

Abstract
Fibrosis is an unregulated tissue repair process whose predominant characteristics are the proliferation of myofibroblasts and an excessive deposition of extracellular matrix. Transforming growth factor (TGF)-β1 is considered as one of the most fibrogenic cytokines. However, the molecular mechanisms involved in its profibrotic role are not fully understood. Here, we addressed the role of TGF-β1 on cell proliferation and intracellular signal transduction in a pig model of skin fibrosis induced by γ-irradiation. Primary myofibroblasts were isolated from the fibrotic tissue and their response to TGF-β1 was compared to that of normal skin fibroblasts. The present results show that the differentiation of myofibroblasts involves a lack of TGF-β1 growth inhibition and an impaired TGF-β1 signaling. Receptor activity and Smad2/4 or Smad3/4 complex formation were similar in both cell types after TGF-β1 treatment. However, the translocation of Smad3 protein into the nucleus was reduced in myofibroblasts as compared to that in fibroblasts, as well as its binding to target DNA sequences and the activation of the Smad binding elements found in the PAI-1. Interestingly, Smad2 was translocated similarly to the nucleus in both cell types suggesting that this protein may function normally in myofibroblasts. We propose that uncoupling of antiproliferative and profibrotic actions of TGF-β1 in fibrosis may occur through differential regulation of the activities of Smad2 and Smad3 transcription factors.
Fibrotic disorders can affect various organs, including skin, lung, kidney, and liver. A major process in fibrosis development is the transdifferentiation of fibroblastic cells into myofibroblasts. Myofibroblasts acquire contractile properties like smooth muscle cells and are defined morphologically through identification of expressed cytoskeletal proteins, and particularly α-smooth muscle actin, which is not expressed by fibroblasts in vivo. 1,2
Fibrosis reflects a pathological excess of biological events involved in normal tissue repair rather than specific events. Both in wound healing and fibrosis, the major role of myofibroblasts is to secrete matrix molecules and proteases that allow repair and remodeling, and to contract the scar. In normal wound healing, feed-back mechanisms down-regulate the repair process after scar contraction, whereas in fibrosis this does not occur. For example, fibroblast activation is a transitory process in wound healing, as myofibroblasts disappear by apoptosis in the last steps of the repair process, 3 whereas in fibrosis activation of fibroblasts and myofibroblast proliferation are chronic processes.
Among the various factors that have been implicated in the development of fibrosis, including platelet-derived growth factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), and endothelin, transforming growth factor (TGF)-β1 seems to be the most potent fibrogenic cytokine. 4-6 Several lines of evidence point to a causal relation between elevated production of TGF-β1 and tissue fibrosis. 7,8 First, in vivo administration of TGF-β1 in healthy animals produces tissue fibrosis. Second, transgenic mice that overexpress TGF-β1 in specific organs develop severe fibrotic diseases in these organs. On the contrary, down-regulation of TGF-β1 by neutralizing antibodies or by antisense can inhibit the development of a fibrotic tissue.
TGF-β1 is a multifunctional cytokine exerting in vivo three main biological activities. 9 It is a strong inhibitor of cell growth, it exerts immunosuppressive activities, and it regulates the deposition of extracellular matrix components. As regards the fibrotic process, important actions of TGF-β1 are related to the deposition of extracellular matrix and to cell differentiation. TGF-β1 causes the deposition and remodeling of extracellular matrix by simultaneously stimulating cells to increase the synthesis of most matrix proteins, decrease the production of matrix-degrading proteases and increase the production of inhibitors of these proteases, and modulate the expression of integrins. 4 When fibroblasts are treated with TGF-β1, they adopt the morphology of an activated myofibroblast, express specific markers such as α-smooth muscle actin, and secrete high levels of collagen matrix. 10
From all these results, TGF-β1 is now considered as a key molecule and a master switch for the general fibrotic program. However, the molecular mechanisms involved in its profibrotic role are not fully understood. For example, the effect of TGF-β1 on cell proliferation is unclear in the fibrotic process. One important function of TGF-β1 is to inhibit cell cycle progression by blocking the late G1 activation of cyclin-dependent kinases, thereby preventing phosphorylation of the retinoblastoma protein and S phase entry. 11 This physiological function of TGF-β1 does not fit well with the development of the fibrotic process, which implies overproliferation of various cell types.
New tools are now available to better understand the cellular responses to TGF-β1, such as those provided by the discovery of the Smad signaling pathway specific to TGF-β1. 12-15 The activities of TGF-β1 are mediated by signaling through a unique set of receptors having intrinsic serine-threonine kinase activity, the type I and II receptors. Activation of the receptor complex is initiated on binding of the ligand to TGF-β1 receptor II (TβR-II), which then recruits the TGF-β1 receptor type I (TβR-I) and transphosphorylates its glycine-serine-rich domain. The activated TβR-I subsequently propagates the signal to downstream intracellular targets, which are a new family of intracellular proteins termed “ Smads.” Smad2 and Smad3 are receptor-activated Smad proteins (R-Smads) that are phosphorylated by TβR-I kinase and subsequently hetero-oligomerize with the common mediator Smad4. The activated Smad complex translocates to the nucleus, where it activates the transcription of target genes.
Comparisons among Smads have revealed the presence of conserved amino (MH1) and carboxy-terminal (MH2) domains, which both mediate interactions with a number of DNA binding proteins, and a central linker region. The MH1 domain is important for mediating the DNA-binding activity of Smads, whereas the MH2 domain has transcriptional activating activity. Smad3 and Smad4 have been shown to bind to the specific nucleotide sequence AGAC, called the SBE for Smad binding element. A number of such Smad3/Smad4 binding elements have been characterized in the promoters of TGF-β1 responsive genes. These include genes encoding the plasminogen activator inhibitor PAI-1, 16 SMAD7, 17,18 type I and VII collagens, c-jun, and junB.
