protein level in the epidermis of transgenic mice was affected as a compensatory mechanism. Western blotting analysis showed that the level of CDK6 protein was not affected in either of the transgenic lines (Figure 6) ▶ . Thus, there was no apparent compensatory reduction of CDK6 expression in the epidermis. No changes in the protein levels of CDK2, a protein kinase that is sequentially activated after CDK4,6 during G1/S interphase 7 were found in the two transgenic lines compared with the normal siblings (Figure 6) ▶ .
It is accepted that pRb is a negative regulator that acts in the G1 phase of the cell cycle 1,3,4,40 and it is a substrate of CDK4,6/D-type cyclins. p130 and p107 are pRb-related proteins that may have similar functions and also have been described as substrates of CDK4,6. 40-42 Therefore the phosphorylation status and protein level of the pRb family from epidermal lysates of transgenic and normal sibling mice were also analyzed. No evident changes in mobility that would be consistent with phosphorylation were detected in pRb, p107, or p130 (Figure 6) ▶ . Moreover, p130 and p107 protein levels did not increase in transgenic animals compared with the wild-type animals. On the other hand, pRb protein levels increased in both transgenic lines compared to normal siblings. This result is consistent with elevated pRb protein levels detected after proliferation induced by phorbol ester in mouse epidermis. 25 Taken together, these results showed that overexpression of CDK4 did not influence the protein level of its regulatory subunits (D-type cyclins) and did not increase the phosphorylation pattern of the pRb family, although, an increase in the level of pRb protein was detected.
CDK Activities and Complex Formation in Epidermal Tissue
To study whether CDK4 overexpression resulted in functional changes in the CDKs, we analyzed the kinase activity of CDK4 and CDK2 in the epidermis of transgenic and wild-type mice using pRb as a substrate. Figure 7A ▶ shows elevated CDK4 and CDK2 activity in K5CDK4 transgenics compared with normal sibling mice. No modification in the level of CDK6 activity was observed (data not shown). CDK4 and CDK2 activity increased 7- and 2.5-fold, respectively, in transgenic lines. The CDKs activities of wild-type epidermis were almost undetectable (Figure 7A) ▶ , therefore the increased activity in transgenic mice was easily observed.
Figure 7.
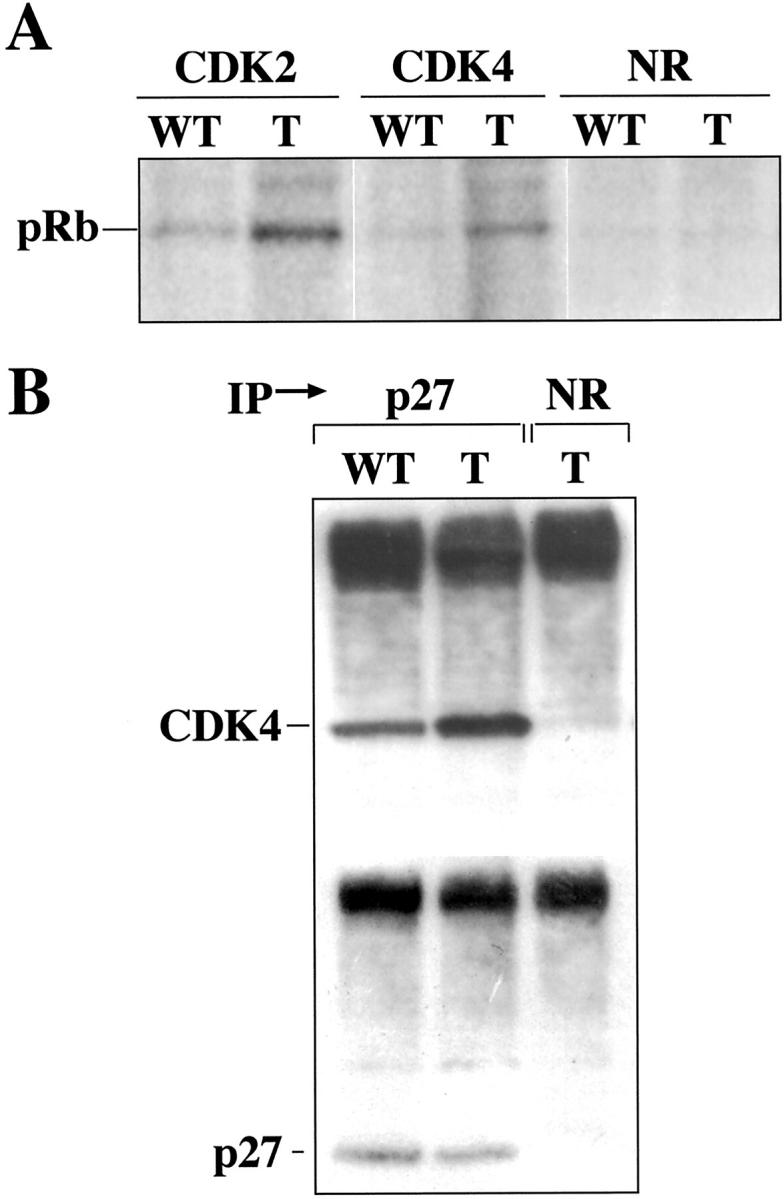
