

Abstract
Myocarditis in humans is often associated with an autoimmune process in which cardiac myosin (CM) is a major autoantigen. Experimental autoimmune myocarditis (EAM) is induced in mice by immunization with CM. We found that EAM in A/J mice exhibits a Th2-like phenotype demonstrated by the histological picture of the heart lesions (eosinophils and giant cells) and by the humoral response (association of IgG1 response with disease and up-regulation of total IgE). Blocking interleukin (IL)-4 with anti-IL-4 monoclonal antibody (mAb) reduced the severity of EAM. This reduction in severity was associated with a shift from a Th2-like to a Th1-like phenotype represented by a reduction in CM-specific IgG1; an increase in CM-specific IgG2a; an abrogation of total IgE response; a decrease in IL-4, IL-5, and IL-13; as well as a dramatic increase in interferon (IFN)-γ production in vitro. Based on the latter finding, we hypothesized that IFN-γ limits disease. Indeed, IFN-γ blockade with a mAb exacerbated disease. The ameliorating effect of IL-4 blockade was abrogated by co-administration of anti-IFN-γ mAb. Thus, EAM represents a model of an organ-specific autoimmune disease associated with a Th2 phenotype, in which IL-4 promotes the disease and IFN-γ limits it. Suppression of IFN-γ represents at least one of the mechanisms by which IL-4 promotes EAM.
Myocarditis is a major cause of heart failure and sudden death among adolescents and young adults. 1 Although some myocarditis patients recover, many progress to dilated cardiomyopathy, an often fatal condition and a frequent reason for cardiac transplantation. 2 Many cases of myocarditis in humans are associated with an autoimmune process in which cardiac myosin (CM) is a major autoantigen. 3,4 Treatment of autoimmune myocarditis is challenging, with immunosuppressive therapies giving mixed results. 5 The design of better treatment regimens depends on understanding the mechanisms leading to the immune-mediated damage to the heart. To study these mechanisms, we have previously established a murine model of experimental autoimmune myocarditis (EAM) induced by immunization with CM. 6 In susceptible mouse strains, such as A/J and BALB/c, EAM represents a model of acute myocarditis with the peak of inflammation in the heart corresponding to day 21 after immunization.
Cytokines, such as tumor necrosis factors and interleukin (IL)-1, have been shown to play a key role in the development of EAM. 7-9 The role of other cytokines, including those characteristic of Th1 and Th2 immune responses, is unclear. Depending on the cytokine milieu, T-helper lymphocytes can differentiate into two subsets, Th1 and Th2, which represent functionally polarized and mutually antagonistic immune responses. 10,11 Interferon (IFN)-γ and IL-4 are prototypic Th1- and Th2-type cytokines, respectively. IFN-γ stimulates Th1 T-cell development, activates macrophages, induces MHC class I and II expression, promotes delayed-type hypersensitivity reactions, induces Ig class switching to IgG2a in mice, recruits Th1 T cells to the site of the inflammation, plays an important role in clearing intracellular bacteria and intracellular parasites, and exhibits antiviral activity. 12,13 IL-4, on the other hand, stimulates Th2 T-cell development, activates B cells, induces MHC class II expression on B cells, promotes allergic reactions, induces Ig class switching to IgE and IgG1 in mice, recruits eosinophils and Th2 cells to the site of inflammation, and is important in clearing extracellular parasites. 13-15 Extensive studies using other models of autoimmune disease have led to the conclusion that Th1 responses promote autoimmune processes whereas Th2 responses may suppress them. 10,11
The goal of this study was to investigate the balance of Th1 and Th2 responses in the development of EAM. We found that EAM exhibits a Th2-like phenotype both in terms of the histological composition of the heart infiltrate and of the humoral response. Based on these findings, we hypothesized that IL-4 plays a critical role in promoting EAM. Indeed, neutralization of IL-4 with anti-IL-4 monoclonal antibody (mAb) during the course of EAM induction significantly reduced the severity of disease as well as the markers of a Th2 response, and enhanced the production of IFN-γ by splenocytes in vitro. Blocking IFN-γ with a mAb markedly exacerbated disease providing evidence for a limiting effect of IFN-γ on the disease process. We conclude that the suppression of a disease-limiting factor, IFN-γ, represents a plausible pathogenetic mechanism of IL-4 action.
Materials and Methods
Induction of Myocarditis by Immunization
Myocarditis was induced in 5- to 7-week-old female A/J mice obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained in The Johns Hopkins University School of Medicine conventional animal facility. CM was purified from murine hearts according to the procedure by Shiverick and colleagues. 16 On days 0 and 7, mice received subcutaneous injections of either 250 μg of CM or 100 nmol of a peptide from cardiac α-myosin heavy chain [myhcα(334-352), synthesized by Macromolecular Resources, Colorado State University, Department of Biochemistry, Fort Collins, CO] emulsified in Complete Freund’s Adjuvant (CFA) (Sigma Chemical Co., Saint Louis, MO) supplemented with 5 mg/ml of Mycobacterium tuberculosis strain H37Ra (Difco, Detroit, MI). On day 0, mice received an intraperitoneal injection of 500 ng of pertussis toxin (List Biological Laboratories, Campbell, CA). Mice were sacrificed on day 21. All animal work was approved by the Animal Care and Use Committee of The Johns Hopkins University.
Induction of Myocarditis by Splenocyte Transfer
Donor A/J mice were immunized with either CM or myhcα(334-352) as described above. On day 21, spleens were aseptically removed and splenocytes were collected. Red blood cells were lysed by incubation with a lysing buffer (Quality Biological Inc., Gaithersburg, MD) for 5 minutes. Splenocytes were cultured for 3 days in RPMI 1640 (Life Technologies, Rockville, MD) with additional supplementation, as we have previously described, 17 in the presence of 10 μg/ml of CM. After stimulation, splenocytes were washed in phosphate-buffered saline (PBS) and counted by trypan blue exclusion. Recipient female 5- to 7-week-old A/J mice were irradiated (500 rads) 1 day before the transfer. Splenocytes were injected into a tail vein in a dose of 5 × 10 7 cells/mouse. Control mice received similarly stimulated splenocytes from donors that received adjuvants without CM immunization. Mice were sacrificed on day 14 after transfer.
Histological Assessment of Myocarditis
Immediately after euthanasia, mouse hearts were excised, fixed in 10% phosphate-buffered formalin, and embedded in paraffin. Five-μm-thick sections were cut from base to apex and stained with hematoxylin and eosin (H&E). Every fifth section (a total of five sections from each heart) was examined by two independent investigators in a blinded manner. Severity of myocarditis was assessed on a scale from 0 to 5 based on the percentage of the heart section involved: grade 0, no disease; grade 1, up to 10% of the heart section; grade 2, 11 to 30%; grade 3, 31 to 50%; grade 4, 51 to 90%; and grade 5, 90 to 100%. A microscope with a grid was used to estimate the percentage of the heart section involved.
Congo Red Staining for Eosinophils
Five-μm-thick sections of paraffin-embedded hearts were stained with 0.5% Congo Red in 50% glycine buffer (pH 10)/ethanol for 30 minutes and counterstained with hematoxylin.
Immunohistochemical Staining for Antibody Deposition in the Heart
Murine hearts were collected on day 21 after immunization, embedded in Tissue-Tek OCT (Miles Inc., Elkhart, IN), and frozen at −70°C. Five-μm-thick sections were dehydrated in a desiccator at 37°C for 1.5 hours and then fixed in chilled acetone for 10 minutes. Fixed sections were washed for 5 minutes in each of the three solutions in the following order: 1) PBS; 2) PBS containing 1% normal goat serum and 1.5% hydrogen peroxide; and 3) PBS containing 1% bovine serum albumin. Staining was performed as described. 17 Incubation with each antibody was for 1 hour at room temperature. An anti-mouse IgG1 mAb (a rat IgG2a isotype) (clone G1-6.5; BD PharMingen, San Diego, CA) was used as a detection antibody. As an isotype control, heart sections were incubated with a rat IgG2a mAb of irrelevant specificity (clone R35-95, BD PharMingen). Peroxidase-conjugated streptavidin (P 0397; DAKO, Carpinteria, CA) in 1:500 dilution in PBS was used as a tertiary antibody. The positive stain was visualized by incubation with 3-amino-9-ethyl-carbazole (no. A-5754; Sigma Chemical Co.) substrate solution for 20 minutes.
Detection of IgG and IgE in Sera
Mice were bled on days 0, 9, 16, and 21 from the retro-orbital venous plexus using heparinized tubes. Serum levels of CM-specific IgG and its subclasses were determined using CM-coated microtiter plates as we previously described. 17 Adjusted optical density (OD) was calculated as follows: adjusted OD = (mean OD of a sample − mean OD of a negative control)/(mean OD of a positive control − mean OD of a negative control). Total serum IgE was determined by a specific sandwich enzyme-linked immunosorbent assay (ELISA) using microtiter plates precoated with anti-mouse IgE capture mAb (clone R35-72, BD PharMingen).
