

Abstract
Viral infection is one environmental factor that has been implicated as a precipitating event that may initiate β-cell damage during the development of diabetes. This study examines the mechanisms by which the viral replicative intermediate, double-stranded (ds) RNA impairs β-cell function and induces β-cell death. The synthetic dsRNA molecule polyinosinic-polycytidylic acid (poly IC) stimulates β-cell DNA damage and apoptosis without impairing islet secretory function. In contrast, the combination of poly IC and interferon (IFN)-γ stimulates DNA damage, apoptosis, and necrosis of islet cells, and this damage is associated with the inhibition of glucose-stimulated insulin secretion. Nitric oxide mediates the inhibitory and destructive actions of poly IC + IFN-γ on insulin secretion and islet cell necrosis. Inhibitors of nitric oxide synthase, aminoguanidine, and NG-monomethyl-l-arginine, attenuate poly IC + IFN-γ-induced DNA damage to levels observed in response to poly IC alone, prevent islet cell necrosis, and prevent the inhibitory actions on glucose-stimulated insulin secretion. NG-monomethyl-l-arginine fails to prevent poly IC- and poly IC + IFN-γ-induced islet cell apoptosis. PKR, the dsRNA-dependent protein kinase that mediates the antiviral response in infected cells, is required for poly IC- and poly IC + IFN-γ-induced islet cell apoptosis, but not nitric oxide-mediated islet cell necrosis. Alone, poly IC fails to stimulate DNA damage in islets isolated from PKR-deficient mice; however, nitric oxide-dependent DNA damage induced by the combination of poly IC + IFN-γ is not attenuated by the genetic absence of PKR. These findings indicate that dsRNA stimulates PKR-dependent islet cell apoptosis, an event that is associated with normal islet secretory function. In contrast, poly IC + IFN-γ-induced inhibition of glucose-stimulated insulin secretion and islet cell necrosis are events that are mediated by islet production of nitric oxide. These findings suggest that at least one IFN-γ-induced antiviral response (islet cell necrosis) is mediated through a PKR-independent pathway.
Insulin-dependent diabetes mellitus is an autoimmune disease that is characterized by the selective destruction of insulin-secreting β-cells found in pancreatic islets of Langerhans. 1 Although β-cells are destroyed during the development of diabetes, the molecular mechanisms responsible for this destruction have remained elusive. Recent studies have shown that cytokine-induced islet cell destruction is mediated by β-cell production of nitric oxide. 2-4 We and others 5-7 have shown that treatment of rat islets with interleukin (IL)-1 results in the expression of inducible nitric oxide synthase (iNOS), production of nitric oxide, and inhibition of insulin secretion. IL-1-induced islet damage is potentiated by interferon (IFN)-γ, a cytokine that reduces the concentration of IL-1 required to stimulate iNOS expression by β-cells. 8 One mechanism by which nitric oxide impairs β-cell function is the inhibition of islet oxidative metabolism resulting in reduced cellular levels of ATP. 9,10 IL-1 treatment results in an inhibition of islet and primary β-cell glucose oxidation to CO2 and aconitase activity, effects that are prevented by the inhibition of iNOS. 7,11 In addition, prolonged treatment of rat islets for 36 hours with IL-1 results in an irreversible inhibition of both islet cell aconitase activity and glucose-stimulated insulin secretion. 12
Cytokines and nitric oxide also stimulate DNA damage in islets. IL-1, alone or in combination with IFN-γ and/or tumor necrosis factor, has been reported to induce islet cell DNA damage that is prevented by the inhibition of iNOS, 13,14 and nitric oxide donor compounds induce DNA strand breaks in islet cells. 15,16 Apoptosis, a mechanism of programmed cell death that results in the removal of unwanted or damaged cells in the absence of inflammation, has been suggested as the mechanism by which cytokines and nitric oxide mediate β-cell death. 17-19 However, apoptosis is an energy-requiring event, 20,21 whereas nitric oxide impairs oxidative metabolism and reduces islet ATP levels, effects consistent with β-cell necrosis. In addition, nitric oxide also has been shown to attenuate apoptosis by preventing caspase activation. 22,23
There are at least two different stages of β-cell destruction during the development of diabetes: 1) a precipitating event that stimulates the initial destruction of β-cells; and 2) a later antigen-driven, T-cell-dependent destruction of β-cells. 24,25 Viral infection is one precipitating event that has been implicated in the induction of autoimmunity directed against β-cells in genetically susceptible individuals. 24,26 Viruses have been isolated from pancreata, and virus-specific IgM antibodies have been identified in newly diagnosed diabetic patients. 24,27 Also, diabetes can be induced by infection with encephalomyocarditis virus, Coxsackie B4 virus, Kilham’s rat virus, rubella virus, and retrovirus in genetically susceptible strains of rodents and primates. 28-31 Double-stranded (ds) RNA, formed during viral replication, activates the antiviral response in infected cells. 32,33 Recently, we have shown that treatment of rat islets with dsRNA and IFN-γ results in the inhibition of insulin secretion and islet degeneration, effects that correlate with iNOS expression and that are prevented by the inhibition of iNOS enzymatic activity. 34
Antiviral responses in infected cells are mediated in part by PKR, the dsRNA-dependent protein kinase. 32,33 PKR is a 65- to 68-kd ubiquitously expressed serine/threonine kinase that is activated by binding to dsRNA. After binding to dsRNA, PKR dimerizes, is activated by autophosphorylation, and phosphorylates cellular substrates such as the protein synthesis initiation factor eIF2α, the nuclear factor-κB inhibitory protein IκB, and IκB kinase. 32,33,35 Phosphorylation of eIF2α results in inhibition of protein synthesis because of the sequestration of eIF2B and subsequent inhibition of GDP for GTP exchange. 36,37 dsRNA-induced phosphorylation of IκB or IκB kinase results in IκB degradation, nuclear factor-κB nuclear localization, and nuclear factor-κB-dependent gene transcription. 38,39 PKR also seems to play a fundamental role in the regulation of apoptosis. 40,41 Expression of inactive PKR, or PKR depletion, prevents the induction of apoptosis by viral infection, dsRNA, tumor necrosis factor, and serum starvation. 42-45 In this study, the mechanisms by which dsRNA, alone or in combination with IFN-γ, stimulates β-cell death have been examined. Alone, dsRNA induces islet cell apoptosis, an event that correlates with normal glucose responsiveness of islets. In combination, dsRNA + IFN-γ stimulates apoptosis, necrosis, and inhibits insulin secretion by rat islets. Nitric oxide is not required for dsRNA- or dsRNA + IFN-γ-induced islet cell apoptosis; however, nitric oxide seems to mediate both dsRNA + IFN-γ-induced islet cell necrosis and inhibition of insulin secretion. Using islets isolated from PKR-deficient mice, we also show that dsRNA fails to induce islet cell apoptosis, whereas dsRNA + IFN-γ-induced islet cell necrosis and nitric oxide production are not affected. These results show that islet secretory function is not adversely affected under conditions in which islet cells undergo PKR-dependent apoptosis in response to dsRNA. However, insulin secretion is impaired under conditions in which islet cells undergo necrosis, a mechanism of cell death that seems to be mediated by nitric oxide and that does not require the presence of PKR.