CDK activities and CDK/p27 complex formation in K5CDK4 mouse epidermis. A: Kinase activity of CDK4 and CDK2 from K5CDK4 (T) and normal sibling animals (wt). Fresh epidermal proteins were immunoprecipitated with specific antibodies and in vitro kinase assays were performed with a pRb peptide as substrate. B: Fresh epidermal proteins from transgenic (T) and wild-type mice (wt) were immunoprecipitated with a polyclonal antibody against p27Kip1 and immunoblotted with polyclonal antibody for CDK4 or p27Kip1. The control was immunoprecipitated with normal rabbit serum (NR).
Several reports have previously shown that p27Kip1 is associated with CDK activation by its redistribution between CDK4,6 and CDK2. 43,44 To study the participation of p27Kip1 in CDK activation, analysis of CDK4/p27Kip1 complexes was performed. We analyzed complex formation by immunoprecipitating with an antibody against p27Kip1 and detecting CDK4 by Western blot. Figure 7B ▶ shows that p27Kip1 increased binding to CDK4 1.5-fold in transgenic mice compared with wild-type animals. Therefore, the excess of CDK4 could release CDK2 from the inhibitory effect of p27Kip1, resulting in the observed increase of CDK2 activity (Figure 7A) ▶ . This result shows for the first time, in an in vivo system, that the noncatalytic function of CDK4, sequestering p27Kip1, may play an important role in hyperproliferation. p21Cip1 was not detected in these complexes, and its presence was almost nondetectable in normal mouse skin. 25
Discussion
In previous studies, we have shown that the components of the cdk-holoenzymes (cyclins and CDKs) were differentially regulated. Whereas phorbol ester (TPA) induced cyclin D1 expression, cyclin D2, D3, and CDK4 protein levels remained constant. 25 Also, cyclin D1 and cyclin D2 are overexpressed during mouse skin tumor development, whereas CDK4 and CDK6 protein levels do not change. 26 However, CDK4 and CDK6 activities are elevated in TPA-induced epidermal proliferation and in mouse skin tumors during progression (Rodriguez-Puebla and Conti, unpublished results). These data, together with CDK4 overexpression in certain human tumors, 23,45 led us to hypothesize that deregulated expression of G1 CDKs synergize with D-type cyclins during normal and neoplastic epidermal proliferation and likely take part in tumor progression. Then, our initial prediction was that CDK4 overexpression should result in hyperproliferation only if any of the D-type cyclins were also overexpressed. In this report, however, we showed that the severe hyperproliferative phenotype was independent of D-type cyclin overexpression.