Treatment with Anti-Cytokine Antibodies
Rat anti-mouse IL-4 mAb 11B.11 (IgG1) was obtained as a gift from the National Cancer Institute, Frederick, MD. A hybridoma cell line producing rat anti-mouse IFN-γ mAb R4-6A2 (IgG1) was purchased from the American Tissue Culture Collection (Manassas, VA). Rat anti-Escherichia coli (E. coli) β-galactosidase mAb GL113 (IgG1) (the hybridoma cell line was kindly provided by Fred Finkelman, University of Cincinnati, Cincinnati, OH) was used as an isotype control. Hybridoma cell lines were grown by the Core Facility of The Johns Hopkins University. Monoclonal Abs were purified from the concentrated hybridoma supernatants using a HiTrap protein G column (Supelco Chromatography, Bellefonte, PA). Mice were immunized with CM and received anti-IL-4 mAb in a dose of 4 mg of mAb on days −1 and 6 (1 day before each CM injection) and in a dose of 2 mg of mAb on days 2, 9, 12, 15, and 18. Anti-IFN-γ mAb was administered in a dose of 1 mg on days −1, 6, and 12. GL113 mAb was used as an isotype control for both anti-IL-4 and anti-IFN-γ mAbs. All Abs were administered intraperitoneally. Monoclonal Abs were dissolved in a vehicle buffer (50 mmol/L sodium phosphate, 300 mmol/L sodium chloride, pH 6.8, for anti-IL-4 mAb and PBS for anti-IFN-γ and isotype control mAbs).
Detection of Cytokines in Splenocyte Culture
On day 21 after immunization, spleens were removed aseptically. Splenocytes were cultured at the initial cell density of 5 × 10 6 cells/ml for 48 hours in RPMI 1640 medium (Life Technologies, Inc.) with additional supplementation in the presence of media alone, different concentrations of CM, or Con A (Sigma Chemical Co.) as we previously described. 17 Levels of IL-4, IL-5, IL-10, IL-12, IL-13, and IFN-γ in the splenocyte culture supernatants were measured using Quantikine murine cytokine ELISA kits (R&D Systems, Minneapolis, MN).
Statistical Analyses
Antibody and cytokine data were analyzed using the Student’s t-test. The Mann-Whitney nonparametric test was used to compare EAM severity scores between different treatment groups. The strength of the association between CM-specific IgG1 levels and EAM severity was assessed by regression analysis (Statview software, Abacus Concepts, Inc., Berkeley, CA). P values of <0.05 were considered statistically significant.
Results
Cellular Composition of the Heart Infiltrate
A/J mice were immunized with CM on days 0 and 7. At sacrifice on day 21, hearts were removed for histological examination. Depending on the experiment, the prevalence of myocarditis varied from 50 to 100%. Histologically, myocarditis was characterized by the presence of foci of myocardial infiltration and cardiomyocyte death, involving mainly the subepicardial regions of the myocardium in mild to moderate cases and affecting the full thickness of the myocardium in more severe disease. Myocardial infiltrate often involved the apical area and was frequently accompanied by pericardial infiltration.
The heart infiltrate in EAM consisted of many macrophages and CD4+ T cells with some CD8+ T cells and B220+ B cells, as we have previously demonstrated by immunohistochemistry. 17 Additionally, the heart infiltrate contained many eosinophils (Figure 1, B and C) ▶ with a typical eosinophilic cytoplasm and a donut-shaped nucleus (characteristic of mouse eosinophils). The abundance of eosinophils was more prominent in severe cases of myocarditis (lesion scores ≥3). We observed a large number of eosinophils in EAM induced either by active immunization (Figure 1, B and C) ▶ or by splenocyte transfer (Figure 1, G and H) ▶ . Furthermore, EAM induced by immunization with a 19 amino acid peptide from cardiac α-myosin heavy chain [myhcα(334-352)] 18 was characterized by the presence of eosinophils in the heart lesions. A similar histological picture characterized by many eosinophils was observed in a different strain of mice, BALB/c, on CM immunization (data not shown). Another striking feature of the myocardial infiltrate in EAM was the presence of multinucleated giant cells (Figure 1D) ▶ . Giant cells were typically seen in more severe cases of EAM. The presence of eosinophils suggests a Th2-like phenotype of the disease. 19-21 Furthermore, macrophage fusion leading to the formation of multinucleated giant cells is induced by IL-4 in both humans 22 and mice. 23
Figure 1.
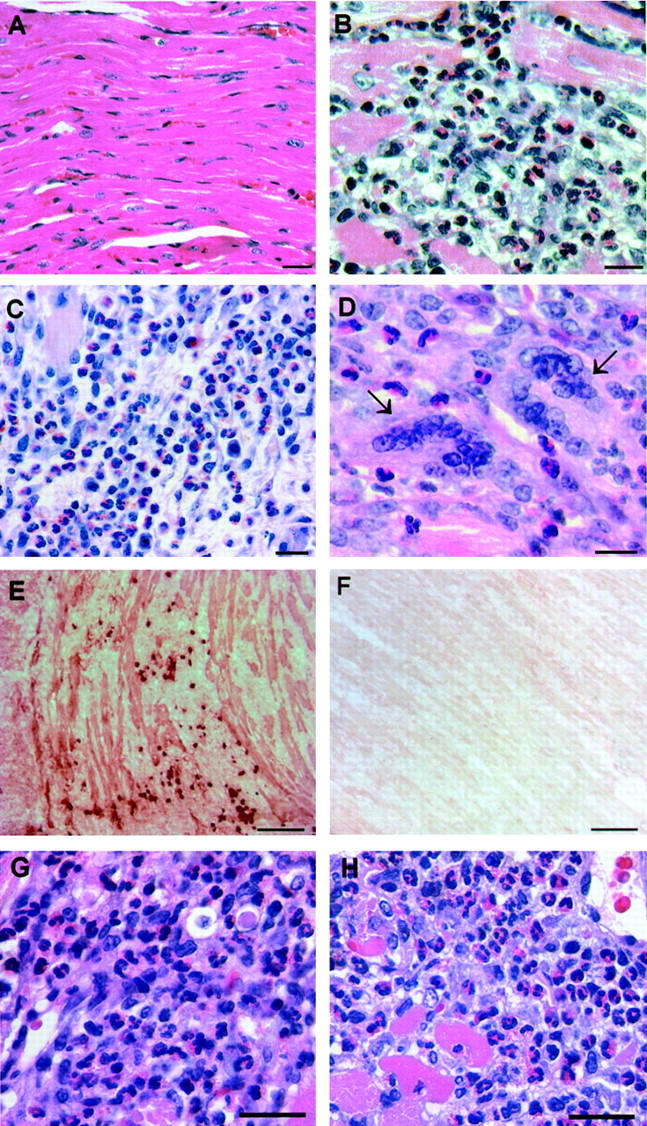
Th2-like characteristics of the heart infiltrate in EAM. A/J mice immunized with CM were sacrificed on day 21 and their hearts were removed for histological examination. A: Normal heart. B and C: Heart infiltrate in EAM is characterized by the presence of abundant eosinophils. D: Heart infiltrate contains giant cells (arrows); eosinophils are seen in the vicinity of giant cells. E: IgG1 deposition and IgG1-positive cells in EAM were detected by immunohistochemistry. The positive stain was visualized with 3-amino-9-ethyl-carbazole substrate solution. Shown heart has a grade 3 lesion. F: Background staining of the same heart using a rat IgG2a isotype control mAb. G and H: Eosinophils in heart lesions in a splenocyte transfer model of EAM. Donor mice were immunized with either CM (G) or with myhcα(334-352) peptide (H). Their splenocytes were collected on day 21 after immunization, stimulated in culture for 3 days with 10 μg/ml of either CM (G) or the peptide (H), and transferred into the recipient mice in a dose of 5 × 10 7 cells. Recipients were sacrificed on day 14 after transfer and their hearts were assessed for the presence of myocarditis. H&E stain (A–D, G, and H); Congo Red (C). Scale bars: 25 μm (A–D), 100 μm (E and F), and 20 μm (G and H).
Antibody Responses in EAM
To further examine the immune response to CM, we measured serum levels of CM-specific antibody. We found that CM-specific IgG (all subclasses) response was positively associated with disease (P < 0.0001). Further, by examining the IgG subclasses we found that CM-specific IgG1 (Figure 2, A and C) ▶ but not IgG2a (Figure 2, B and D) ▶ , or any other IgG subclass (data not shown) correlated with the presence of disease on CM immunization. Day 9 is the earliest time point at which we could detect CM-specific antibody responses. On day 9, IgG2a, IgG2b, and IgG3 responses were still absent whereas the IgG1 response was already detectable in those mice that developed disease (Figure 2, A and B) ▶ . IgG1 levels were also positively associated with the severity of disease (Figure 2E) ▶ (P value for the regression coefficient was 0.003). On the other hand, CM-specific IgG2a tended to be lower in mice with more severe disease (data not shown). A positive association between disease and CM-specific IgG1 was present when EAM was induced using a smaller dose of CM, 100 instead of 250 μg (data not shown).