Materials and Methods
Materials and Animals
CMRL-1066 tissue culture medium, l-glutamine, penicillin, streptomycin, and rat recombinant IFN-γ were from Life Technologies, Inc. (Grand Island, NY). Fetal calf serum was obtained from Hyclone (Logan, UT). Male Sprague-Dawley rats (250 to 300 g) were purchased from Harlan (Indianapolis, IN). PKR−/− mice in a C57BL/6 × 129 background 46 were bred at Saint Louis University. C57BL/6 × 129 mice were obtained from Jackson Laboratories (Bar Harbor, ME). Polyinosinic-polycytidylic acid (poly IC), aminoguanidine hemisulfate (AG), and collagenase type XI were from Sigma Chemical Co. (St. Louis, MO). The In situ cell death detection kit: fluorescein was from Boehringer Mannheim (Indianapolis, IN), mouse IFN-γ from R & D Systems, Inc. (Minneapolis, MN), and horseradish peroxidase-conjugated donkey anti-rabbit IgG and CY3-conjugated donkey anti-guinea pig IgG from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). All other reagents were from commercially available sources.
Islet Isolation, Cell Dispersion, and Fluorescence-Activated Cell Sorting (FACS) Purification of β Cells
Islets were isolated from male Sprague-Dawley rats (250 to 300 g), PKR−/−, and PKR+/+ mice by collagenase digestion as described previously. 47,48 Islets were dispersed into individual cells by treatment with trypsin (1 mg/ml) in Ca2+- and Mg2+-free Hanks’ solution at 37°C for 3 minutes as described previously. 48 Islets were cultured overnight at 37°C in an atmosphere of 95% air and 5% CO2 in complete CMRL-1066 tissue culture medium (CMRL-1066 containing 2 mmol/L l-glutamine, 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) before each experiment. For FACS purification of β-cells, islets isolated from 12 rats were cultured overnight (∼1200 islets per 3 ml) in complete CMRL-1066 media under an atmosphere of 95% air and 5% CO2 at 37°C. Islets were then dispersed into individual cells as described above, and incubated for 60 minutes at 37°C in complete CMRL-1066 before cell sorting. β-cells are purified based on size and endogenous Flavin Adenine Dinucleotide (FAD) fluorescence as described previously 11,49 using a FACStar + flow cytometer (Becton Dickinson, San Jose, CA). The cells were illuminated at 488 nm, and emission was monitored at 515 to 535 nm. This purification results in β- and α-cell purity of 90 to 95% and 80 to 85%, respectively. 11
Glucose-Stimulated Insulin Secretion
Islets were cultured at 37°C for 48 hours with the indicated concentrations of poly IC, IFN-γ, and AG. Islets were isolated, washed three times with Krebs-Ringer bicarbonate buffer (KRB) (25 mmol/L HEPES, 115 mmol/L NaCl, 24 mmol/L NaHCO3, 5 mmol/L KCl, 1 mmol/L MgCl2, 2.5 mmol/L CaCl2, and 0.1% bovine serum albumin, pH 7.4) containing 3 mmol/L d-glucose, aliquoted into siliconized 10-mm × 75-mm borosilicate culture tubes (20 islets/200 μl of KRB containing 3 mmol/L d-glucose), and preincubated for 30 minutes at 37°C with shaking. Insulin secretion was initiated by replacing the preincubation buffer with 200 μl of KRB containing either 3 mmol/L or 20 mmol/L d-glucose. Islets were incubated for 30 minutes at 37°C in an atmosphere of 95% air and 5% CO2, the supernatant was then removed and analyzed for insulin by radioimmunoassay. 50
Nitrite Determination
Nitrite production was determined by mixing 50 μl of culture medium with 50 μl of Griess reagent. 51 The absorbance at 540 nm was measured and nitrite concentrations were calculated from a sodium nitrite standard curve.
Electron Microscopy
Rat islets (400 per ml of complete CMRL-1066) were cultured for 48 hours with or without 50 μg/ml poly IC and 150 U/ml rat recombinant IFN-γ. The islets were fixed in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.3, containing 2% sucrose and 1 mmol/L calcium chloride overnight at 4°C, washed several times in cold cacodylate buffer containing 5% sucrose, and postfixed with 1% osmium tetroxide in 0.1 mol/L cacodylate buffer containing 2% sucrose for 3 hours at 4°C. After washing with distilled water, the tissue was stained en bloc with uranyl acetate, dehydrated through graded ethanols and propylene oxide, and infiltrated, embedded, and polymerized in Polybed resin (Polysciences, Inc., Warrington, PA). The islets were then infiltrated for 6 hours with 100% Polybed resin, embedded in fresh resin in Better Equipment for Electron Microscopy (BEEM) capsules, and polymerized overnight at 70°C. Thin sections were cut from the trimmed tissue blocks with a Reichert Ultracut E ultramicrotome using a diamond knife and were collected on 200-mesh copper grids. The sections were stained with uranyl acetate and lead citrate and viewed and photographed with a JEOL 100 CX electron microscope at 60 kV.