Epidermal and Dermal Phenotype
Previously, we reported on the generation of cyclin D1, cyclin D2, and cyclin D3 transgenic mice that expressed each of the D-type cyclins under the bovine keratin 5 promoter. 27,29 These transgenic mice exhibited epidermal hyperplasia and, in some cases, thymic hyperplasia. These phenotypes are consistent with a positive regulator role of D-type cyclins in CDK4,6/cyclin complexes. The data shown here indicate that forced expression of CDK4 in mouse epidermal tissue results in strong skin abnormalities, specifically, interfollicular epidermal hyperplasia and hypertrophy, and severe dermal fibrosis. The stronger K5CDK4 phenotype compared with K5D1, K5D2, or K5D3 mice was not expected because D-type cyclins are rate-limiting factors in CDK complex formation in mouse epidermis. 25,26 In this sense, the protein level of cyclin D1 governs the CDK4/cyclin D1 complex formation in normal epidermis and murine skin tumors. 25,26 The epidermis thickness of the K5CDK4 animals was increased twofold whereas in the K5-D-type cyclin mice it increased an average of 0.25-fold. 29 Moreover, no development of dermal fibrosis was detected in any K5-D-type cyclin transgenic animals.
The hyperplastic epidermal phenotype of K5CDK4 mice seems to be very similar to that observed in hyperproliferative skin generated by chemical treatment. In these cases, an increase in the rate of proliferation is follow by a mild expansion of the proliferative compartment that is compensated by a concomitant expansion of the differentiated compartments. 46 However, the K5CDK4 transgenic epidermis also showed areas of parakeratosis (nucleated cells in the stratified layer), basal and suprabasal cells loosely packed, and increased cellular size (hypertrophy). Interestingly, this phenotype was unique for K5CDK4 mice because hyperplasia, but not hypertrophy was observed in the three D-type cyclin transgenic mice. 27,29 In fact, overexpression of CDK4 in mouse astrocytes also results in a unique phenotype that includes an increase in cell size as well as hyperploidy. 47 In support of these events, Cdk4/cyclin D overexpression in Drosophila caused hyperplasia in wing imaginal cells and hypertrophy in salivary glands and differentiating eyes. 11,12 Thereafter, future studies will be directed toward investigating whether the hypertrophy observed in K5CDK4 mouse keratinocytes is associated with hyperploidy. Another interesting hypothesis to be tested is whether members of the HDAC family are involved in the increase in nuclear size. The role of HDAC is to remove acetyl groups from the amino-terminal regions of histone octamers, thereby promoting nucleosome assembly 48 and inhibition of gene expression. Recent studies indicate that HDAC interacts with pRb; phosphorylation of pRb by CDK4 releases HDAC from this complex. 5,49,50 Whether CDK4 overexpression, phosphorylation of pRb, and HDAC release have any role in the nuclear structure observed in K5CDK4 transgenic mice requires further investigation.
Although the transgene is expressed in the epidermis and in hair follicles, we have also observed gross abnormalities in the dermis, which is characterized by replacement of adipose tissue with connective tissue. In addition, the dermal thickness increased 2.8-fold in transgenic compared with normal sibling mice. Munz and colleagues 51 reported on a similar phenotype in activin A (a member of transforming growth factor-β family) transgenic mice, which affects keratinocyte proliferation and also produces dermal fibrosis. This might be because of the diffusion of cytokines from the keratinocytes into the underlying dermis as has been reported. 51 This data demonstrated that dermal fibrosis could be produced by secretion of growth factors from the epidermis. Thus, whether expression of CDK4 in epidermis results in modifications in the expression of cytokines requires further investigation. If this is the case, up-regulation of a positive regulator of cell cycle (CDK4) could have important consequences in fibrotic diseases such as fibrotic kidneys and cirrhotic livers.
Modifications in Keratin Expression
Here, we showed that K5CDK4 transgenic mice express keratin 6 in the interfollicular epidermis and this event correlates well with the hyperproliferative phenotype. Keratin 6 is expressed in cell culture, benign and malignant epidermal tumors, and hyperplasia induced by the tumor promoter TPA, or wounding, 36,52 but not in normal mouse epidermis. Also, transgenic mice that overexpressed keratin 16 (the partner of keratin 6) in the basal-cell layer showed keratinocyte hypertrophy and hyperplasia with a significant increase in epidermal thickness. 53
One of the earliest changes associated with commitment to differentiation and migration into the spinous layer is induction of the differentiation-specific pair of keratins, K1/K10. 31,32 Keratin 1 was detected in the suprabasal cell layers, demonstrating that the cells entered into the differentiation program and that the additional layers seen in the epidermis of the transgenic mice consist, at least partially, of differentiated cells. Keratin 5 is normally expressed in the basal layer of wild-type and transgenic mice. Histological section staining with anti-loricrin antibody showed that this marker of terminal differentiation was normally distributed in the granular layer (data not shown). Taken together, these results demonstrate that CDK4 overexpression induced a marker of proliferation (K6), but did not influence the pattern of terminal differentiation.