Although Smad2 is structurally highly similar to Smad3, and both Smad2 and Smad3 are phosphorylated by Tβ-RI receptor, they do not share similar DNA binding activity. This notable difference in the mechanisms of transcriptional activation by Smad2 and Smad3 involves an insertion of 30 amino acids in the N-terminal MH1 domain of Smad2, which precludes its binding to DNA. 19 Accordingly, Smad2 is involved in transcriptional activation only by associating with other regulatory proteins, capable of DNA binding. An example of this is found in the activation of the goosecoid gene. The transcription factor FAST2 binds to the promoter of this gene, but efficient transcriptional activation requires TGF-β1 stimulation and the formation of a complex containing FAST2, Smad2, and Smad4. 20
In addition to these positively acting Smads, the inhibitory Smad7 antagonizes the TGF-β1 signal by interacting with the receptor complex, thus preventing access and phosphorylation of R-Smads. 21
Fibrosis can be induced by radiotherapy. We previously reported that TGF-β1 is involved in the progression of radiation-induced fibrosis. 8 This result was obtained in a standard model of cutaneous and muscular fibrosis developed in the pig.
This model has been characterized using histology, biochemistry, and molecular biology techniques. 22-24 In the present study, we wanted to address in the same model the role of TGF-β1 on cell proliferation, as well as the profibrogenic molecular mechanisms of TGF-β1 at the level of signal transduction. For that purpose, we isolated myofibroblasts and fibroblasts from fibrous and normal pig skin, respectively. We previously demonstrated that the pig myofibroblasts isolated from the fibrotic skin retain most of their specific phenotype when they are studied in primary culture, including increased synthesis of extracellular matrix components, 25,26 altered cytoskeleton, 27 and high TGF-β1 secretion 8,27 as compared to normal fibroblasts. In this work, we show that the specific differentiation of myofibroblasts also involves alterations of the TGF-β1 response. These cells exhibit a lack of TGF-β1 growth inhibition associated with a down-regulation of Smad3 activity.
Materials and Methods
Cell Culture and Chemicals
Cells were maintained in a water-saturated 5% CO2/95% air atmosphere at 37°C. The medium used was Dulbecco’s modified Eagle’s medium (4.5 g/L glucose, ICN, Costa Mesa, CA) routinely supplemented with 10% fetal calf serum, 10 mmol/L HEPES, 4 mmol/L l-glutamine, and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin, ICN). Recombinant human TGF-β1 was purchased from R&D systems (Oxon, UK) (no. 240-B-002).
Experimental Model
Studies were performed on cells isolated from a pig experimental model of skin and muscular fibrosis induced by high-dose exposure to γ-rays. 22 Large White pigs were irradiated on the external side of the right thigh. Six months later, the fibrotic tissue was surgically removed from the thigh and the normal skin was removed from the left flank of each animal. Myofibroblasts and fibroblasts were enzymatically isolated from fibrotic or normal tissue, respectively, and directly frozen. Primary cultures (passage 0) were established from the frozen pool as previously described 28 with some modifications. Cells were seeded on collagen I-coated flasks (Biocoat, Falcon) at a density of 5000 cells/cm 2 and cultivated for 4 days in Dulbecco’s modified Eagle’s medium supplemented with 20% fetal calf serum, 10 mmol/L HEPES, 4 mmol/L l-glutamine, and antibiotics (500 U/ml penicillin and 500 μg/ml streptomycin, ICN) and from then until confluence in standard medium (∼100,000 cells/cm2). After trypsin detachment, fibroblasts were seeded on classic flasks (Nunclon) at 4000 cells/cm 2 and cultured in the standard medium (passage 1). All experiments were performed on cells at passage 1, because we previously found that these cells still exhibited a phenotype close to the one found in vivo in the tissues. This phenotype was progressively lost in later passages as regards for example matrix production or proliferation rate. 28,25 In the present work, cells from five animals were used.
Statistical Analyses
All experiments presented were done in triplicate with cells from at least two different animals. Results were expressed as the mean ± SD. Statistical comparison of two means was done by Student’s unpaired t-test. P < 0.05 was considered to be statistically significant.
Immunohistochemistry
Samples of normal and fibrotic pig skin were fixed in 10% formalin-buffered solution and embedded in paraffin wax. Serial 5-μm sections were cut, dewaxed, and treated for immunohistochemical detection. To unmask antigen, a chemical and thermic treatment was performed by incubating sections in a 0.01 mol/L sodium citrate buffer, pH 6, at 95°C for 5 minutes. Then, sections were incubated in a 0.3% H2O2 in phosphate-buffered saline (PBS) solution for 10 minutes and in a 3% bovine serum albumin in PBS solution for 30 minutes, respectively, to quench endogenous peroxidases and to block nonspecific sites. Sections were incubated overnight at 4°C in a PBS solution containing a mouse monoclonal anti proliferating cell nuclear antigen (DAKO, Glostrup, Denmark) at a dilution of 1:500. This primary antibody was revealed by the LSAB 2 kit (DAKO). Briefly, slides were submitted to sequential 10-minute incubations with biotinylated link antibody and peroxidase-labeled streptavidin followed by a diaminobenzidine (DAB+, DAKO) revelation and a counterstaining by hematoxylin.
Plasmids
pTK-RL allows expression of the Renilla luciferase from the constitutive tyrosine kinase promoter and was used as an internal control in the transfection assay. pCAGA-MLP allows expression of the Firefly luciferase (luciferase) reporter gene from a synthetic promoter containing nine SMAD3-responsive CAGA elements. 16 This vector was kindly supplied by Dr. Gauthier (Laboratoire Glaxo Wellcome, Les Ulis, France). pSBE-S7x1 and pSBE-S7x3 allow regulation of the luciferase gene from one or three SMAD7 SBE, respectively. They were constructed as follows: oligonucleotides TCGACAGGGTGTCTAGACGGCCACG and TCGACGTGGCCGTCTAGACACCCTG were annealed, leading to a double-stranded DNA containing one SMAD7 SBE 18 and TCGA single-strand extremity, and cloned into pGL3-E1bTATA 29 digested with XhoI. PCS2/Flag-Smad2, pCMV5/Flag-DPC4, pRK5Flag-Smad3, pcDNA3Flag-Smad7, pcDNA3-TβR-I/HA, and pCMV5-TβR-II/HIS are expression vectors for human Smad2, Smad4, Smad3, Smad7, TβR-I (ALK5), and TβR-II proteins, respectively. These vectors were kindly supplied by Drs. Massagué (Memorial Sloan Kettering Cancer Center, New York), Derynck, and ten Dijke (The Netherlands Cancer Institute, Amsterdam, The Netherlands).