Figure 2.
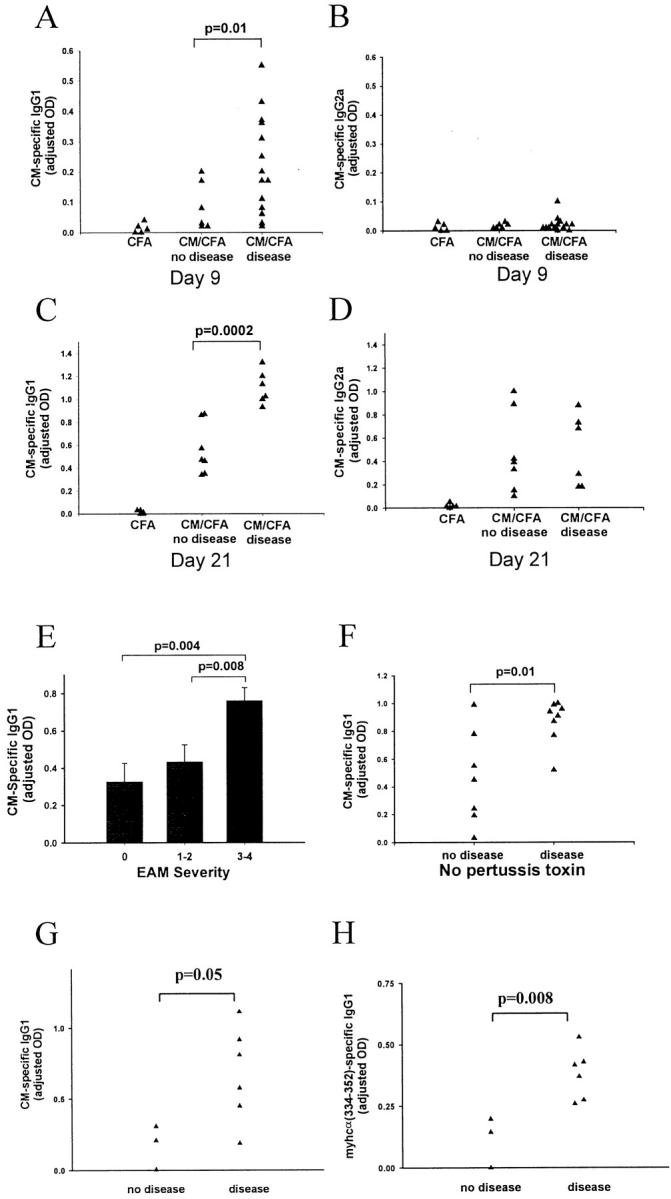
Association of EAM with CM-specific IgG1 response. Treated A/J mice received immunization with CM in CFA plus pertussis toxin. Control mice received the vehicle buffer in CFA plus pertussis toxin. On day 21 after immunization, mice were sacrificed and their hearts were removed for histological examination. Antibody levels were assessed by ELISA and presented as adjusted OD. A: CM-specific IgG1 on day 9. B: CM-specific IgG2a on day 9. C: CM-specific IgG1 on day 21. D: CM-specific IgG2a response on day 21. E: CM-specific IgG1 response on day 21. F: CM-specific IgG1 response on day 14 in mice that received CM immunization without pertussis toxin. G and H: CM-specific and myhcα(334-352)-specific IgG1 responses on day 21 in mice immunized with myhcα(334-352) peptide. A–D and F–H: Each data point represents a value from an individual mouse. E: Data are presented as mean value ± SEM of at least six individual mice per disease severity group. A–E: Data are representative of five experiments. F–H: Data are representative of two experiments. P values were calculated using the Student’s t-test.
It has been reported that IgG1 production in mice is enhanced by IL-4, whereas IgG2a response is associated with IFN-γ production. 13 As a surrogate measure of a Th2 to Th1 ratio, we examined an IgG1 to IgG2a ratio and found that it correlated with both the presence and severity of EAM (P value for the regression coefficient was 0.003). The addition of pertussis toxin to the treatment regimen may skew the immune response toward Th2. To exclude the possibility that the predominance of IgG1 response in our model was simply a pertussis toxin-associated phenomenon, we repeated the experiment omitting pertussis toxin from the treatment regimen. Histologically identical EAM was still induced in the absence of pertussis toxin, although the disease was less severe. Most important, the IgG1 response remained the predominant response and was associated with disease (Figure 2F) ▶ . By examining Ab responses in mice immunized with myhcα(334-352) peptide, we found that disease is associated with both CM-specific IgG1 (Figure 2G) ▶ and myhcα(334-352)-specific IgG1 (Figure 2H) ▶ . In these mice there was no association of disease with other IgG subclasses. We also found the same association of CM-specific IgG1 with disease in EAM induced by splenocyte transfer, in which recipients received no adjuvants (data not shown). This further supports the conclusion that the predominance of IgG1 response in EAM is not dependent on adjuvants.
We found that IgG1 is deposited along the cardiomyocytes in the diseased heart (Figure 1, E and F) ▶ but not in the normal heart. In addition, IgG1-positive cells were found in the cardiac lesions. These cells, which are most likely to be IgG1-producing plasma cells, were present in clusters throughout the heart infiltrate. By contrast, there was a very little deposition of IgG2a in the heart in EAM (data not shown).
Because IL-4 also drives IgE response, 13 we measured the serum levels of total IgE in EAM and found that they are increased on CM immunization (Figure 4A ▶ , day 21 versus day −1 and day 9). Total IgE response started rising after day 9, was present on day 16, and further increased until day 21 when the experiment was terminated. However, total IgE response did not significantly correlate with the presence of disease (data not shown). Measurement of CM-specific IgE production was not possible because of the lack of sensitivity of our ELISA method. Thus, both the histological findings and the humoral immune response in EAM are consistent with a Th2 type of immune response, implicating IL-4 as a pathogenetic factor in this disease.
Figure 4.
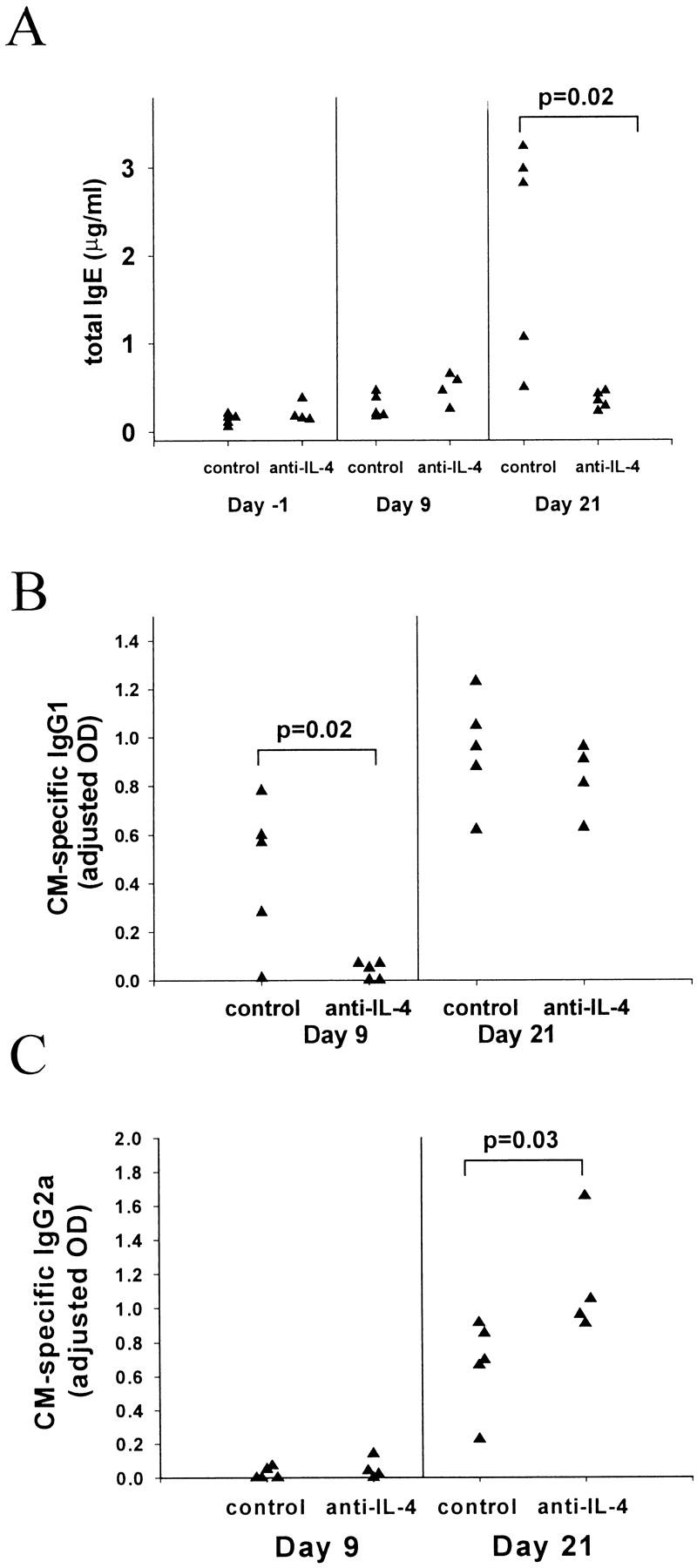
Effects of IL-4 blockade on antibody responses. All mice received CM immunization on days 0 and 7. Anti-IL-4 and the isotype control mAbs were given as described in Figure 3 ▶ . Sera were collected on days −1, 9, 16, and 21. A: Total IgE levels on days −1 (1 day before the first CM injection), 9, and 21 as measured by a sandwich ELISA (described in Materials and Methods). B: CM-specific IgG1 response on days 9 and 21. C: CM-specific IgG2a response on days 9 and 21. Both CM-specific IgG1 and IgG2a responses were measured using ELISA. All data points represent values from individual mice. Data are representative of three experiments. P values were calculated using the Student’s t-test.