Model of Cell Proliferation
We can postulate several mechanisms that could be responsible for the hyperproliferative phenotype of the mouse epidermis of K5CDK4 transgenic mice. First, CDK4 expression can act in a catalytic fashion and bind to D-type cyclins and/or other G1/S cyclins (cyclin E, cyclin A), and further increase the level of phosphorylation of the pRb family. This classic model suggests that overexpression of CDK4 can displace other CDKs and be activated by different types of cyclins. However, we were unable to detect CDK4 binding to other G1 cyclins. In support of these results, a recent report showed that cyclin E, in the knock-in cyclin D1/cyclin E mice, does not bind to CDK4. 54 Second, CDK4-D-type cyclins can also function in a noncatalytic fashion. As was demonstrated, CDK4 binds and sequesters CKI p27Kip1. Thus the CDK2-cyclin E and/or CDK2-cyclin A complexes are not inhibited by p27Kip1, and remain activated. 55 Likewise, transforming growth factor-β growth arrest depends on the inhibition of CDK4 synthesis and further inhibition of CDK2 activity by CKIs. 56 Also, CDK4 disruption was associated with increased binding of p27Kip1 to cyclin E/CDK2 and diminished activation of CDK2 was accompanied by impaired pRb phosphorylation. 44 Third, p16Ink4a and D-type cyclins bind to the same site on CDK4. 57 Thus, CDK4 protein is normally distributed in complexes with D-type cyclins or with the product of the tumor suppressor gene p16Ink4a. Thus, the excess of CDK4 protein in transgenic epidermis could result in sequestration of p16Ink4a and further formation of more CDK4-D-type cyclins complexes. In fact, the opposite effect was observed when p16Ink4a expression was induced in U2-OS cells. In this case, both CDK4- and CDK2-associated kinase activities were inhibited by redistribution of cyclin-CDK inhibitor complexes. 43,58 All of these models require phosphorylation of pRb by CDKs. The in vitro kinase assays showed that both CDK4 and CDK2 activities increased in the transgenic epidermis. Our assays of CDK4-CKI complex formation demonstrated that at least one CKI (p27Kip1) is sequestered by the increased levels of CDK4 and this event may be responsible for the elevated CDK2 activity and, in part, for the epidermis proliferation.
The fact that the level of D-type cyclin remained constant in transgenic mice compared with wild-type mice suggests that no elevations in the regulatory subunit levels are required to produce the hyperproliferative phenotype. Thus, the increased activity of CDK4 is not because of activation of D-type cyclins, so, the mechanism responsible for CDK4 activation must involve events other than simple binding to D-type cyclins. In accordance with these data, amplification of CDK4 in glioblastomas or glioma cell lines is not followed by amplification or elevated expression of cyclin D1. 23,45
Previously, we have reported that in murine skin tumors, p16Ink4a is overexpressed without changes in CDK4 expression. 26 On the other hand, in glioma cell lines, CDK4 amplification is an alternative mechanism to p16Ink4a deletion. 23 Thus, overexpression of CDK4 could overcome the increased level of p16Ink4a and play an important role in tumor development.
Together, our results showed that the noncatalytic or stoichiometric function of CDK4 sequestering p27Kip1 plays an important role in epidermal proliferation in vivo. Finally, biochemical studies and detailed analysis of CDK complex formation in the epidermis of transgenic and knockout mice should help in understanding how the absence or overexpression of CDK4 can affect the constitution of these complexes and how they affect cell growth and cell-cycle progression.
Acknowledgments
We thank April Weiss for help with the mouse experiments, the Science Park histology service for assistance with the immunohistochemical staining, Cassie Bigbee for technical support, Dr. Susan M. Fischer for helpful reading this paper, Melissa Bracher for secretarial assistance, and Sharon Stockman for editing the paper.