Transfection of Cells and Reporter Gene Assay
Transient transfections were performed using the FuGENE™6 reagent (no.1 814 443; Boehringer Mannheim, Mannheim, Germany) according to the manufacturer and supercoiled DNA was isolated with an endo-free Qiagen column kit (Qiagen, Inc., Valencia, CA). Unless otherwise indicated, cells were seeded in 24-well (2 cm2/well) plates at 20,000 cells per well. Twenty-four hours later, medium was changed with 0.5 ml of fresh complete or otherwise indicated medium and 10 μl of medium were added, containing 1 μl of FuGENE™6, 500 ng of reporter plasmid, if necessary, 5 ng of each expression vector used, and 80 ng of the internal control pRL-TK. At 24 hours after transfection, cells were at 60% confluence and were incubated with TGF-β1. Luciferase activity was measured 24 hours later using the Dual-luciferase reporter assay system (Promega, Madison, WI) and a Berthold Microplaque luminometer EG&G LB 96 V. The Renilla luciferase activity was used to normalize the transfection efficiency. Fold activation is the ratio between the normalized luciferase activity obtained after TGF-β1 treatment and the corresponding value obtained without TGF-β1 treatment.
Protein Extraction
Culture flasks (75 cm2) were emptied of their medium, placed on ice, and the cell monolayers rinsed twice with 5 ml of PBS before adding 500 μl of RIPA buffer containing anti-proteases and antiphosphatases, to lyse the cells during 20 minutes on a 3D agitator. Lysed cells were scraped from the flasks and centrifuged in Eppendorf tubes for 20 minutes at 4°C, 13,000 × g and the supernatants were aliquoted and frozen at −80°C until use. Nuclear protein extracts were prepared as described by Schreiber and colleagues. 30 Using this widely used technique to prepare the nuclear extracts, the highly expressed Hsp47, a protein that is linked to cell membranes, 31 is present in the nuclear fraction of cellular proteins and thus we used Hsp47 in each experiment to control protein loadings.
Electrophoretic Mobility Shift Assay
The double-strand oligonucleotide used is that used to construct pCAGA-MLP. 16 It contains three repetitions of the AGCCAGAC Smad-binding element. Electrophoretic mobility shift assay was performed basically as previously described. 32 DNA-protein interactions were performed in a final volume of 23.5 μl containing 10 μl of nuclear extracts containing 5 μg of proteins, 10 μl of binding buffer (20 mmol/L HEPES, 8 mmol/L MgCl2, glycerol 20%, 8 mmol/L spermidine, 11.1 ng/μl of single-strand sonicated salmon sperm DNA, 0.1 μg/μl of poly-dIdC), 1 ng of radiolabeled probe resuspended in 1 μl and 2 μl of antibodies (400 ng) where necessary. This mixture was incubated for 20 minutes at room temperature and DNA-protein complexes were separated by electrophoresis on a 4% polyacrylamide gel in 0.5× Tris borate-ethylenediaminetetraacetic acid.
Protein Analysis
Twenty μg of proteins were separated under reduced conditions on 10% polyacrylamide gels according to Laemmli. 33 Western blotting was done on Protran BA83 (0.2-μm membranes; Schleicher & Schuell, Keene, NH). These membranes were blocked overnight at 4°C in PBS with 5% nonfat milk, then incubated at room temperature for 90 minutes with primary antibodies, washed 2 × 7 minutes with PBS/0.2% Tween 20, and then incubated with the appropriate secondary antibody coupled to horseradish peroxidase. Specific protein bands were revealed by enhanced chemiluminescence (Amersham-Pharmacia, Buckinghamshire, UK) on Kodak BioMax film (Eastman-Kodak, Rochester, NY). The following primary antibodies were used: goat polyclonal anti-Smad2 (sc6200; Santa Cruz Biotechnology, Santa Cruz, CA), Smad3 (sc6202, Santa Cruz), Smad2/3 (sc6033, Santa Cruz), TβR-II (AF 241-NA, R&D), mouse monoclonal anti-Smad4 (sc7966, Santa Cruz), rabbit polyclonal anti-TβR-I (sc399, Santa Cruz), phospho-Smad2 (06–829, Upstate Biotechnology, Lake Placid, NY). Sample deposits on gels were controlled using the glyceraldehyde-3-phosphate dehydrogenase marker. Molecular weight standards were Rainbow markers (Amersham-Pharmacia, Orsay, France).
Immunoprecipitation of total cell extracts was performed overnight with the indicated antibodies in 1.8-ml Eppendorf tubes on a slowly rotating wheel. The immune-complexes were recovered on Protein-G Sepharose beads (Amersham-Pharmacia), washed in RIPA buffer, and then heated for 5 minutes in Laemmli buffer, followed by electrophoresis on 10% polyacrylamide gels. These gels were then treated as for the Western blotting experiments using anti-Smad 4.
Results
Proliferative Response to TGF-β1 Is Altered in Myofibroblasts
High expression of TGF-β1 has been previously described in the radiation-induced skin fibrosis model used in this study. 8,23 To reveal the proliferative status of myofibroblasts in this particular environment, sections of fibrotic tissue were labeled with an antibody against proliferating cell nuclear antigen (PCNA), a proliferative marker. This tissue exhibited a high density of myofibroblasts, and a large majority of them were PCNA-positive as compared to fibroblasts from normal skin (Figure 1a) ▶ . This indicated that, despite the presence of an abnormally high level of TGF-β1, myofibroblasts were proliferating. To evaluate the TGF-β1 response of myofibroblasts, we compared the proliferative effect of exogenous TGF-β1 on this cell type and the fibroblasts. In this experiment, as in the other in vitro experiments presented in this article, cells were used at passage 1. Proliferation of the fibroblasts was inhibited by high (4 ng/ml) as well as low (0.1 ng/ml) concentrations of TGF-β1 (Figure 1) ▶ . In contrast, proliferation of the myofibroblasts was not inhibited by the low dose of TGF-β1 and only weakly by the high dose of TGF-β1 (Figure 1) ▶ . These results indicated that the proliferative response to TGF-β1 is altered in myofibroblasts as compared to fibroblasts.