Immunohistochemistry and Terminal dUTP Nick-End Labeling (TUNEL) Staining
Islets (100 per 400 μl of complete CMRL-1066), cultured for 24 or 48 hours with poly IC, IFN-γ, AG, or NG-monomethyl-l-arginine (NMMA) were isolated and dispersed into individual cells as described above. FACS purified β-cells (50,000 cells per 400 μl of complete CMRL-1066) were also treated under the same conditions. The islet cells and primary β-cells were washed three times with 0.1 mol/L of phosphate-buffered saline (PBS) (pH 7.4) and then transferred to microscope slides by centrifugation (∼40,000 cells/slide). The cells were fixed in 4% paraformaldehyde for 30 minutes at room temperature, washed with PBS, and permeabilized (0.1% Triton X-100 and 0.1% sodium citrate) on ice for 2 minutes. Islet cells and β-cells containing DNA damage were detected by TUNEL staining according to manufacture specifications (In situ cell death detection kit, fluorescein; Boehringer Mannheim). For co-localization of TUNEL staining and insulin, slides were blocked for 15 minutes with 10% normal goat serum after TUNEL detection, and then incubated for 1 hour at room temperature with guinea pig anti-human insulin (1:200 dilution; Linco Inc., Saint Charles, MO). The slides were washed three times with PBS and then incubated for 1 hour at room temperature with a 1:200 dilution of CY3-conjugated donkey anti-guinea pig secondary antibody. Immunofluorescence microscopy was used for detection of insulin and TUNEL-positive cells.
Cell Viability and Apoptosis
Islets (25 per 400 μl of complete CMRL-1066) cultured for 24 or 48 hours with poly IC, IFN-γ, and NMMA, were isolated and dispersed into individual cells as described above. Cells were diluted to a concentration of 2.5 × 10 5 cells/ml of PBS. One μl of 1:1 mixture of acridine orange and ethidium bromide (100 μg/ml each) was added to 25 μl of the cell suspension and the cells were gently mixed. The cell suspension (10 μl) was transferred to a microscope slide, covered, and cell viability was examined at ×40 by fluorescein fluorescence. More than 400 cells per condition were scored in a double-blind manner using the following previously published criteria: 52 1) early apoptotic cells contain bright-green highly condensed or fragmented chromatin and red cytoplasm; 2) late apoptotic cells contain bright-orange highly condensed or fragmented chromatin and red cytoplasm; and, 3) necrotic cells contain uniform bright-orange chromatin with organized structure. Live cells contain uniform green nucleus with red cytoplasm. Apoptosis is presented as the total number of cells demonstrating features of early and late apoptosis. Control untreated islet cells contained ∼2% apoptotic and ∼8% necrotic cells, and this level of damage seems to be associated with islet dispersion into individual cells.
Statistics
Statistical comparisons were made between groups using a one-way analysis of variance. Significant differences between treatment groups compared with untreated controls (*; P < 0.05) were evaluated using a Bonferroni’s post hoc analysis.
Results
dsRNA Stimulates β-Cell DNA Damage
The effects of dsRNA, alone and in combination with IFN-γ, on the integrity of islet cell and β-cell DNA were evaluated by TUNEL staining. Treatment of rat islets for 24 hours with poly IC stimulates a threefold increase in the number of islet cells that contain DNA strand breaks (Figure 1b) ▶ , as evidenced by the green nuclear fluorescence in a limited number of islet cells (Figure 1a) ▶ . Alone, IFN-γ does not stimulate islet cell DNA damage; however, the combination of poly IC + IFN-γ stimulates a fourfold increase in the number of islet cells containing DNA damage. To determine whether dsRNA stimulates β-cell DNA damage, TUNEL staining was co-localized with insulin-containing cells (red fluorescence, Figure 1a ▶ ). In response to poly IC or poly IC + IFN-γ, >85% of the TUNEL-positive cells contained insulin (data not shown). To confirm that dsRNA stimulates DNA damage in β-cells, the effects of poly IC alone, and in combination with IFN-γ, on DNA damage in primary β-cells purified by FACS were examined. Treatment of FACS-purified β-cells for 24 hours with poly IC and poly IC + IFN-γ results in an approximate threefold increase in the number of cells containing DNA damage (Figure 1, c and d) ▶ . Alone, IFN-γ does not stimulate DNA damage, nor does it enhance DNA damage induced by poly IC in FACS-purified β-cells.
Figure 1.

Effects of poly IC and IFN-γ on islet cell DNA integrity. Rat islets (100 per 400 μl of complete CMRL-1066) (a) or FACS-purified β-cells (50,000 per 400 μl of complete CMRL-1066) (c) were treated for 24 hours with 50 μg/ml of poly IC, or rat islets (b) and FACS-purified β-cells (d) were treated with the indicated concentrations of poly IC, rat IFN-γ, and NMMA. The islets and β-cells were isolated and DNA damage was analyzed by TUNEL staining (green fluorescence) as described in Materials and Methods. Insulin-containing cells were identified using a guinea pig anti-human insulin and CY3-conjugated donkey anti-guinea pig secondary antibodies (a). The arrows indicate TUNEL-positive β-cells (c). The percentage of TUNEL-positive cells was determined in a double-blind manner. Results are representative of three independent experiments (a and c) or the average ± SEM of three independent experiments (b and d).