Footnotes
Address reprint requests to Marcelo L. Rodriguez-Puebla. The University of Texas, M. D. Anderson Cancer Center, Science Park–Research Division, Smithville, TX 78957. E-mail: mlrodri@odin.mdacc.tmc.edu.
Supported by National Institutes of Health grants CA 42157 and CA 57596; Center grant CA 16672 to M. D. Anderson Cancer Center; National Institute of Environmental Health Sciences grant P30-E507784-01; and funds from the University Cancer Foundation at the University of Texas M. D. Anderson Cancer Center.
References
- 1.Sherr CJ: G1 phase progression: cyclin on cue. Cell 1994, 79:551-555 [DOI] [PubMed] [Google Scholar]
- 2.Sherr CJ: Mammalian G1 cyclins. Cell 1993, 73:1059-1065 [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ: D-type cyclins. Trends Biochem Sci 1995, 20:187-190 [DOI] [PubMed] [Google Scholar]
- 4.Nevins JR: E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258:424-429 [DOI] [PubMed] [Google Scholar]
- 5.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC: Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98:859-869 [DOI] [PubMed] [Google Scholar]
- 6.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC: Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 2000, 101:79-89 [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa MH, Higasashi HK, Jung I, Suzuki-Takahashi M, Ikeda K, Tamai I, Kato K, Segawa E, Yoshida S, Nishimura S, Taya Y: The consensus motif for phosphorylation by cyclin D1-CDK4 is different from that for phosphorylation by cyclin A/E-CDK2. EMBO J 1996, 15:7060-7069 [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong Y, Hannon G, Zhang H, Casso D, Kobayashi R, Beach D: p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366:701-704 [DOI] [PubMed] [Google Scholar]
- 9.Xiong Y: Why are there so many CDK inhibitors? Biochim Biophys Acta 1996, 1288:1-5 [DOI] [PubMed] [Google Scholar]
- 10.Hirama T, Koeffler HP: Role of the cyclin dependent kinase inhibitors in the development of cancer. Blood 1995, 86:841-854 [PubMed] [Google Scholar]
- 11.Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF: Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J 2000, 19:4533-4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA: The Drosophila cyclinD-Cdk4 complex promotes cellular growth. EMBO J 2000, 19:4543-4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sicinski P, Donaher J, Parker S, Li T, Fazeli A, Gardener H, Haslam S, Bronson R, Elledge S, Weinberg R: Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995, 82:621-630 [DOI] [PubMed] [Google Scholar]
- 14.Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C: Mice lacking cyclin d1 are small and show defects in eye and mammary gland development. Genes Dev 1995, 9:2364-2372 [DOI] [PubMed] [Google Scholar]
- 15.Sicinski P, Donaher J, Geneg Y, Parker S, Garder H, Park M, Robker R, Richards J, McGinnis L, Biggers J, Epping J, Bronson R, Elledege S, Weinberg R: Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 1996, 384:470-474 [DOI] [PubMed] [Google Scholar]
- 16.Zwijsen R, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides R: CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997, 88:405-415 [DOI] [PubMed] [Google Scholar]
- 17.Knudsen K, Cavenee W, Arden K: D-type cyclins complex bind with the androgen receptor and inhibits its transcriptional transactivation ability. Cancer Res 1999, 59:2297-2301 [PubMed] [Google Scholar]
- 18.McHannon C, Suthiphongchai T, DiRenzo J, Ewen M: P/CAF associates with cyclin D1 and potentiates its activation of the estrogen receptor. Proc Natl Acad Sci USA 1999, 96:5382-5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuman E, Ladha M, Lin N, Upton T, Miller S, DiRenzo J, Pestell R, Hinds P, Dowdy S, Brown M, Ewen M: Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol 1997, 17:5338-5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohta M, Nagai H, Shimizu M, Rasio D, Berd D, Mastrangelo M, Singh AD, Shields JA, Shields CJ, Croce CM: Rarity of somatic and germline mutations of the cyclin-dependent kinase 4 inhibitor gene, CDK4I, in melanoma. Cancer Res 1994, 54:5269-5272 [PubMed] [Google Scholar]