Figure 1.

Regulation of myofibroblast proliferation by TGF-β1. a: Immunostaining of normal dermis or fibrotic tissue with an antibody directed against PCNA. White and black arrows show examples of PCNA-negative and -positive cells, respectively. b: Proliferation in primary cell culture was estimated by measuring the BrdU incorporation during DNA synthesis. Cells (3500 per well) were seeded in 96-well plates and cultivated for 24 hours in 0.2 ml of medium containing 10% serum. Then cells were maintained for 48 hours in medium containing 1 or 2.5% of serum. Medium was changed every 24 hours. Thereafter, cells were incubated in fresh similar medium for 24 hours with or without TGF-β1 and for an additional 8 hours with BrdU. Cell proliferation was measured with a cell proliferation ELISA BrdU kit. Results are expressed as the percentage of BrdU incorporation in treated cells as compared to the nontreated cells. Percent noted on the right indicates the percentage of serum used. For a given dose of TGF-β1, asterisks indicate that the difference of response between fibroblasts and myofibroblasts was significant (P > 0.05).
TGF-β1-Dependent Activation of the PAI-1 SBE Is Impaired in Myofibroblasts
Because TGF-β1 acts mainly through activation of the Smad pathway, we examined the ability of exogenous TGF-β1 to activate a Smad-responsive promoter in myofibroblasts. We used the well-characterized synthetic Smad3/Smad4-responsive promoter containing a repetition of the PAI-1 SBE sequence described by Dennler and colleagues. 16 Fibroblasts and myofibroblasts were transfected with the luciferase reporter plasmid pCAGA-MLP and incubated for 24 hours with increasing doses of TGF-β1. In fibroblasts, TGF-β1 induced a dose-dependent activation of the luciferase reporter gene that rose to a 40-fold up-regulation. In myofibroblasts, this up-regulation was, whatever the dose of TGF-β1, 4–6-fold less important (Figure 2A) ▶ . The response of cells from five different animals has been analyzed without notable variation from one animal to another (Table 1) ▶ . To avoid circumstantial influence, the same experiment was performed with cells at different stages of proliferation, with different concentrations of serum, and on collagen I-coated plastic. Whatever the conditions used, the up-regulation of the reporter gene in response to TGF-β1 was 4–6-fold less in myofibroblasts as compared to fibroblasts (Figure 2B) ▶ . To test directly the interaction of the Smad proteins with the SBE, a gel retardation assay was performed (Figure 2C) ▶ . Nuclear proteins from untreated fibroblasts exhibited no SBE-binding activity. After 30 minutes of TGF-β1 treatment, a strong SBE-binding activity appeared and led to the formation of a major retarded complex. Addition of specific antibodies revealed that this complex contained Smad3 and Smad4 but not Smad2. In myofibroblasts, a TGF-β1-inducible SBE-binding activity was also seen, but the extent of Smad3/Smad4-SBE complex formation was strongly reduced as compared to fibroblasts. From these results, we concluded that myofibroblasts exhibited an impaired activation of the Smad pathway in response to TGF-β1.
Figure 2.
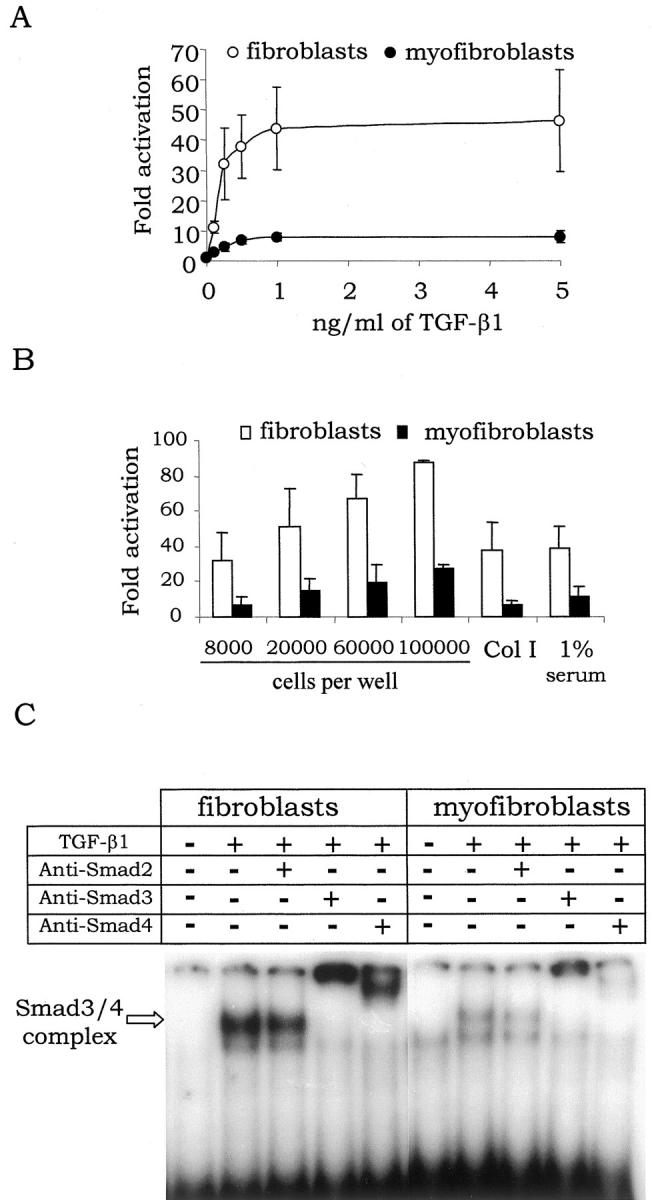
TGF-β1-dependent activation of PAI-1 SBE in fibroblasts and myofibroblasts. A: Fibroblasts and myofibroblasts were transfected with the pCAGA-MLP reporter plasmid and stimulated for 24 hours with the indicated dose of TGF-β1 before measurement of the luciferase activity. For each animal used, transfection efficiency of fibroblasts and myofibroblasts was similar, as revealed by an equal expression in both cell types of the Renilla luciferase from the internal control vector. Furthermore, for each experiment, basal expression level of the Firefly luciferase from the reporter vector was also similar in both cell types. B: TGF-β1 response of cells was measured as in A but with different growth conditions: with cells seeded at different concentrations before the transfection step (8000, 20,000, and starting from 60,000 cells per well correspond, respectively, to nonconfluent, semiconfluent, and confluent before TGF-β1 was added); with cells seeded on plastic coated or not with collagen I (Col I); and with cells incubated in 1% serum starting from the transfection. C: Confluent fibroblasts or myofibroblasts were incubated in 1% serum for 24 hours and then treated 30 minutes with 2 ng/ml of TGF-β1. Nuclear extracts from these cells were used to perform a gel retardation assay with the SBE probe. C is a mount of a single film.