Previous studies have shown that treatment of rat islets with poly IC + IFN-γ results in the time-dependent expression of iNOS and production of nitric oxide that is first apparent after a 24-hour incubation, and maximal after a 48-hour incubation. 34 In addition, nitric oxide has been shown to directly stimulate DNA strand breaks in a number of cell types including islets. 13,53 Importantly, poly IC-induced DNA damage seems to occur by nitric oxide-independent mechanisms, as the iNOS inhibitor NMMA does not attenuate poly IC-induced TUNEL-positive staining. However, NMMA reduces poly IC + IFN-γ-induced DNA strand breaks to levels induced by poly IC alone (Figure 1c) ▶ . These findings suggest that a 24-hour exposure of rat islets to poly IC results in nitric oxide-independent β-cell DNA damage, and that poly IC + IFN-γ-induced DNA damage is both nitric oxide-dependent and nitric oxide-independent.
Although poly IC + IFN-γ stimulates iNOS expression by FACS-purified β-cells, 34 the results presented in Figure 1 ▶ indicate that poly IC and poly IC + IFN-γ stimulate primary β-cell DNA damage to similar levels after a 24-hour incubation. The lack of an increase in DNA damage of primary β-cells in response to poly IC + IFN-γ is not believed to be because of a lack of nitric oxide production, but is most likely associated with proximity of β-cells in intact islets as compared to β-cells free-floating in solution. In islets, β-cells are in close physical contact such that nitric oxide produced from one β cell may influence the integrity of DNA in adjacent cells by simple diffusion from the producing cell. However, the distance between FACS-purified β-cells free-floating in culture is much larger and thereby reducing the effective concentration of nitric oxide and potential paracrine actions of this free radical on DNA integrity.
In addition to insulin-secreting β-cells, islets contain large numbers of glucagon-secreting α cells (∼25 to 30%). Treatment of FACS-purified α cells for 24 hours with poly IC or poly IC + IFN-γ does not stimulate α-cell DNA damage as assessed by TUNEL staining (data not shown). These findings are also consistent with an inability of α cells to express iNOS or produce nitric oxide in response to dsRNA, IFN-γ, or dsRNA + IFN-γ. 34
Effects of dsRNA and IFN-γ on Islet Cell Apoptosis and Necrosis
Although TUNEL staining identifies cells containing DNA damage, this method does not discriminate between apoptotic and necrotic cell death. To determine the mode of islet cell death in response to dsRNA, vital dye staining (acridine orange and ethidium bromide) of isolated islet cells was performed. Treatment of rat islets with poly IC stimulates an approximate sixfold to sevenfold increase in apoptosis after 24-hour and 48-hour incubations, and islet cell apoptosis in response to poly IC is not further enhanced by IFN-γ (Figure 2, a and b) ▶ . Poly IC-induced islet cell apoptosis does not seem to require the production of nitric oxide, as poly IC fails to stimulate iNOS expression by rat islets, 34 and NMMA does not attenuate poly IC-induced islet cell apoptosis. In contrast, poly IC + IFN-γ stimulates an approximate twofold and fivefold increase in islet cell necrosis after a 24- and 48-hour incubation, respectively, and an approximate sixfold to sevenfold increase in islet cell apoptosis at both time points. NMMA prevents islet cell necrosis, but fails to prevent poly IC + IFN-γ-induced islet cell apoptosis. The lower level of islet cell necrosis after a 24-hour incubation (twofold increase) as compared to a 48-hour incubation (fivefold) is consistent with the time-dependent effects of poly IC + IFN-γ on iNOS expression by rat islets. 34 These findings indicate that poly IC- and poly IC + IFN-γ-induced islet cell apoptosis occurs by nitric oxide-independent mechanisms and that islet cell necrosis in response to poly IC + IFN-γ is dependent on the production of nitric oxide.
Figure 2.
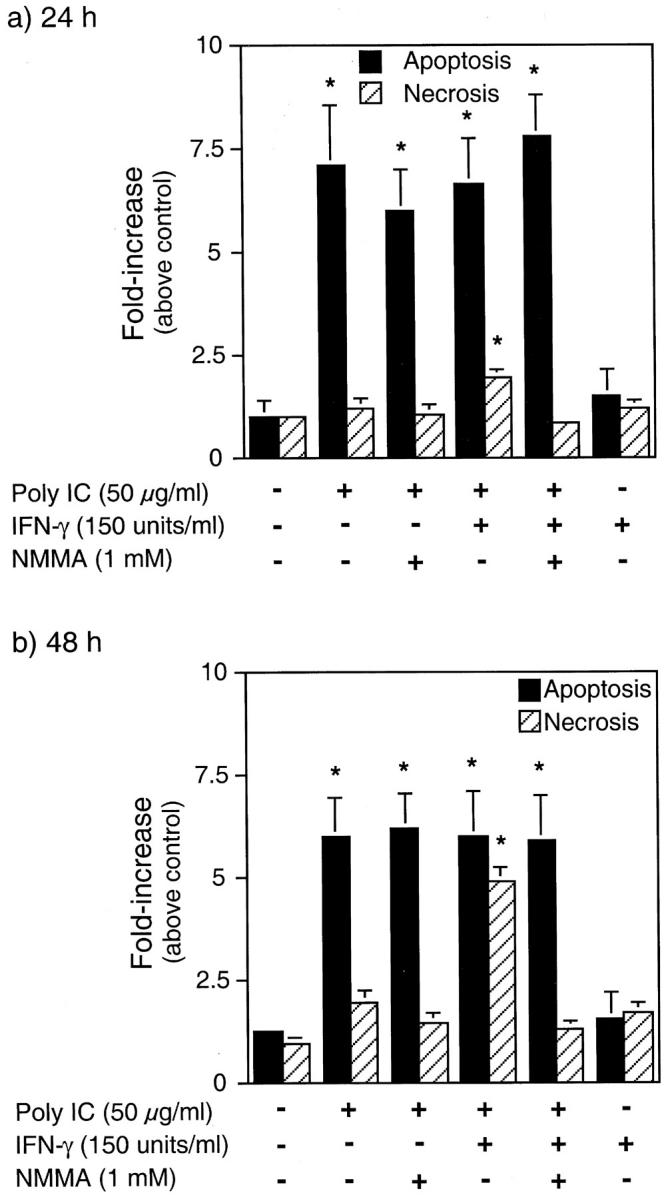
Effects of dsRNA and IFN-γ on islet cell apoptosis and necrosis. Rat islets (25 per 400 μl of complete CMRL-1066) were treated for 24 or 48 hours with the indicated concentrations of poly IC, rat IFN-γ, and NMMA. The islets were isolated, dispersed into individual cells, and apoptosis and necrosis determined by acridine orange and ethidium bromide staining as described in Materials and Methods. Results are the average ± SEM of three independent experiments.