- 21.Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa-Luonnet D, Bernard J, Bressac-de Paillerets R: Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. Hum Mol Genet 1998, 7:209-216 [DOI] [PubMed] [Google Scholar]
- 22.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Büschenfelde KH, Beach D: A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269:1281-1284 [DOI] [PubMed] [Google Scholar]
- 23.He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day RS, James CD: CDK4 amplification is an alternative mechanism to p16 homozygous deletion in glioma cell lines. Cancer Res 1994, 54:5804-5807 [PubMed] [Google Scholar]
- 24.Ichimura K, Schmidt EE, Goike HM, Collins VP: Human glioblastomas with no alterations of the CDKN2A and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996, 13:1065-1072 [PubMed] [Google Scholar]
- 25.Rodriguez-Puebla ML, Robles AI, Johnson DG, LaCava M, Conti CJ: Synchronized proliferation induced by TPA treatment of mouse skin: an in vivo model for cell cycle regulation. Cell Growth Differ 1998, 9:31-39 [PubMed] [Google Scholar]
- 26.Rodriguez-Puebla ML, LaCava M, Gimenez-Conti IB, Jonhson DG, Conti CJ: Deregulated expression of cell-cycle proteins during premalignant progression in SENCAR mouse skin. Oncogene 1998, 17:2251-2258 [DOI] [PubMed] [Google Scholar]
- 27.Robles AI, Larcher F, Whalin RB, Murillas R, Richie E, Gimenez-Conti IB, Jorcano JL, Conti CJ: Expression of cyclin D1 in epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad Sci USA 1996, 93:7634-7638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez-Puebla ML, LaCava M, Conti C: Cyclin D1 overexpression in mouse epidermis increases cyclin-dependent kinase activity and cell proliferation in vivo but does not affect skin tumor development. Cell Growth Differ 1999, 10:467-472 [PubMed] [Google Scholar]
- 29.Rodriguez-Puebla ML, LaCava M, Miliani de Marval PL, Jorcano JL, Richie E, Conti CJ: Cyclin D2 overexpression in transgenic mice induces thymic and epidermal hyperplasia whereas cyclin D3 expression results only in epidermal hyperplasia. Am J of Pathol 2000, 157:1039-1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramirez A, Bravo A, Jorcano J, Vidal M: Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation 1994, 58:53-64 [DOI] [PubMed] [Google Scholar]
- 31.Woodcock-Mitchell J, Eichner R, Nelson WG, Sun T: Immunolocalization of keratin polypeptides in human epidermis using monoclonal antibodies. J Cell Biol 1982, 95:580-588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roop D, Hawley-Nelson P, Cheng CK, Yuspa SH: Keratin gene expression in mouse epidermis and cultured epidermal cells. Proc Natl Acad Sci USA 1983, 80:716-720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schweizer J, Kinjo M, Furstenberger G, Winter H: Sequential expression of mRNA-encoded keratin sets in neonatal mouse epidermis: basal cells with properties of terminally differentiating cells. Cell 1984, 37:159-170 [DOI] [PubMed] [Google Scholar]
- 34.Knapp B, Rentrop M, Schweizer J, Winter H: Three cDNA sequences of mouse type I keratins. Cellular localization of the mRNAs in normal and hyperproliferative tissues. J Biol Chem 1987, 262:938-945 [PubMed] [Google Scholar]
- 35.Moll R, Moll I, Franke W: Differences of expression of cytokeratin polypeptides in various epithelial skin tumors. Arch Dermatol Res 1984, 276:349-363 [DOI] [PubMed] [Google Scholar]
- 36.Weiss RA, Eichner R, Sun T: Monoclonal antibody analysis of keratin expression in epidermal diseases: a 48- and 56-kdalton keratin as molecular markers for hyperproliferative keratinocytes. J Cell Biol 1984, 98:1397-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsushine H, Ewen ME, Strom DK, Kato J-Y, Hanks SK, Roussel MF, Sherr CJ: Identification and properties of an atypical catalytic subunit (p34/cdk4) for mammalian D type G1 cyclins. Cell 1992, 71:323-334 [DOI] [PubMed] [Google Scholar]