Table 1.
Luciferase Activation in Response to TGF-β1
| Animal | Fibroblast | Myofibroblasts | Ratio | n | P |
|---|---|---|---|---|---|
| 1 | 47.6 ± 15.7 | 7.7 ± 3.1 | 6.2 | 8 | 0.001 |
| 2 | 79.9 ± 21 | 18.2 ± 7.5 | 4.4 | 4 | 0.01 |
| 3 | 28.5 ± 4.1 | 7.9 ± 0.4 | 3.6 | 3 | 0.001 |
| 4 | 36.6 ± 8.5 | 8.5 ± 2.8 | 4.3 | 4 | 0.001 |
| 5 | 33.8 ± 5.3 | 7 ± 2.6 | 4.8 | 5 | 0.001 |
Level of Expression of the Smad Pathway Elements Is Not Affected in Myofibroblasts
The low response of the myofibroblasts to TGF-β1 could be because of an impaired constitutive expression of one or several elements of the Smad pathway. As shown by Western blotting analysis of the total protein extracts, the amount of TβR-I and TβR-II, of Smad2, Smad3, and Smad4 was similar in myofibroblasts and fibroblasts (Figure 3) ▶ . Variation of the amount of these proteins after TGF-β1 treatment was tested. We found that after 24 hours of incubation with 1 ng/ml of TGF-β1, the amount of TβR-I, TβR-II, Smad2, Smad3, and Smad4 was unchanged. Therefore, the low TGF-β1 response of myofibroblasts is not because of an altered expression of one of the TGF-β1 pathway elements.
Figure 3.

Level of expression of the Smad pathway elements in fibroblasts and myofibroblasts. Total protein extracts from fibroblasts and myofibroblasts cultivated to 70 to 80% confluence were used to compare by Western blotting the amount of the indicated Smad pathway elements. Glyceraldehyde-3-phosphate dehydrogenase was used as a control of protein loading. The positions of the molecular weight markers are indicated on the right. For each protein analyzed, the figure represents a mount of a single film.
Smad3 Is a Limiting Factor of the PAI-1 SBE Activation in Myofibroblasts
To determine the factors that limit the response of the myofibroblasts to TGF-β1, we overexpressed each element of the Smad pathway and looked at the TGF-β1-dependent activation of the PAI-1 SBE in myofibroblasts and in fibroblasts. As expected, overexpression of Smad2, which is not involved in the PAI-1 SBE activation, had no effect on the reporter gene expression in both cell types (Figure 4A) ▶ . Overexpression of Smad4, TβR-I, TβR-II, or co-overexpression of both subunits of the TGF-β1 receptor had no or little effect on the response of both cell types to TGF-β1 (Figure 4A) ▶ . Overexpression of Smad7, the Smad inhibitor of the TGF-β1 response, reduced to a similar extent (∼50%) the TGF-β1 response of fibroblasts and myofibroblasts, indicating that this protein, despite its inhibitor function, was probably not the limiting factor of the myofibroblasts response to TGF-β1. Overexpression of Smad3 had a strong and differential effect on myofibroblasts and fibroblasts: Smad3 overexpression enhanced the TGF-β1 response of both cell types but this effect was threefold more important in myofibroblasts than in fibroblasts (Figure 4A) ▶ . Therefore, it seemed that Smad3 was a limiting factor of the myofibroblasts response to TGF-β1. The observation that Smad activation could be restored in myofibroblasts suggested that the TGF-β1 receptor was functional in myofibroblasts. To verify this point, we next examined the phosphorylation of Smad2 in response to TGF-β1. For this purpose, we used an antibody specifically directed against the receptor-dependent phosphorylated form of Smad2. As shown in Figure 4B ▶ , this protein was similarly phosphorylated in myofibroblasts and in fibroblasts. Taken together, these results indicated that Smad3 limited the activation of the PAI-1 SBE despite the presence of a functional receptor.
Figure 4.

Analysis of the Smad pathway activity. A: Effect of overexpression of the Smad pathway elements on TGF-β1 response. Cells were co-transfected with the pCAGA-MLP reporter plasmid and with vectors allowing the expression of the indicated proteins under the control of a CMV promoter and were stimulated or not for 24 hours with 1 ng/ml of TGF-β1 before luciferase measurement. Using a CMV-luciferase construct, we verified that this promoter exhibited a similar activity in the two cell types. For each cell type and protein studied, results are expressed as the ratio between the TGF-β1-dependent activation factor found with the expression vector and that found with the empty vector. The effect of overexpression was significantly different (P < 0.05) between both cell types only when Smad3 was used. B: TGF-β1-dependent phosphorylation of Smad2. Total protein extracts from fibroblasts and myofibroblasts cultivated in 10% serum up to 70 to 80% of confluence and treated for 0 or 2 hours with 2 ng/ml of TGF-β1 were used to detect by Western blotting the induction of C-terminal phosphorylation of Smad2 with an anti-phospho-Smad2 antibody. For each protein, the figure shown is a mount of a single film.