Electron Microscopic Analysis of Islet Cell Viability in Response to dsRNA and IFN-γ
To confirm that β-cells undergo apoptosis and necrosis in response to dsRNA and dsRNA + IFN-γ, respectively, islet cell morphological damage was assessed by electron microscopy. Treatment of rat islets for 48 hours with poly IC results in β-cell apoptosis that is characterized by chromatin condensation in insulin-containing β-cells (Figure 3b) ▶ . In combination, dsRNA + IFN-γ stimulates β-cell apoptosis as evidenced by β-cell chromatin condensation in insulin-containing cells (Figure 3c) ▶ , and β-cell necrosis that is characterized by the loss of both plasma membrane and nuclear membrane integrity (Figure 3d) ▶ . Morphological damage in islet cells treated for 48 hours with IFN-γ alone was not observed (data not shown). These findings are consistent with results presented in Figures 1 and 2 ▶ ▶ , and indicate that poly IC stimulates islet cell apoptosis, whereas the combination of poly IC + IFN-γ stimulates both islet cell apoptosis and necrosis.
Figure 3.
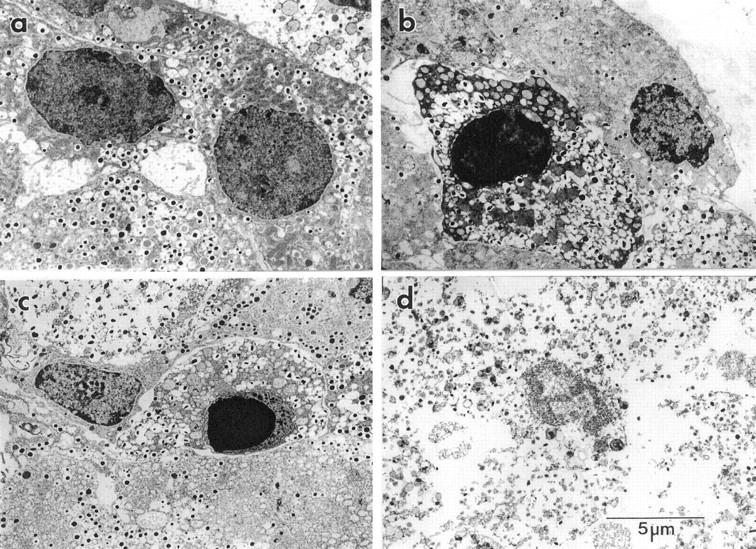
Effects of dsRNA and IFN-γ on β-cell morphology as determined by electron microscopy. Rat islets (400 per ml of complete CMRL-1066) were untreated (a), treated for 48 hours with 50 μg/ml poly IC (b), or treated with 50 μg/ml of poly IC and 150 U/ml rat IFN-γ (c and d). The islets were isolated, fixed, and morphological damage assessed by electron microscopy as stated in Materials and Methods. β-cell apoptosis is characterized by chromatin condensation (b and c) and islet cell necrosis is characterized by the loss of both plasma membrane and nuclear membrane integrity (d). Results are representative of four independent experiments.
Effects of dsRNA and IFN-γ on Glucose-Stimulated Insulin Secretion by Rat Islets
To determine whether dsRNA-induced islet cell death has functional consequences on β-cells, the effects of poly IC and IFN-γ, alone and in combination, on glucose-stimulated insulin secretion were examined. A 48-hour incubation of rat islets with dsRNA + IFN-γ results in a near complete inhibition of glucose-stimulated insulin secretion (Figure 4a) ▶ . The inhibitory effects of poly IC + IFN-γ on glucose-stimulated insulin secretion are associated with a more than threefold increase in nitric oxide production (Figure 4b) ▶ . AG, a selective inhibitor of iNOS, prevents the inhibitory actions of poly IC + IFN-γ on glucose-stimulated insulin secretion, and the stimulatory actions of these agents on nitric oxide production by rat islets. Alone, neither dsRNA nor IFN-γ inhibit glucose-stimulated insulin secretion or induce nitric oxide production by rat islets after 48 hours of incubation. The lack of nitric oxide production by rat islets in response to poly IC or IFN-γ is consistent with our previous studies showing that these agents fail to stimulate iNOS mRNA accumulation (Northern analysis) or protein expression after 24- and 40-hour incubations, respectively. 34
Figure 4.