- 38.Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G: Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene 1994, 9:1633-1640 [PubMed] [Google Scholar]
- 39.Bates S, Bonetta L, MacAllan D, Parry D, Holder A, Dickson C, Peters G: CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct set of the cyclin-dependent kinases that associate with cyclin D1. Oncogene 1994, 9:71-79 [PubMed] [Google Scholar]
- 40.Ewen M: The cell cycle and the retinoblastoma protein family. Cancer Metastasis Rev 1994, 13:45-66 [DOI] [PubMed] [Google Scholar]
- 41.Hannon GJ, Demetrick D, Beach D: Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev 1993, 7:2378-2391 [DOI] [PubMed] [Google Scholar]
- 42.Ewen ME, Xing YG, Lawrence JB, Livingston DM: Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell 1991, 66:1155-1164 [DOI] [PubMed] [Google Scholar]
- 43.McConnell B, Gregory F, Stott F, Hara E, Peters G: Induced expression of p16Ink4a inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol 1999, 19:1981-1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsutsui T, Hesabi B, Moons DS, Pandolfi P, Hansel K, Koff A, Kiyokawa H: Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity. Mol Cell Biol 1999, 19:7011-7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmit E, Ichimura K, Reifenberger G: CdkN2 (p16/MTST1) gene deletion or Cdk4 amplification occurs in the majority of glioblastomas. Cancer Res 1994, 54:6321-6324 [PubMed] [Google Scholar]
- 46.Klein-Szanto AJP: Morphological evaluation of tumor promoter effects on mammalian skin. Slaga TJ eds. Mechanisms of Tumor Promotion, Tumor Promotion and Skin Carcinogenesis. 1984, :pp 42-72 CRC Press, Boca Raton [Google Scholar]
- 47.Holland EC, Hively WP, Gallo V, Varmus HE: Modeling mutations in the G1 arrest pathway in human gliomas: overexpression of CDK4 but not loss of INK4a-ARF induces hyperploidy in cultured mouse astrocytes. Genes Dev 1998, 12:3644-3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE: Histone deacetylase activity is required for full transcriptional repression by msin3a. Cell 1997, 89:341-347 [DOI] [PubMed] [Google Scholar]
- 49.Brehem A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T: Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391:597-601 [DOI] [PubMed] [Google Scholar]
- 50.Magnaghi JL, Groisman R, Naguibnueva I, Robin P, Lorain S, Le VJ, Troalen F, Trouche D, Harel BA: Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 1998, 391:601-605 [DOI] [PubMed] [Google Scholar]
- 51.Munz B, Smola H, Engelhard F, Bleuel K, Brauchle M, Lein I, Evans LW, Huylebroeck D, Balling R, Werner S: Overexpression of activin A in the skin of transgenic mice reveals new activities of activin in epidermal morphogenesis, dermal fibrosis and wound repair. EMBO J 1999, 18:5205-5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roop DR, Mehrel T, Krieg K, Nakazawa H, Cheng CK, Yuspa SH: Keratin expression in mouse epidermal tumors. Carcinog Compr Surv 1989, 11:257-271 [PubMed] [Google Scholar]
- 53.Paladini RD, Coulombe PA: Directed expression of keratin 16 to the progenitor basal cells of transgenic mouse skin delays skin maturation. J Cell Biol 1998, 142:1035-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P: Rescue of cyclin D1 deficiency by knockin cyclin E. Cell 1999, 97:767-777 [DOI] [PubMed] [Google Scholar]
- 55.Sherr CJ, Roberts JM: CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999, 10:1491-1502 [DOI] [PubMed] [Google Scholar]
- 56.Ewen ME, Sluss HK, Whitehouse L, Livingston DM: TGFB inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell 1993, 74:1009-1020 [DOI] [PubMed] [Google Scholar]
- 57.Coleman KG, Wautlet BS, Morrissey D, Mulheron J, Sedman SA, Brinkley P, Price S, Webster KR: Identification of CDK4 sequences involved in cyclin D1 and p16 binding. J Biol Chem 1997, 272:18869-18874 [DOI] [PubMed] [Google Scholar]
- 58.Parry D, Mahony D, Wills K, Lees E: Cyclin-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol 1999, 19:1775-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]