Anti-IL-4 Treatment Reduces the Severity of EAM
Based on the presence of eosinophils and giant cells in the heart infiltrate, the strong association of CM-specific IgG1 with disease, and the up-regulation of the IgE response, we hypothesized that IL-4 is involved in the development of EAM. Therefore, we examined the role of IL-4 in EAM by treating mice with anti-IL-4 mAb during the course of disease induction. A/J mice were immunized with CM as described previously. In addition, mice received anti-IL-4 mAb (11B.11) intraperitoneally on days −1, 2, 6, 9, 12, 15, and 18. A control group received equivalent amounts of isotype control mAb (GL113). Additionally, we included a control group that received the equivalent volumes of a vehicle buffer and a control group that received CM immunization alone. All mice were sacrificed on day 21. We found that the anti-IL-4 treatment significantly reduced the severity of myocarditis (Figure 3 ▶ and also see Figure 7A ▶ ). The prevalence of severe disease (grade ≥ 3) was 20% in the anti-IL-4-treated group and 80% in the isotype control group. In repeated experiments, the severity grade of 4 or higher was never observed in any of the anti-IL-4-treated mice. All of the control groups (isotype control, vehicle buffer control, and the group that received CM without any other treatment) did not differ in terms of disease prevalence or severity. The reduction in severity observed in the anti-IL-4-treated group was associated with smaller numbers of eosinophils present in the heart infiltrate.
Figure 3.

IL-4 blockade reduces the severity of EAM. Mice immunized with CM received anti-IL-4 mAb (11B.11 rat IgG1 anti-mouse IL-4) in a dose of 4 mg of the mAb on days −1 and 6 (one day before each CM injection) and in a dose of 2 mg of the mAb on days 2, 9, 12, 15, and 18. The isotype control group received the same regimen (same time and dose) of GL113 rat IgG1 anti-E. coli β-galactosidase mAb. The buffer controls received the same regimen of a vehicle buffer for anti-IL-4 mAb. Mice were sacrificed on day 21. Data points represent individual mice and are plotted against percentages of myocardial involvement representing the midpoints of intervals that correspond to the assigned severity scores (right vertical axis) (score 0, 0%; score 1, 5%; score 2, 20%; score 2.5, 25%; score 3, 40%; score 3.5, 45%; score 4, 70%; score 4.5, 80%; and score 5, 95%). Data are representative of three experiments. P values were calculated using the Mann-Whitney nonparametric test.
Figure 7.
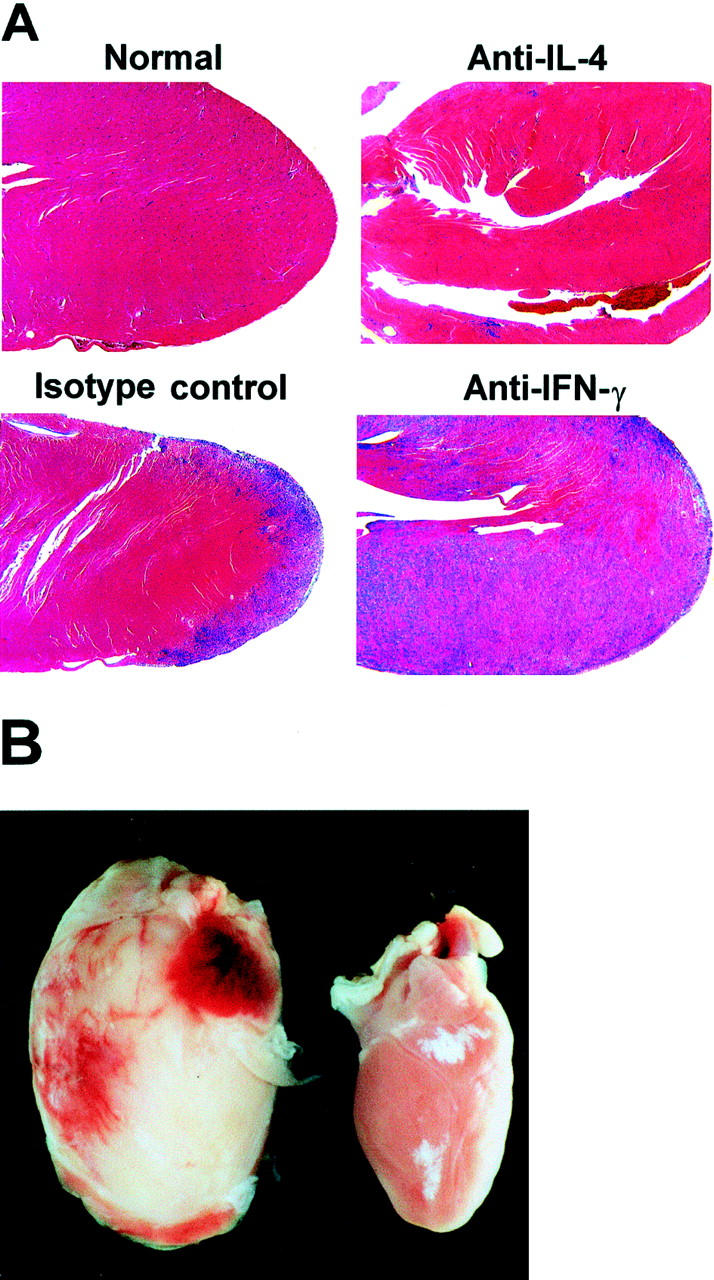
IL-4 blockade reduces and IFN-γ blockade exacerbates the severity of EAM. A: Normal heart is from an untreated A/J mouse. CM-immunized mice received anti-IL-4 (11B.11 rat IgG1 anti-mouse IL-4 mAb), or anti-IFN-γ (R4-6A2 rat IgG1 anti-mouse IFN-γ mAb), or the isotype control mAb (GL113 rat IgG1 anti-E. coli β-galactosidase mAb) as described in Figures 3 and 6 ▶ ▶ . Mice were sacrificed on day 21. Hearts shown have EAM severity grades that correspond to the median grade from each group (anti-IL-4 heart has grade 1, isotype control heart has grade 3, and anti-IFN-γ heart has grade 4). Original magnification, ×15. H&E stain. B: A representative heart from the anti-IFN-γ-treated group (left) compared to a control mouse heart with no disease (right).
Anti-IL-4 Treatment Abrogates IgE, Reduces IgG1, and Enhances IgG2a Antibody Responses
We examined the humoral immune response for changes associated with the reduction in severity of EAM on anti-IL-4 treatment. First, we measured the levels of total IgE and found that anti-IL-4 treatment abrogated the total IgE response (Figure 4A) ▶ . This result confirmed the effectiveness of the blockade of IL-4 because there is a very strong association between the IgE levels and IL-4 activity. Next, we examined IgG subclasses and found that anti-IL-4 treatment significantly suppressed CM-specific IgG1 levels early during the course of EAM (day 9), but this suppression became nonsignificant later (day 21) compared to the control groups (Figure 4B) ▶ . The fact that the anti-IL-4-treated group had a substantial IgG1 response on day 21 was consistent with incomplete abrogation of the disease by anti-IL-4 treatment; there was still a positive correlation between the severity of disease and IgG1 levels (P value for the regression coefficient was 0.007).