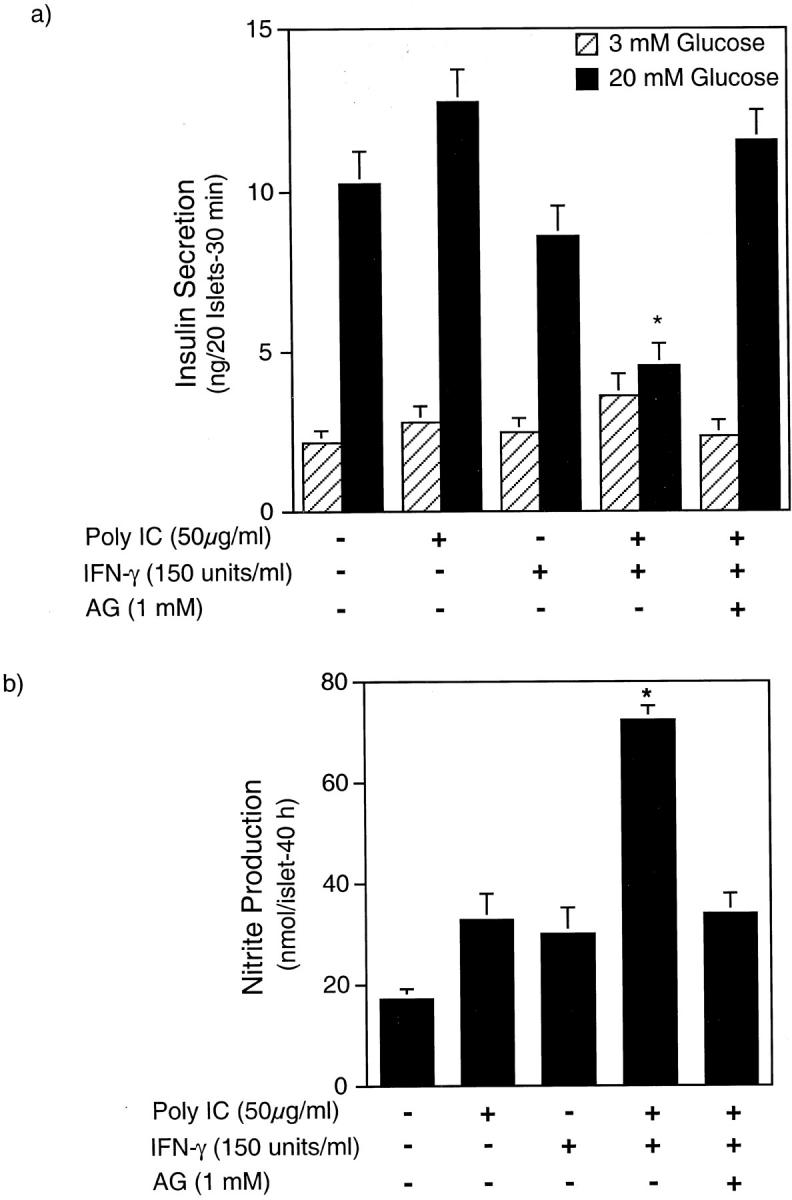
Effects of poly IC and IFN-γ on glucose-stimulated insulin secretion and nitric oxide production by rat islets. Rat islets (220 per ml of complete CMRL-1066) were treated for 48 hours with the indicated concentrations of poly IC, rat IFN-γ, and AG. The islets were isolated and glucose-stimulated insulin secretion was examined (a), and nitrite production (b) was determined on the culture supernatant. Results for insulin secretion and nitrite production are the average ± SEM of three independent experiments.
Role of PKR in dsRNA- and dsRNA and IFN-γ-Induced Islet Cell DNA Damage
PKR has been implicated as a primary regulator of cell death induced by viral infection, or by the viral replicative intermediate dsRNA. 40,41 To examine the role of PKR in islet death, the effects of dsRNA, alone and in combination with IFN-γ, on DNA damage and nitric oxide production by islets isolated from PKR−/− and PKR+/+ mice were examined (Figure 5, a and b) ▶ . Treatment of islets isolated from PKR+/+ mice for 48 hours with poly IC results in an approximately threefold increase in the number of cells containing DNA damage. Poly IC-induced DNA damage is significantly enhanced in the presence of IFN-γ, an effect that is mediated by nitric oxide, as NMMA reduces poly IC + IFN-γ-induced DNA damage to levels similar to those induced by poly IC alone. PKR seems to mediate dsRNA-induced islet cell DNA damage, as poly IC fails to induce DNA strand breaks in islet cells isolated from PKR−/− mice (Figure 5a) ▶ . However, PKR is not required for dsRNA + IFN-γ-induced islet cell DNA damage, as poly IC + IFN-γ induce an approximate threefold increase in TUNEL-positive cells, an effect that is prevented by NMMA. Figure 5b ▶ confirms that poly IC + IFN-γ stimulates similar levels of nitrite production by islets isolated from PKR−/− and PKR+/+ mice, and that poly IC and IFN-γ alone fail to induce nitric oxide production by these islets. These findings indicate that poly IC-induced DNA damage is PKR-dependent and nitric oxide-independent; however, poly IC + IFN-γ-induced DNA damage does not require the presence of PKR, but is dependent on islet production of nitric oxide.
Figure 5.