The Activity of Smad3 Is Affected in Myofibroblasts
Despite a normal level of expression for the Smad3 protein in total cellular extracts, Smad3 limited the TGF-β1-dependent activation of the PAI-1 SBE. Activation of Smad3 by the TGF-β1 receptor leads to its phosphorylation, its association with Smad4, and to the translocation of this complex into the nucleus, where it regulates gene transcription. To study Smad3 activity, we investigated the formation of Smad3 and Smad2 complexes with Smad4 and the nuclear translocation of these proteins in response to TGF-β1. Co-immunoprecipitation of Smad4 with either Smad3 or Smad2 in total cell proteins after TGF-β1 treatment showed that Smad3 and Smad2 associated with Smad4 to the same level in both cell types (Figure 5A) ▶ . However, nuclear translocation of Smad proteins occurred differently in myofibroblasts and in fibroblasts (Figure 5B) ▶ . Quantification of Smad translocation indicated that Smad3 was 2.5-fold less translocated in myofibroblasts than in fibroblasts, whereas Smad2 translocation was similar (Figure 5C) ▶ . This result indicated that Smad3 may be specifically affected in myofibroblasts. To test this hypothesis, we studied the responsive elements that enable an assessment of Smad2 transcriptional activity. To our knowledge, only two elements of this type have been characterized. The first one is ARE-1, which is specifically activated by Smad2 and the Fast-1 co-factor. 20 The second one is the SBE found in SMAD7, which is activated by both Smad2 and Smad3. 18,34 Unfortunately, ARE-1 was not functional in the pig fibroblasts, so we have used the Smad7 SBE. Fibroblasts and myofibroblasts were transfected with luciferase reporter plasmids containing the SMAD7 SBE and incubated with TGF-β1. Similarly to PAI-1, myofibroblasts exhibited a reduced response to TGF-β1 as compared to fibroblasts. However, the difference was only twofold (Figure 5D) ▶ , as compared to fivefold for PAI-1 SBE. These results show that the difference of response to TGF-β1 between fibroblasts and myofibroblasts depends on the Smads implicated in the regulation of the SBE used.
Figure 5.

Analysis of Smad2 and Smad3 activities. A: Association of R-Smads with Smad4 after TGF-β1 treatment. Confluent fibroblasts and myofibroblasts cultured in 10% serum were serum-starved for 12 hours and treated for 30 minutes with 2 ng/ml of TGF-β1. Total protein extracts were immunoprecipitated using antibody against either Smad2 or Smad3. The immunoprecipitates were used to detect by Western blotting the presence of the Smad4 protein. Western blots show first Smad4 detected from the immunoprecipitate obtained with an antibody against Smad2 (IP Smad2), then Smad4 after IP against Smad3 (IP Smad3). The last mount is a control showing Smad4 detection by direct Western blotting of the total protein extracts and the loading control glyceraldehyde-3-phosphate dehydrogenase. B: Nuclear translocation of Smad proteins after TGF-β1 treatment. Nuclear extracts were used to measure Smad translocation in the nucleus by Western blotting. Smad2 and Smad3 were simultaneously revealed by the Smad2/3 sc6033 antibody. Hsp47 was used as a loading control. The same results were obtained with nonconfluent cells or with cells cultivated in 1% serum (data not shown). C: Quantification of R-Smad translocation. Nuclear translocation from five independent experiments on six cell lines were quantified using Molecular Analyst. The amounts of Smad proteins translocated in the nucleus of fibroblasts were used as the reference (100%). The differences concerning Smad3 were found significant. (P < 0.05). D: TGF-β1-dependent activation of SMAD7 SBE. Fibroblasts and myofibroblasts cultivated in 10% serum were transfected with pSBE-S7x1 or pSBE-S7x3, containing either one or three copies of the SMAD7 SBE upstream of the luciferase reporter gene, and stimulated or not for 24 hours with 1 ng/ml of TGF-β1 before measurement of the luciferase activity.
Discussion
TGF-β1 has been described as a key molecular switch for fibroproliferative diseases. Some of its effects on the fibrotic process, although incompletely described, are clearly profibrotic, such as the increase of extracellular matrix deposition. However, other known effects of TGF-β1, such as inhibition of proliferation, do not fit well with the development of the fibrotic process, which is characterized by cell overproliferation.
To address this issue in a model of skin fibrosis, we studied the effect of this cytokine on a specific cell type, the myofibroblast, which is the major effector cell type in the fibrotic tissue, and in primary cultures, to retain its differentiation status. Thus myofibroblasts isolated from skin fibrotic tissue were incubated with TGF-β1, and their proliferative response was compared to that of fibroblasts isolated from normal skin. We found that fibroblasts were significantly inhibited by TGF-β1. Thus, concerning these cells, TGF-β1 exerted its widespread inhibitory role. By contrast, myofibroblasts seemed to be almost unresponsive to TGF-β1, as they were not growth inhibited by low doses of the cytokine and reacted poorly to high doses.
To understand the molecular mechanisms of this lack of response to TGF-β1, we studied the activation of the Smad binding element of the PAI-1 promoter in both cell types. This SBE is known to respond specifically to the Smad3/Smad4 complex. 16 Using a gel retardation assay, we found a significantly reduced DNA-binding activity of Smad3/Smad4 proteins in TGF-β1-treated myofibroblasts as compared to fibroblasts. The functional consequences of these altered interactions, assessed by a gene reporter assay, were that Smad-binding elements were highly activated in fibroblasts stimulated by TGF-β1, whereas myofibroblasts exhibited a fivefold reduced response. These results were highly reproducible, as whichever animal provided the primary cells and whatever the culture conditions used, myofibroblasts always exhibited a similar reduced binding activity and an impaired PAI-1 SBE activation.
We made several hypotheses to explain this result: the impaired TGF-β1 signaling could be related to activation of the inhibitors of the pathway or to alterations of an activating component of the pathway, by mutation or deregulation.
Inhibitory Smads act by preventing activation of the receptor-activated Smads (R-Smads) by competition for association with the receptor complex, thereby preventing phosphorylation and association of R-Smads with Smad4. 21 In our myofibroblast model, Smad2 phosphorylation occurred normally after TGF-β1 treatment as compared to fibroblasts, as well as the association of both Smad2 and Smad3 with Smad4. Furthermore, overexpression by transfection of Smad7, which is the major TGF-β1 inhibitory Smad protein, induced a similar 50% decrease of the PAI-1 SBE activation in both cell types, suggesting that Smad7 regulation was similar in both cell types. Taken together, these results indicate that the impaired TGF-β1 signaling in myofibroblasts was not because of an overactivation of a Smad inhibitory protein and notably of Smad7.