Anti-IL-4-treated mice had higher levels of CM-specific IgG2a on day 21 compared to the control groups (Figure 4C) ▶ . Anti-IL-4-treated mice also had higher levels of specific IgG2b and IgG3 but the differences were not statistically significant (data not shown). This increase in the IgG2a response can be explained by the fact that IL-4 and IFN-γ have an antagonistic relationship. Neutralization of IL-4 leads to the up-regulation of IFN-γ and therefore enhances IFN-γ-mediated effects such as the production of IgG2a antibody.
Effects of Anti-IL-4 Treatment on Cytokine Production by Cultured Splenocytes
To examine the effect of anti-IL-4 treatment on cytokine production, we collected splenocytes from the individual mice on day 21 after immunization and cultured them for 48 hours in the presence of CM. Splenocyte supernatants were examined for the presence of different cytokines. Anti-IL-4 treatment decreased IL-4, IL-5, and IL-13 production by splenocytes in response to CM (Figure 5, A–C) ▶ and dramatically enhanced the ability of the splenocytes to produce IFN-γ (Figure 5D) ▶ as compared to the control groups. We also looked at levels of IL-10 and IL-12 in the supernatants and found no difference between the anti-IL-4-treated group and the control groups (data not shown). IL-12 levels were below the lower limit of detection in the majority of mice. Taken together, our findings demonstrate that anti-IL-4 treatment resulted in diminished total IgE and CM-specific IgG1 responses as well as decreased production of IL-4, IL-5, and IL-13 by cultured splenocytes, indicating a suppression of a Th2 response. Additionally, the reduction in severity of EAM on the anti-IL-4 treatment was associated with both increased levels of CM-specific IgG2a antibody and enhanced ability of splenocytes to produce IFN-γ in response to CM stimulation, indicating a greater Th1 response.
Figure 5.

IL-4 blockade decreases IL-4, IL-5, and IL-13 but enhances IFN-γ production by cultured splenocytes. Mice were sacrificed on day 21; splenocytes were cultured for 48 hours in the presence of 10 μg/ml of CM. Supernatants were tested for the presence of IL-4 (A), IL-5 (B), IL-13 (C), and IFN-γ (D) by ELISA. Cytokine levels in supernatants from mice treated with anti-IL-4 are compared to those from mice treated with the isotype control mAb. Data are presented as mean value ± SEM for four individual mice per group, and are representative of two experiments. P values were calculated using the Student’s t-test.
Anti-IFN-γ Treatment Exacerbates EAM
We hypothesized that the switch to a Th1 response and a greater production of IFN-γ may account for the ameliorating effect of the anti-IL-4 treatment. To study the role of IFN-γ in EAM we blocked IFN-γ with mAb R4-6A2 during the course of EAM induction. Control groups received either isotype control mAb GL113 or a vehicle buffer. Mice were sacrificed on day 21 and the hearts were removed for histological examination. Anti-IFN-γ treatment markedly exacerbated disease compared to the control mice (Figures 6 and 7A) ▶ ▶ with extensive heart lesions rich in eosinophils. Some mice from the anti-IFN-γ-treated group had a histological score of 5, indicating involvement of 90 to 100% of the heart. The exacerbation of disease in anti-IFN-γ-treated mice was also obvious on gross examination of the hearts. The hearts from anti-IFN-γ-treated mice had an intense white discoloration and were grossly enlarged indicating progression to dilated cardiomyopathy (Figure 7B) ▶ . Such severe pathology, both gross and microscopic, has never been observed in our laboratory on the standard CM immunization regimen.
Figure 6.

Administration of anti-IFN-γ mAb exacerbates EAM. All mice received CM immunization on days 0 and 7. The anti-IFN-γ group received 1 mg of R4-6A2 rat IgG1 anti-mouse-IFN-γ mAb on days −1, 6, and 12. Isotype control group received the same regimen of GL113 rat IgG1 anti-E. coli β-galactosidase mAb. Anti-IL-4 group was treated as described in Figure 3 ▶ . The control group in B received the isotype control mAb simulating the anti-IL-4 mAb regimen. The anti-IL-4 + anti-IFN-γ group received a combination of the anti-IL-4 and anti-IFN-γ mAb regimens. All mice were sacrificed on day 21. Data points represent individual mice and are plotted against percentages of myocardial involvement representing the midpoints of intervals that correspond to the assigned severity scores (right vertical axis) as described in Figure 3 ▶ . Data are representative of two experiments. P values were calculated using the Mann-Whitney test.
Simultaneous Blockade of IL-4 and IFN-γ Does Not Alter the Disease Severity
Because anti-IL-4 and anti-IFN-γ treatments had opposing effects on the severity of EAM, we examined the effect of a simultaneous blockade of both cytokines on EAM. The following four treatment groups were run in parallel: a group that received both anti-IL-4 and anti-IFN-γ mAbs, anti-IL-4 mAb alone group, anti-IFN-γ mAb alone group, and isotype control group. Consistent with our previous results, we found that anti-IL-4 treatment reduced the severity of EAM and anti-IFN-γ exacerbated EAM. When we blocked both cytokines together, mice exhibited the same severity of disease as the control mice (Figure 6B) ▶ . This further confirmed the finding that IL-4 and IFN-γ have opposing effects on EAM and suggested that at least one of the mechanisms by which anti-IL-4 treatment can reduce disease is through the enhancement of a protective IFN-γ response.
Discussion
In this report, we demonstrate that IL-4 mediates the development of EAM in A/J mice and is critical for the progression to severe forms of acute myocarditis. Anti-IL-4 treatment markedly reduced the severity of EAM and this reduction in severity was associated with a switch from a Th2-like to a Th1-like phenotype. These effects of anti-IL-4 seem to be dependent on increased levels of IFN-γ. Indeed, the blockade of IFN-γ exacerbated the disease. Taken together, these data support a pathogenetic role of the defining Th2 cytokine, IL-4, and a protective role of the prototypic Th1 cytokine, IFN-γ, in EAM.
These results are somewhat surprising in view of the prevailing opinion that organ-specific autoimmunity is a Th1-driven process whereas Th2 responses play a protective role. 10-12 The results of our cytokine-blocking experiments become less surprising if one takes into account the Th2-like phenotype associated with EAM. In this study, we demonstrate that the disease is associated with the presence of eosinophils and giant cells in the heart infiltrate, elevation of total IgE levels, and predominance of an IgG1 response that strongly correlates with both the presence and severity of disease. In addition, the IgG1 to IgG2a ratio correlates with both the presence and severity of EAM, serving as a surrogate for a Th2 to Th1 ratio. The presence of the Th2-like phenotype was independent of the protocol used to induce EAM. The omission of pertussis toxin from the treatment regimen, induction of EAM with a splenocyte transfer, the use of porcine CM 17 or a pathogenic CM peptide instead of intact murine CM, did not alter the Th2-like phenotype. The reduction in disease severity on anti-IL-4 treatment was associated with a decrease in Th2 markers. This was manifested by the reduction in CM-specific IgG1 antibody production early in the course of disease, abrogation of total IgE antibody response, a decrease in the number of eosinophils in the heart infiltrate, and a diminished in vitro production of IL-4, IL-5, and IL-13 by spleen cells. The reduction in disease severity on anti-IL-4 treatment was also associated with up-regulation of Th1 markers such as CM-specific IgG2a antibody response and IFN-γ production by splenocytes in vitro. Cytokine production by splenocytes may not reflect the cytokine profile in the heart. However, we were unable to look at the cytokine production by the cells infiltrating the heart because of the difficulty in obtaining a sufficient number of viable lymphocytes. The exacerbation of EAM on IFN-γ blockade demonstrates that IFN-γ has a disease-limiting role. The ameliorating effect of IL-4 blockade was abrogated by co-administration of anti-IFN-γ mAb further confirming the opposing roles of these two cytokines in EAM. These results demonstrate that the balance between IL-4 and IFN-γ determines the progression of the disease process.
Here, we present a definitive report that the prototypic Th2 cytokine, IL-4, plays a disease-promoting role in an autoimmune process directed against an organ-specific antigen in an immunocompetent host. As mentioned earlier, a great deal of evidence in recent literature points to a Th1 response, and Th1 cytokines in particular, as pathogenic in organ-specific autoimmunity. Th2 cytokines, on the other hand, are considered to be protective. These conclusions come mainly from the studies on experimental autoimmune encephalomyelitis 24-26 and murine models of type 1 diabetes. 27,28 Few reports suggest that a Th2 response, and IL-4 in particular, can promote an immune-mediated disease. Disease-promoting abilities of IL-4 have been shown in a murine model of oxasolone-induced colitis, 29 and in a murine model of arthritis induced by an irrelevant antigen. 30 IL-4 has been reported to promote disease in a rat model of experimental autoimmune uveoretinitis induced by the administration of bovine S antigen. 31 Surprisingly, the authors demonstrated that IL-4 enhanced IFN-γ production in their model and suggested that this increase in IFN-γ was responsible for the proinflammatory activity of IL-4. However, Caspi and colleagues 32 have demonstrated a protective role of IFN-γ in experimental autoimmune uveoretinitis. IL-4 has been shown to play a critical role in the induction of collagen-induced arthritis, but this effect was only present when anti-IL-4 mAb was administered early during the course of the disease induction; administration of anti-IL-4 mAb for a longer period of time exacerbated disease. 33 Additionally, Th2 cells were able to transfer experimental autoimmune encephalomyelitis 34 and acute pancreatitis with diabetes 19 in immunocompromised recipient mice.