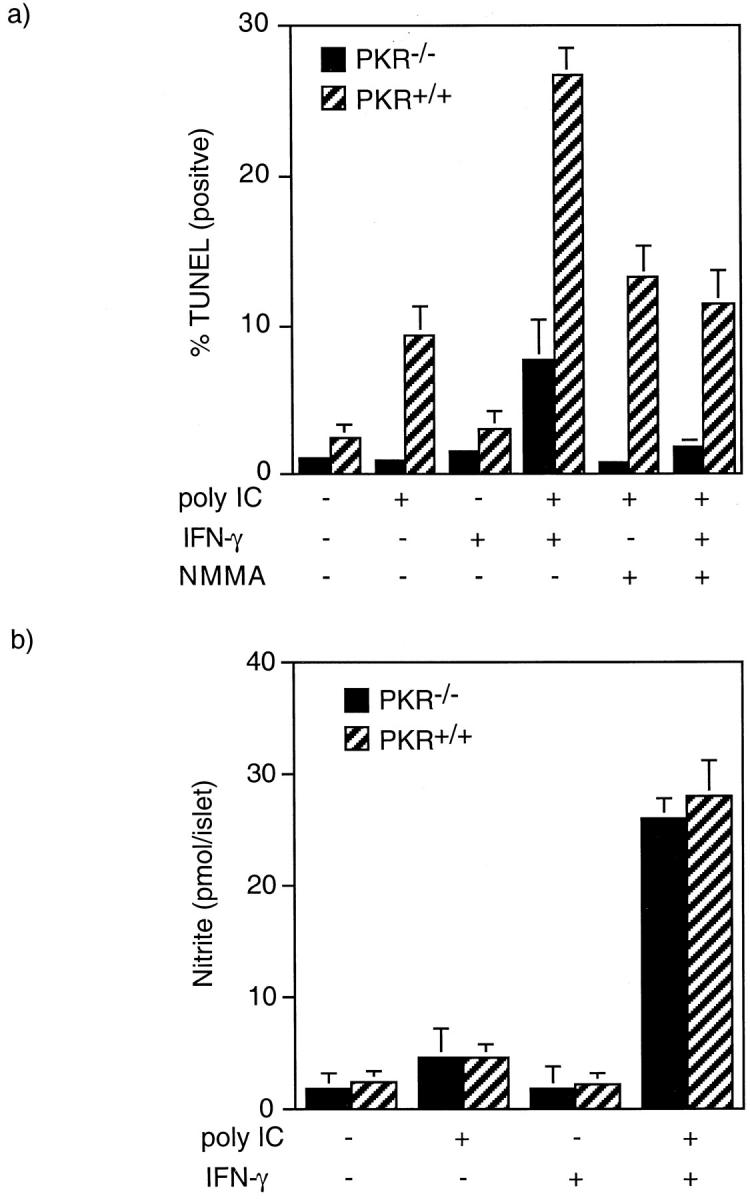
Effects of poly IC and IFN-γ on DNA integrity of islet cells isolated from PKR−/− and PKR+/+ mice. Islets (100 per 400 μl of complete CMRL-1066) isolated from PKR−/− and PKR+/+ mice were treated for 48 hours with 400 μg/ml of poly IC, 150 U/ml mouse IFN-γ and 1 mmol/L NMMA as indicated. The islets were then isolated, dispersed into individual cells, and DNA damage was determined by TUNEL staining (a). For nitrite production, islets (100 per 400 μl of complete CMRL-1066) isolated from PKR−/− and PKR+/+ mice were treated for 48 hours with 400 μg/ml of poly IC and 150 U/ml of mouse IFN-γ as indicated. The supernatant was removed and nitrite production determined (b). Results are the average ± SEM of three (a) and two (b) independent experiments.
The absolute level of DNA damage induced by poly IC + IFN-γ is significantly lower in islet cells isolated from PKR−/− mice as compared to islet cells isolated from PKR+/+ mice. This result suggests that PKR may participate in poly IC + IFN-γ-induced necrosis in addition to its role in the regulation of poly IC-induced islet cell apoptosis. However, because poly IC-induced islet cell apoptosis is mediated by PKR (Figure 5a) ▶ and occurs by nitric oxide-independent mechanisms (Figure 2) ▶ , and TUNEL staining detects both apoptosis and necrosis, one would expect to observe lower levels of poly IC + IFN-γ-induced TUNEL-positive staining in islet cells isolated from PKR−/− mice because only nitric oxide-dependent DNA damage would be detected by TUNEL staining under these conditions. Therefore, the reduced level of poly IC + IFN-γ-induced TUNEL staining in islet cells isolated from PKR−/− mice is most likely because of the lack of PKR-dependent apoptosis.
Discussion
Viral infection is one precipitating event that has been implicated in the induction of autoimmune diabetes. Nevertheless, it has been difficult to study mechanisms of viral-induced diabetes because multiple viruses from both RNA and DNA viral families have been implicated in the development of this disease. 27 In this study, the synthetic dsRNA molecule poly IC has been used to examine the response of β-cells to a viral infection. Poly IC has been shown to mimic the antiviral response of infected cells. 54 Treatment of cells with poly IC results in the induction of type I interferons, activation of PKR, and the inhibition of translation because of eIF2α phosphorylation. 33,41 Poly IC has also been shown to induce diabetes in diabetes-resistant BioBreeding (BB) rats, and to accelerate diabetes in diabetes-prone BB rats. 55,56 Similar to viral-induced diabetes, the development of diabetes in response to poly IC seems to be strain-dependent. Rats containing MHC class IIu genes develop diabetes whereas poly IC fails to induce insulitis or diabetes in rats expressing MHC class IIa genes. 57 Poly IC-induced diabetes is characterized by islet inflammation comprised of macrophages, monocytes, and CD8+ T cells, and requires the generation of autoreactive T cells as evidenced by the induction of diabetes after the transfer of conA-activated nondiabetic spleen cells isolated from poly IC-treated rats to naive syngeneic recipients. 57 Also, poly IC-induced diabetes is associated with an increase in class I MHC gene expression. 57 IFN-γ seems to play a key role in regulating β-cell expression of class I MHC, 58 suggesting that increased levels of IFN-γ may be present in islets during the development of poly IC-induced diabetes. Importantly, these features of poly IC-induced diabetes are characteristics associated with the development of diabetes in the NOD mouse, BB rat, viral-induced diabetes in animal models, and insulin-dependent diabetes mellitus.