Concerning the mutational hypothesis, we have not sequenced the corresponding genes because we had several results that did not support this explanation. First, the myofibroblast response to TGF-β1 was only partially inhibited and this altered response was found in myofibroblasts from all five animals tested, indicating that it was a stable phenomenon that seems incompatible with the low probability of partial inactivation by specific mutations. Secondly, we previously described that the specific differentiation of fibroblasts and myofibroblasts, for example concerning collagen synthesis, 25 is not stable when cells are cultivated for several passages. Concerning the SBE activation, we observed that the difference of response to TGF-β1 between myofibroblasts and fibroblasts tended to disappear after five passages, such that a similar response was observed at passage 10 (data not shown). This result indicates that this phenomenon is more probably linked to the specific differentiation status of the myofibroblasts rather than to a mutation.
So we addressed the hypothesis that the impaired TGF-β1 signaling in myofibroblasts is related to the down-regulation of an activating component of the pathway, and we tried to identify this component. In other studies, impaired cell responses to TGF-β1 have been associated with alterations in TGF-β1 receptor activity. 35,36 This is not the case in our model for several reasons. Both subunits of the receptor were equally expressed in the two cell types, overexpression of these receptor subunits did not modify the activation of PAI-1 SBE in myofibroblasts and, more importantly, Smad2 phosphorylation and association of Smad2 and Smad3 with Smad4 occurred normally in myofibroblasts after TGF-β1 treatment, as compared to fibroblasts. For the elements of the Smad pathway located downstream of the receptor (Smad2, Smad3, and Smad4), similar protein amounts were found in total proteins of both cell types. However, when nuclear proteins were studied, Smad3 amount was found 2.5-fold decreased in myofibroblasts after TGF-β1 treatment, as compared to fibroblasts. Moreover, only the overexpression of the Smad3 protein in myofibroblasts could correct the altered activation of PAI-1 SBE in response to TGF-β1, which then became similar to that of fibroblasts. As all of the processes linked to Smad3 signaling in the cell cytoplasm were found normal, whereas Smad3 amounts and activity were altered in the nucleus, these results indicate that nuclear activity of Smad3 is a limiting factor of the myofibroblast response to TGF-β1. The results that we obtained concerning Smad2, the other R-Smad activated by TGF-β1, leave many questions open. Our results indicate that this protein seems to function normally in myofibroblasts: its phosphorylation, association with Smad4 complexes, and more importantly its nuclear translocation were similar in both cell types in response to TGF-β1. To address Smad2 transactivating activity, we used the SMAD7 SBE, which is an element responsive to both Smad2/Smad4 and Smad3/Smad4 complexes. 18,34 We found that the activation of this SBE was less altered in myofibroblasts than that of the PAI-1 SBE, which is responsive only to Smad3/Smad4 complex. This result fits well with the hypothesis that Smad2 activity is normal in myofibroblasts, whereas Smad3 activity is reduced. Experiments using responsive elements specific to Smad2/Smad4 complexes could allow a better demonstration of this point. However, there is only one such published element, found in the FAST-1 promoter, 20 and we found that this element was not functional in the pig fibroblasts.
What could be the consequences of a specific alteration of Smad3 activity for the fibrotic process? The present results on fibrosis cells can be compared with those recently published on wound healing. When mice are inactivated for the Smad3 gene, they show accelerated cutaneous wound healing characterized by an increased rate of re-epithelialization, a reduced local inflammatory infiltrate, and a normal matrix deposition by fibroblasts. 37 From these studies, a specific role for Smad3 was proposed, based on an uncoupling of antiproliferative and profibrotic TGF-β1 effects. Inhibition of proliferation and induction of the early inflammatory reaction could be Smad3-dependent processes, whereas matrix deposition could be a Smad3-independent process. The authors proposed that, in later phases of normal wound healing, increased levels of TGF-β1 in the wound might lead to suppression of Smad3 activity. 38 A parallel can be made between these wound repair studies in mutant mice and fibrosis, as it is thought that the fibrotic process implies deregulation of the processes involved in wound repair. In fibrosis, uncoupling of antiproliferative and profibrotic actions of TGF-β1 may occur through differential regulations of Smad2 and Smad3 activities. Our results obtained on normal primary cells support the view that such uncoupling may occur in vivo during the differentiation of normal cells. We propose that inactivation of Smad3 in myofibroblasts allows hyperproliferation of these cells in the fibrotic environment with high TGF-β1 concentrations, whereas normal Smad3 activation is necessary for the inhibition of fibroblast proliferation in normal skin. Concerning Smad2, retention of the normal Smad2 activity could allow the myofibroblasts to respond to other stimulatory effects of TGF-β1 related to matrix deposition and cell differentiation, so that Smad2 might be sufficient to permit the chronic deposition of extracellular matrix in fibrosis conditions. An important issue will be to define the particular target genes of these transcription factors, to better understand their respective role in the control of matrix deposition and cell proliferation.
Acknowledgments
We thank J.-M. Gauthier for his help, support and comments concerning this work. We thank J Massagué, R. Derynck, C.-H. Heldin, and P. ten Dijke for kindly providing vectors. We also thank G. Frelat for his support of this work, J.-L. Lefaix for his help and comments, as well as J.-J. Leplat and Y. Tricaud for technical assistance.
Footnotes
Address reprint requests to Dr. Michèle Martin, Service de Génomique Fonctionnelle, Départment de Radiobiologie et Radiopathologie, Direction des Sciences du Vivant, Commissariat à l’Energie Atomique, 2, rue Gaston Crémieux-CP 5722, 91057 Evry Cedex, France. E-mail: martin@dsvidf.cea.fr.
Supported by European Community research grant n°FI4P-CT95-0029 and by the Comité de Radioprotection d’Electricité de France.