In this report, we demonstrate that IL-4 inhibits the disease-limiting factor, IFN-γ, providing a likely mechanism for the pathogenic activity of IL-4. There have been conflicting reports on the action of IFN-γ in organ-specific autoimmune diseases. Some reports demonstrated a disease-promoting role of IFN-γ, 35-37 whereas others reported that IFN-γ plays a protective role. 32,38,39 IFN-γ exerts well-established proinflammatory activities, including up-regulation of MHC class II, recruitment of Th1 T cells to the site of inflammation, and enhancement of IL-12 production. 12 Despite these proinflammatory effects, IFN-γ has potent abilities to limit an autoimmune response. 40 The exact mechanisms of this disease-limiting action of IFN-γ are not known. The anti-inflammatory actions of IFN-γ that may explain its disease-limiting role in EAM include suppression of IL-4-induced activation of B cells, 12 inhibition of eosinophil and Th2 T-cell recruitment to the site of inflammation, 41 and suppression of Th2 T-cell proliferation. 42 Preferential suppression of Th2 T cells by IFN-γ has been explained by the presence of IFN-γ receptor β chain on Th2 but not on Th1 T cells. 43 Th2 T cells do not seem, however, to be the exclusive target of the suppressive effect of IFN-γ because IFN-γ was shown to induce cell death in a Th1 T-cell clone. 44 Albina and colleagues 45 proposed that so-called “suppressor” macrophages could inhibit T-cell proliferation through IFN-γ-induced nitric oxide that possesses potent cytotoxic and anti-proliferative effects. It has been recently demonstrated that IFN-γ eliminates activated CD4+ as well as CD8+ T cells that expand during immune responses. 46-48 Studies using IFN-γ knockout mice are underway in our laboratory and the preliminary results are consistent with the findings described in this report. Furthermore, reports by Smith and colleagues 8 and by Eriksson and colleagues 49 support the protective role of IFN-γ in EAM.
Another potentially important mechanism of IL-4 action is the recruitment of eosinophils and Th2 cells to the site of inflammation via up-regulated endothelial expression of VCAM-1 14 and increased production of eotaxin. 50 In this regard, we have previously reported an up-regulation of VCAM-1 on cardiac endothelium in EAM. 17 Recruited through binding to VCAM-1, activated eosinophils can mediate cardiomyocyte destruction by releasing the cytotoxic contents of their intracellular granules, including eosinophilic cationic protein, major basic protein, eosinophil-derived neurotoxin, and eosinophil peroxidase. In fact, IL-4-induced tumor eradication was mediated by activated eosinophils. 51,52 Eosinophilia in humans is associated with endomyocardial damage mediated by eosinophil degranulation. 53
IL-4 may contribute to disease by activating B cells and thereby enhancing IgG1 and IgE production. 13,54 Here, we demonstrate that the IgG1 response is the predominant antibody response that correlates with the severity of EAM. The fact that IgG1 is deposited locally in the heart and that IgG1-positive cells are present in the infiltrate further substantiates the potential ability of IgG1 antibody to mediate local damage in the heart. Locally deposited IgG1 may contribute to cardiomyocyte destruction by triggering antibody-dependent cell-mediated cytotoxicity reactions via the Fcγ receptor III on cytotoxic cells. 55 Cardiomyocyte-bound IgG1 may induce eosinophil degranulation and subsequent cardiomyocyte destruction. 56,57 De Andres and colleagues 58 have shown that activated murine eosinophils express Fcγ receptor III on their surface providing a link through which IgG1 and eosinophils may come together to destroy the cardiomyocytes. Transfer of IgG antibody caused myocarditis in some strains of mice but not in others. 59,60 In a pilot experiment using A/J mice, we found that purified IgG1 from mice with myocarditis transferred pericardial lesions with infiltrating cells and calcification (unpublished observations). These lesions, however, differed in distribution from those induced by active immunization. Future experiments are needed to prove the ability of IgG1 antibody to initiate a disease identical to that induced by active immunization. Finally, IgE antibody may contribute to the local tissue damage. The role of IgE in EAM should be investigated in the future.
IL-4 blockade significantly reduced the severity of EAM, but it did not completely abrogate the disease. One possible explanation is that the anti-IL-4 mAb could not completely block all of the actions of IL-4 particularly those mediated in an autocrine and paracrine manner. We did show, however, that the blockade was effective in abrogating the total IgE response. In addition, it caused a significant reduction in CM-specific IgG1 antibody production on day 9, increased CM-specific IgG2a antibody levels, diminished production of Th2 cytokines, and a dramatic enhancement of IFN-γ production. All these actions support the effectiveness of the anti-IL-4 blockade but do not prove that the blockade was complete. Another possible explanation is that IL-4 only partially mediates the disease and some other cytokine or cytokines may promote the disease in the absence of IL-4. It is plausible that such Th2-type cytokines as IL-13 and IL-5 could compensate for the absence of IL-4. IL-13 closely resembles IL-4 in many of its actions. 61 The absence of either IL-4 or IL-13 significantly impairs Th2 responses, but only the absence of both cytokines has been shown to completely abrogate IgE and IgG1 responses and abolish pulmonary granuloma formation and eosinophil infiltration in response to Schistosoma mansoni egg immunization. 62 IL-5 is important for eosinophilic responses 63 and may contribute to the pathogenesis of EAM via activation and recruitment of eosinophils to the heart. Further studies are in progress to determine the role of these cytokines in EAM.
Another potential candidate that may mediate disease in the absence of IL-4 is tumor necrosis factor (TNF)-α, which has been suggested as an important pathogenic factor in EAM. 8,9 The in vitro production of TNF-α correlates with the presence of disease on CM immunization. 17,64 Smith and colleagues 8 have demonstrated that anti-TNF mAb reduced the severity of EAM but did not completely abrogate the disease. TNF-α and IL-4 synergize in the up-regulation of VCAM-1 expression on endothelium that is important for the T cell and eosinophil recruitment. 65,66 Both cytokines are important for the maturation of dendritic cells, 67 which may be important antigen-presenting cells in EAM. The presence of resident dendritic cells in normal myocardium has been reported. 68 A molecular basis for the synergism between TNF-α and IL-4 has been demonstrated by Shen and Stavnezer. 69 TNF-α can be associated not only with Th1 but also with Th2 immune responses. 11 It has been shown to promote the development of asthma, a Th2-mediated disease. 70
EAM may not be a purely Th2-driven disease; a combination of Th1 and Th2 factors may be necessary for a full-blown disease. These factors may work synergistically or their actions may be temporally and/or spatially distinct. One potential disease-promoting Th1-type cytokine is IL-12. The importance of IL-12 for the induction of autoimmune disease has been demonstrated in a number of animal models. 71 The role of IL-12 in EAM is still unclear. Okura and colleagues 72 have demonstrated that the addition of IL-12 to the cell culture of CM-specific T cells increased their pathogenicity in a T-cell transfer model of EAM in rats. Our preliminary results using IL-12 receptor β1 knockout mice on a BALB/c background demonstrate the importance of IL-12 receptor signaling for the development of EAM. Further in vivo studies are necessary to determine the role of IL-12 in EAM, especially in A/J mice that are known to be poor producers of IL-12. 73
Myocarditis depends on the genetics of the host, with different strains of mice exhibiting different levels of susceptibility to EAM. 6 It has also been shown that even among susceptible strains immunopathogenic factors contributing to the development of myocarditis vary depending on the mouse genetic background in both the Coxsackievirus B3-induced 74 and CM-induced models. 59 Our results are based on studies using highly susceptible A/J mice, which are known to produce relatively strong Th2 responses, 75 are prone to develop experimental allergic asthma, and are partially deficient in IL-12 production. 73 It is plausible that, given their genetic predisposition toward Th2 responses, A/J mice develop a Th2-driven autoimmune myocarditis, whereas a different, more Th1-prone strain of mice might develop a Th1-driven disease. We have found, however, that BALB/c mice also mount a predominant CM-specific IgG1 response, which correlates with disease, and have abundant eosinophils in the heart lesions. Our preliminary studies using IFN-γ knockout mice on a BALB/c background demonstrated that IFN-γ limits disease in this strain as well.