References
- 1.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB: Myofibroblasts. : I. Paracrine cells important in health and disease. Am J Physiol 1999, 277:C1-C9 [DOI] [PubMed] [Google Scholar]
- 2.Serini G, Gabbiani G: Mechanisms of myofibroblast activity and phenotypic modulation. Exp Cell Res 1999, 250:273-283 [DOI] [PubMed] [Google Scholar]
- 3.Desmouliere A, Redard M, Darby I, Gabbiani G: Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995, 146:56-66 [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts AB, Sporn MB (eds): The transforming growth factor-βs. Peptide Growth Factors and Their Receptors, vol I. Berlin, Springer-Verlag, 1990, pp 419–472
- 5.Border WA, Noble NA: Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994, 331:1286-1292 [DOI] [PubMed] [Google Scholar]
- 6.Blobe GC, Schiemann WP, Lodish HF: Role of transforming growth factor beta in human disease. N Engl J Med 2000, 342:1350-1358 [DOI] [PubMed] [Google Scholar]
- 7.Bottinger EP, Kopp JB: Lessons from TGF-beta transgenic mice. Miner Electrolyte Metab 1998, 24:154-160 [DOI] [PubMed] [Google Scholar]
- 8.Martin M, Lefaix J, Delanian S: TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys 2000, 47:277-290 [DOI] [PubMed] [Google Scholar]
- 9.Lawrence DA: Transforming growth factor-beta: a general review. Eur Cytokine Netw 1996, 7:363-374 [PubMed] [Google Scholar]
- 10.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G: Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 1993, 122:103-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saltis J: TGF-beta: receptors and cell cycle arrest. Mol Cell Endocrinol 1996, 116:227-232 [DOI] [PubMed] [Google Scholar]
- 12.Massague J, Wotton D: Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 2000, 19:1745-1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piek E, Heldin CH, ten Dijke P: Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J 1999, 13:2105-2124 [PubMed] [Google Scholar]
- 14.Massague J: TGF-beta signal transduction. Annu Rev Biochem 1998, 67:753-791 [DOI] [PubMed] [Google Scholar]
- 15.Derynck R, Zhang Y, Feng XH: Smads: transcriptional activators of TGF-beta responses. Cell 1998, 95:737-740 [DOI] [PubMed] [Google Scholar]
- 16.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM: Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 1998, 17:3091-3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagarajan RP, Zhang J, Li W, Chen Y: Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J Biol Chem 1999, 274:33412-33418 [DOI] [PubMed] [Google Scholar]
- 18.von Gersdorff G, Susztak K, Rezvani F, Bitzer M, Liang D, Bottinger EP: Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem 2000, 275:11320-11326 [DOI] [PubMed] [Google Scholar]
- 19.Dennler S, Huet S, Gauthier JM: A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 1999, 18:1643-1648 [DOI] [PubMed] [Google Scholar]
- 20.Labbe E, Silvestri C, Hoodless PA, Wrana JL, Attisano L: Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell 1998, 2:109-120 [DOI] [PubMed] [Google Scholar]
- 21.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P: Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389:631-635 [DOI] [PubMed] [Google Scholar]
- 22.Lefaix JL, Martin M, Tricaud Y, Daburon F: Muscular fibrosis induced after pig skin irradiation with single doses of 192Ir gamma-rays. Br J Radiol 1993, 66:537-544 [DOI] [PubMed] [Google Scholar]
- 23.Martin M, Lefaix JL, Pinton P, Crechet F, Daburon F: Temporal modulation of TGF-beta 1 and beta-actin gene expression in pig skin and muscular fibrosis after ionizing radiation. Radiat Res 1993, 134:63-70 [PubMed] [Google Scholar]
- 24.Lefaix JL, Daburon F: Diagnosis of acute localized irradiation lesions: review of the French experimental experience. Health Phys 1998, 75:375-384 [DOI] [PubMed] [Google Scholar]
- 25.El Nabout R, Martin M, Remy J, Kern P, Robert L, Lafuma C: Collagen synthesis and deposition in cultured fibroblasts from subcutaneous radiation-induced fibrosis. Matrix 1989, 9:411-420 [DOI] [PubMed] [Google Scholar]
- 26.Wegrowski J: Fibronectin and glycosaminoglycan synthesis by fibrotic pig fibroblasts in primary culture. Connect Tissue Res 1989, 23:237-249 [DOI] [PubMed] [Google Scholar]
- 27.Vozenin-Brotons MC, Sivan V, Gault N, Renard C, Geffrotin C, Delanian S, Lefaix JL, Martin M: Antifibrotic action of Cu/Zn SOD is mediated by TGF-β1 repression and phenotypic reversion of myofibroblasts. Free Radic Biol Med 2001, 30:30-42 [DOI] [PubMed] [Google Scholar]
- 28.Martin M, Remy J, Daburon F: Abnormal proliferation and aging of cultured fibroblasts from pigs with subcutaneous fibrosis induced by gamma irradiation. J Invest Dermatol 1989, 93:497-500 [DOI] [PubMed] [Google Scholar]
- 29.Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi JJ: Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev 1998, 12:2102-2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber E, Matthias P, Muller MM, Schaffner W: Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 1989, 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tasab M, Batten MR, Bulleid NJ: Hsp47: a molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J 2000, 19:2204-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin M, Vozenin MC, Gault N, Crechet F, Pfarr CM, Lefaix JL: Coactivation of AP-1 activity and TGF-beta1 gene expression in the stress response of normal skin cells to ionizing radiation. Oncogene 1997, 15:981-989 [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 34.Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S: Participation of smad2, smad3, and smad4 in transforming growth factor beta (TGF-beta)-induced activation of smad7. J Biol Chem 2000, 275:29308-29317 [DOI] [PubMed] [Google Scholar]
- 35.McCaffrey TA: TGF-betas and TGF-beta receptors in atherosclerosis. Cytokine Growth Factor Rev 2000, 11:103-114 [DOI] [PubMed] [Google Scholar]
- 36.Roulot D, Sevcsik AM, Coste T, Strosberg AD, Marullo S: Role of transforming growth factor beta type II receptor in hepatic fibrosis: studies of human chronic hepatitis C and experimental fibrosis in rats. Hepatology 1999, 29:1730-1738 [DOI] [PubMed] [Google Scholar]
- 37.Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB: Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1999, 1:260-266 [DOI] [PubMed] [Google Scholar]
- 38.Ashcroft GS, Roberts AB: Loss of Smad3 modulates wound healing. Cytokine Growth Factor Rev 2000, 11:125-131 [DOI] [PubMed] [Google Scholar]