Myocarditis in humans has different forms, ranging from a very acute disease, such as fulminant myocarditis, 76 to a chronic indolent disease that progresses to dilated cardiomyopathy. The murine model described in this report best reflects an acute disease in humans. It is possible that acute and chronic forms of myocarditis are driven by different immunopathological mechanisms. In our IFN-γ blocking experiments, we found that the Th2 phenotype of disease is present as late as day 29 after immunization (unpublished observations). Because we have not looked beyond day 29, our findings may not necessarily represent what happens during later stages of disease or, most importantly, what happens during chronic human myocarditis.
Some investigators have suggested treating autoimmune diseases by deviating the immune response from Th1 to Th2. Yet our findings demonstrate that factors associated with a Th2 response can promote organ-specific autoimmune disease, whereas factors associated with a Th1 response can be protective. This stresses that caution is necessary when applying a Th1/Th2 paradigm in a context of autoimmune disorders.
Acknowledgments
We thank Marsha Wills-Karp, Scheherazade Sadegh-Nasseri, Elizabeth A. Stafford, Mehrdad Hejazi, Michael E. Field, and Jobert G. Barin for critical reading of this manuscript; William M. Baldwin III, C. Lynne Burek, Patrizio Caturegli, David Schwartz, and Hyam I. Levitsky for helpful discussions; Ralph H. Hruban for the help with histopathology; Tom Larsen for the help with obtaining GL113 hybridoma; and the National Cancer Institute for their gift of rat anti-mouse IL-4 mAb 11B.11.
Footnotes
Address reprint requests to Dr. Noel R. Rose, Department of Pathology, The Johns Hopkins University, Ross Building, Room 659, 720 Rutland Ave., Baltimore, MD, 21205. E-mail: nrrose@jhsph.edu.
Supported by National Institutes of Health grants ES07141, HL33878, and HL65100; National Institute of Environmental Health Sciences grant ES03819; and Z.K. was supported by a fellowship of the Deutsche Herzstiftung e.V.
Current address for S. L. H.: Merck Research Laboratories, West Point, PA.
References
- 1.Drory Y, Turetz Y, Hiss Y, Lev B, Fisman EZ, Pines A, Kramer MR: Sudden unexpected death in persons less than 40 years of age. Am J Cardiol 1991, 68:1388-1392 [DOI] [PubMed] [Google Scholar]
- 2.Brown CA, O’Connell JB: Myocarditis and idiopathic dilated cardiomyopathy. Am J Med 1995, 99:309-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caforio AL, Goldman JH, Haven AJ, Baig KM, McKenna WJ: Evidence for autoimmunity to myosin and other heart-specific autoantigens in patients with dilated cardiomyopathy and their relatives. Int J Cardiol 1996, 54:157-163 [DOI] [PubMed] [Google Scholar]
- 4.Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP: Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol 2000, 35:11-18 [DOI] [PubMed] [Google Scholar]
- 5.Mason JW, O’Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, Moon TE: A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med 1995, 333:269-275 [DOI] [PubMed] [Google Scholar]
- 6.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW: Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol 1987, 139:3630-3636 [PubMed] [Google Scholar]
- 7.Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR: Interleukin 1 and tumor necrosis factor can promote coxsackie B3-induced myocarditis in resistant B10.A mice. J Exp Med 1992, 175:1123-1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith SC, Allen PM: Neutralization of endogenous tumor necrosis factor ameliorates the severity of myosin-induced myocarditis. Circ Res 1992, 70:856-863 [DOI] [PubMed] [Google Scholar]
- 9.Bachmaier K, Pummerer C, Kozieradzki I, Pfeffer K, Mak TW, Neu N, Penninger JM: Low-molecular-weight tumor necrosis factor receptor p55 controls induction of autoimmune heart disease. Circulation 1997, 95:655-661 [DOI] [PubMed] [Google Scholar]
- 10.Liblau RS, Singer SM, McDevitt HO: Th1 and Th2 CD4+ cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today 1995, 16:34-38 [DOI] [PubMed] [Google Scholar]
- 11.Mosmann TR, Sad S: The expanding universe of T-cell subsets: th1, Th2, and more. Immunol Today 1996, 17:138-146 [DOI] [PubMed] [Google Scholar]
- 12.Boehm U, Klamp T, Groot M, Howard JC: Cellular responses to interferon-γ. Annu Rev Immunol 1997, 15:749-795 [DOI] [PubMed] [Google Scholar]
- 13.Finkelman FD, Holmes J, Katona IM, Urban JF, Jr, Beckman MP, Park LS, Schooley KS, Coffman RL, Mosmann TR, Paul WE: Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol 1990, 8:303-333 [DOI] [PubMed] [Google Scholar]
- 14.Hickey MJ, Granger DN, Kubes P: Molecular mechanisms underlying IL-4-induced leukocyte recruitment in vivo: a critical role for the α4 integrin. J Immunol 1999, 163:3441-3448 [PubMed] [Google Scholar]
- 15.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE: The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 1999, 17:701-738 [DOI] [PubMed] [Google Scholar]
- 16.Shiverick KL, Thomas LL, Alpert NR: Purification of cardiac myosin. Application to hypertrophied myocardium. Biochim Biophys Acta 1975, 393:124-133 [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Afanasyeva M, Hill SL, Rose NR: Characterization of murine autoimmune myocarditis induced by self and foreign cardiac myosin. Autoimmunity 1999, 31:151-162 [DOI] [PubMed] [Google Scholar]
- 18.Donermeyer DL, Beisel KW, Allen PM, Smith SC: Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J Exp Med 1995, 182:1291-1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pakala SV, Kurrer MO, Katz JD: T helper 2 (Th2) T cells induce acute pancreatitis and diabetes in immune-compromised nonobese diabetic (NOD) mice. J Exp Med 1997, 186:299-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopf M, Le Gros G, Coyle AJ, Kosco-Vilbois M, Brombacher F: Immune responses of IL-4, IL-5, IL-6 deficient mice. Immunol Rev 1995, 148:45-69 [DOI] [PubMed] [Google Scholar]
- 21.Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, Sato T, Habu S, Tashiro H, Sato M, Ohta A: Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med 1999, 190:617-627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNally AK, Anderson JM: Interleukin-4 induces foreign body giant cells from human monocyte/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol 1995, 147:1487-1499 [PMC free article] [PubMed] [Google Scholar]
- 23.McInnes A, Rennick DM: Interleukin-4 induces cultured monocytes/macrophages to form giant multinucleated cells. J Exp Med 1988, 167:598-611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE: Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol 2000, 124:132-143 [DOI] [PubMed] [Google Scholar]
- 25.Shaw MK, Lorens JB, Dhawan A, DalCanto R, Tse HY, Tran AB, Bonpane C, Eswaran SL, Brocke S, Sarvetnick N, Steinman L, Nolan GP, Fathman CG: Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J Exp Med 1997, 185:1711-1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falcone M, Bloom BR: A T helper cell 2 (Th2) immune response against non-self antigens modifies the cytokine profile of autoimmune T cells and protects against experimental allergic encephalomyelitis. J Exp Med 1997, 185:901-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katz JD, Benoist C, Mathis D: T helper cell subsets in insulin-dependent diabetes. Science 1995, 268:1185-1188 [DOI] [PubMed] [Google Scholar]
- 28.Gallichan WS, Balasa B, Davies JD, Sarvetnick N: Pancreatic IL-4 expression results in islet-reactive Th2 cells that inhibit diabetogenic lymphocytes in the nonobese diabetic mouse. J Immunol 1999, 163:1696-1703 [PubMed] [Google Scholar]
- 29.Boirivant M, Fuss IJ, Chu A, Strober W: Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 1998, 188:1929-1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs MJ, van den Hoek AE, van Lent PL, van de Loo FA, van de Putte LB, Van Den Berg WB: Role of IL-2 and IL-4 in exacerbations of murine antigen-induced arthritis. Immunology 1994, 83:390-396 [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanathan S, de Kozak Y, Saoudi A, Goureau O, Van der Meide PH, Druet P, Bellon B: Recombinant IL-4 aggravates experimental autoimmune uveoretinitis in rats. J Immunol 1996, 157:2209-2215 [PubMed] [Google Scholar]
- 32.Caspi RR, Chan C-C, Grubbs BG, Silver PB, Wiggert B, Parsa CF, Bahmanyar S, Billiau A, Heremans H: Endogenous systemic IFN-γ has a protective role against ocular autoimmunity in mice. J Immunol 1994, 152:890-899 [PubMed] [Google Scholar]
- 33.Hesse M, Bayrak S, Mitchison A: Protective major histocompatibility complex genes and the role of interleukin-4 in collagen-induced arthritis. Eur J Immunol 1996, 26:3234-3237 [DOI] [PubMed] [Google Scholar]
- 34.Lafaille JJ, van de Keere F, Hsu AL, Baron JL, Haas W, Raine CS, Tonegawa S: Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J Exp Med 1997, 186:307-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarvetnick N, Shizuru J, Liggitt D, Martin L, McIntyre B, Gregory A, Parslow T, Stewart T: Loss of pancreatic islet tolerance induced by beta-cell expression of interferon-gamma. Nature 1990, 346:844-847 [DOI] [PubMed] [Google Scholar]
- 36.Barret SP, Gleeson PA, de Silva H, Toh BH, van Driel IR: Interferon-γ is required during the initiation of an organ-specific autoimmune disease. Eur J Immunol 1996, 26:1652-1655 [DOI] [PubMed] [Google Scholar]
- 37.Tang H, Mignon-Godefroy K, Meroni PL, Garotta G, Charreire J, Nicoletti F: The effects of a monoclonal antibody to interferon-gamma on experimental autoimmune thyroiditis (EAT): prevention of disease and decrease of EAT-specific T cells. Eur J Immunol 1993, 23:275-278 [DOI] [PubMed] [Google Scholar]
- 38.Lublin FD, Knobler RL, Kalman B, Goldhaber M, Marini J, Perrault M, D’Imperio C, Joseph J, Alkan SS, Korngold R: Monoclonal anti-γ interferon antibodies enhance experimental allergic encephalomyelitis (EAE). Autoimmunity 1993, 16:264-274 [DOI] [PubMed] [Google Scholar]
- 39.Vermeire K, Heremans H, Vandeputte M, Huang J, Billiau A, Matthys P: Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J Immunol 1997, 158:5507-5513 [PubMed] [Google Scholar]
- 40.Matthys P, Vermeire K, Heremans H, Billiau A: The protective effect of IFN-gamma in experimental autoimmune diseases: a central role of mycobacterial adjuvant-induced myelopoiesis. J Leukoc Biol 2000, 68:447-454 [PubMed] [Google Scholar]
- 41.Iwamoto I, Nakajima H, Endo H, Yoshida S: Interferon gamma regulates antigen-induced eosinophil recruitment into the mouse airways by inhibiting the infiltration of CD4+ T cells. J Exp Med 1993, 177:573-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gajewski TF, Fitch FW: Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol 1988, 140:4245-4252 [PubMed] [Google Scholar]
- 43.Pernis A, Gupta S, Gollob KJ, Garfein E, Coffman RL, Schindler C, Rothman P: Lack of interferon gamma receptor beta chain and the prevention of interferon gamma signaling in TH1 cells. Science 1995, 269:245-247 [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Janeway CA, Jr: Interferon gamma plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med 1990, 172:1735-1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albina JE, Abate JA, Henry WL, Jr: Nitric oxide production is required for murine resident peritoneal macrophages to suppress mitogen-stimulated T cell proliferation. Role of IFN-gamma in the induction of the nitric oxide-synthesizing pathway. J Immunol 1991, 147:144-148 [PubMed] [Google Scholar]
- 46.Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S: Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med 2000, 192:117-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu CQ, Wittmer S, Dalton DK: Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med 2000, 192:123-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Badovinac VP, Tvinnereim AR, Harty JT: Regulation of antigen-specific CD8(+) T cell homeostasis by perforin and interferon-gamma. Science 2000, 290:1354-1358 [DOI] [PubMed] [Google Scholar]
- 49.Eriksson U, Kurrer MO, Bingisser R, Eugster HP, Saremaslani P, Follath F, Marsch S, Widmer U: Lethal autoimmune myocarditis in interferon-gamma receptor-deficient mice: enhanced disease severity by impaired inducible nitric oxide synthase induction. Circulation 2001, 103:18-21 [DOI] [PubMed] [Google Scholar]
- 50.Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, Casolaro V, Schleimer RP: Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol 1999, 163:6876-6883 [PubMed] [Google Scholar]
- 51.Tepper RI, Coffman RL, Leder P: An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 1992, 257:548-551 [DOI] [PubMed] [Google Scholar]
- 52.Tepper RI, Pattengale PK, Leder P: Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell 1989, 57:503-512 [DOI] [PubMed] [Google Scholar]
- 53.Patella V, de Crescenzo G, Marino I, Genovese A, Adt M, Gleich GJ, Marone G: Eosinophil granule proteins are selective activators of human heart mast cells. J Immunol 1996, 157:1219-1225 [PubMed] [Google Scholar]
- 54.Vitetta ES, Ohara J, Myers CD, Layton JE, Krammer PH, Paul WE: Serological, biochemical, and functional identity of B cell-stimulatory factor 1 and B cell differentiation factor for IgG1. J Exp Med 1985, 162:726-731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hazenbos WL, Heijnen IA, Meyer D, Hofhuis FM, Renardel de Lavalette CR, Schmidt RE, Capel PJ, van de Winkel JG, Gessner JE, van den Berg TK, Verbeek JS: Murine IgG1 complexes trigger immune effector functions predominantly via Fc gamma RIII (CD16). J Immunol 1998, 161:3026-3032 [PubMed] [Google Scholar]
- 56.Graziano RF, Looney RJ, Shen L, Fanger MW: Fc gamma R-mediated killing by eosinophils. J Immunol 1989, 142:230-235 [PubMed] [Google Scholar]
- 57.Weiler CR, Kita H, Hukee M, Gleich GJ: Eosinophil viability during immunoglobulin-induced degranulation. J Leukoc Biol 1996, 60:493-501 [DOI] [PubMed] [Google Scholar]
- 58.de Andres B, Rakasz E, Hagen M, McCormik ML, Mueller AL, Elliot D, Metwali A, Sandor M, Britigan BE, Weinstock JV, Lynch RG: Lack of Fc-epsilon receptors on murine eosinophils: implications for the functional significance of elevated IgE and eosinophils in parasitic infections. Blood 1997, 89:3826-3836 [PubMed] [Google Scholar]
- 59.Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B: Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med 1995, 181:1123-1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith SC, Allen PM: Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol 1991, 147:2141-2147 [PubMed] [Google Scholar]
- 61.De Vries JE: The role of IL-13 and its receptor in allergy and inflammatory responses. J Allergy Clin Immunol 1998, 102:165-169 [DOI] [PubMed] [Google Scholar]
- 62.McKenzie GJ, Fallon PG, Emson CL, Grencis RK, McKenzie ANJ: Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J Exp Med 1999, 189:1565-1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coffman RL, Seymour BW, Hudak S, Jackson J, Rennick D: Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice. Science 1989, 245:308-310 [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Afanasyeva M, Hill SL, Kaya Z, Rose NR: Nasal administration of cardiac myosin suppresses autoimmune myocarditis in mice. J Am Coll Cardiol 2000, 36:1992-1999 [DOI] [PubMed] [Google Scholar]
- 65.Barks JL, McQuillan JJ, Iademarco MF: TNF-alpha and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol 1997, 159:4532-4538 [PubMed] [Google Scholar]
- 66.Iademarco MF, Barks JL, Dean DC: Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. J Clin Invest 1995, 95:264-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bjorck P: Development of dendritic cells and their use in tumor therapy. Clin Immunol 1999, 92:119-127 [DOI] [PubMed] [Google Scholar]
- 68.Austyn JM, Hankins DF, Larsen CP, Morris PJ, Rao AS, Roake JA: Isolation and characterization of dendritic cells from mouse heart and kidney. J Immunol 1994, 152:2401-2410 [PubMed] [Google Scholar]
- 69.Shen CH, Stavnezer J: Interaction of stat6 and NF-kappaB: direct association and synergistic activation of interleukin-4-induced transcription. Mol Cell Biol 1998, 18:3395-3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barnes PJ: Therapeutic strategies for allergic diseases. Nature 1999, 402(Suppl.):B31-B38 [DOI] [PubMed] [Google Scholar]
- 71.Caspi RR: IL-12 in autoimmunity. Clin Immunol Immunopathol 1998, 88:4-13 [DOI] [PubMed] [Google Scholar]
- 72.Okura Y, Takeda K, Honda S, Hanawa H, Watanabe H, Kodama M, Izumi T, Aizawa Y, Seki S, Abo T: Recombinant murine interleukin-12 facilitates induction of cardiac myosin-specific type 1 helper T cells in rats. Circ Res 1998, 82:1035-1042 [DOI] [PubMed] [Google Scholar]
- 73.Karp CL, Grupe A, Schadt E, Ewart SL, Keane-Moore M, Cuomo PJ, Kohl J, Wahl L, Kuperman D, Germer S, Aud D, Peltz G, Wills-Karp M: Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nature Immunol 2000, 1:221-226 [DOI] [PubMed] [Google Scholar]
- 74.Huber SA: Coxsackievirus-induced myocarditis is dependent on distinct immunopathogenic responses in different strains of mice. Lab Invest 1997, 76:691-701 [PubMed] [Google Scholar]
- 75.Iizawa Y, Wagner RD, Czuprynski CJ: Analysis of cytokine mRNA expression in Listeria-resistant C57BL/6 and Listeria-susceptible A/J mice during Listeria monocytogenes infection. Infect Immun 1993, 61:3739-3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCarthy RE, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK, Hare JM, Baughman KL: Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med 2000, 342:690-695 [DOI] [PubMed] [Google Scholar]