Although poly IC and viral infection induces the development of diabetes in animal models, the direct actions of these agents on β-cell function have been poorly defined. Previous studies have shown that poly IC, in combination with IFN-γ, stimulates iNOS expression and nitric oxide production by isolated rat islets and FACS-purified β-cells, and that nitric oxide mediates islet degeneration and the inhibitory actions of these agents on insulin secretion. 34 Alone, neither poly IC nor IFN-γ impair insulin secretion, induce iNOS expression, or stimulate islet degeneration. In this study the effects of poly IC on β-cell viability have been examined. Poly IC stimulates islet cell apoptosis; however, apoptotic cell death under these conditions does not result in an inhibition of islet function. In contrast, poly IC + IFN-γ stimulates necrosis, in addition to apoptosis, and islet cell necrosis correlates with an inhibition of glucose-stimulated insulin secretion (Figure 6) ▶ . The inhibitory actions of poly IC + IFN-γ on insulin secretion and the induction of islet cell necrosis seem to be nitric oxide-dependent, as inhibitors of iNOS prevent each of these damaging events. Poly IC-induced islet cell apoptosis seems to occur by nitric oxide-independent mechanisms as 1) poly IC fails to stimulate iNOS expression or nitric oxide production by rat islets 34 and 2) inhibitors of iNOS do not attenuate poly IC-induced DNA damage or β-cell apoptosis. These findings suggest that conditions in which islet cells die by apoptosis (treatment with poly IC, or poly IC + IFN-γ + AG/NMMA), sufficient numbers of β-cells within islets remain competent to compensate for the loss of apoptotic β-cells. In contrast, islets are not able overcome the destructive actions of nitric oxide, as islet cell necrosis induced in response to poly IC + IFN-γ results in the complete loss of islet secretory function. In the context of the development of autoimmune diabetes, our findings suggest that viral infection of β-cells may lead to apoptosis, an event that clears virally infected cells and prevents islet inflammation. The remaining uninfected β-cells could then compensate for the apoptotic loss of virally infected cells and diabetes would not develop. However, in the presence of IFN-γ (or the presence of inflammatory T cells capable of producing this cytokine), viral infection would be predicted to induce islet cell necrosis in addition to apoptosis, and the necrotic event may elicit two responses: 1) an increased islet inflammation because of the necrosis of β-cells, and 2) the release of β-cell antigens and induction of autoimmunity directed against remaining β-cells. Clearly, caution should be exercised in the extrapolation of in vitro data to in vivo mechanisms of cellular destruction; however, these findings suggest a novel mechanism that may explain initiation events that could be examined in a number of animal models systems of viral-induced diabetes.
Figure 6.

Mechanism of dsRNA- and IFN-γ-induced islet cell death. The results presented in this study suggest that dsRNA-induced islet cell apoptosis is dependent on PKR and is associated with normal islet insulin secretion. dsRNA- and IFN-γ-induced islet cell necrosis is dependent on islet production of nitric oxide, is associated with inhibition of insulin secretion, and occurs by PKR-independent mechanisms.
The mechanism by which poly IC and poly IC + IFN-γ induce islet cell apoptosis seems to require the presence of functional PKR. Poly IC fails to induce DNA damage in islets isolated form PKR-deficient mice. This finding is consistent with recent studies demonstrating PKR-dependent apoptosis in response to tumor necrosis factor, serum deprivation, and poly IC in cultured fibroblasts. 44,59 Also, PKR seems to participate in vaccinia- and vesicular stomatitis virus-induced apoptosis of HeLa cells and mouse embryonic fibroblasts. 42,45 One mechanism by which PKR stimulates apoptosis is by the up-regulation of FAS expression resulting in FADD/caspase 8-dependent cell death. 43 PKR-mediated apoptosis may also be associated with the phosphorylation state of eIF2α. 40,60 Expression of a Ser51Asp eIF2α mutant, which mimics the actions of phosphorylated eIF2α, induces apoptosis in COS-1 cells, and expression of a nonphosphorylatable Ser51Ala mutant of eIF2α prevents serum deprivation- and tumor necrosis factor-induced apoptosis in NIH 3T3 cells. 44 In our studies, the mechanisms by which PKR mediates dsRNA-induced islet cell apoptosis are unclear. Poly IC stimulates FAS expression by rat islets; however, anti-Fas antibodies do not further potentiate poly IC-induced islet cell apoptosis, and we have been unable to detect Fas ligand expression in islets in response to poly IC (unpublished observation, MA, JC, and JAC). In addition, we have been unable to detect changes in the phosphorylation state of eIF2α in islets treated with poly IC (as determined by immunoprecipitation of eIF2α from 32P-labeled rat islets, data not shown). The lack of eIF2α phosphorylation in response to poly IC treatment is believed to be a consequence of the limited number of islet cells that are responsive to poly IC. Using apoptosis in as an index, ∼10% of islet cells or 200 cells per islet appear to be responsive to dsRNA, a level that seems to fall below the limits of detection of this immunoprecipitation experiment. Therefore, it is not possible to conclude whether eIF2α phosphorylation contributes to poly IC-induced islet cell apoptosis. Although it is clear that PKR is required for islet cell apoptosis, it is not required for dsRNA + IFN-γ-induced islet cell necrosis or nitric oxide production, as poly IC + IFN-γ stimulates a nitric oxide-dependent sevenfold to eightfold increase in DNA damage in islet cells isolated from PKR−/− mice. In addition, we have recently shown that poly IC + IFN-γ stimulates iNOS expression and inhibits insulin secretion to similar levels in islets isolated from PKR−/− and PKR+/+ mice. 61 These findings suggest that PKR mediates apoptotic mechanisms triggered by dsRNA in islets, and that the antiviral responses of necrosis and nitric oxide production activated by the combination poly IC + IFN-γ seem to occur by PKR-independent mechanisms.
Acknowledgments
We thank Joseph Martin and Jessica Gorman for expert technical assistance and the Diabetes Research and Training Center at Washington University School of Medicine (St. Louis, MO) for performing insulin radioimmunoassays.
Footnotes
Address reprint requests to Dr. John Corbett, Saint Louis University, Department of Biochemistry and Molecular Biology, 1402 South Grand Blvd., Saint Louis, MO 63104. E-mail: corbettj@slu.edu.
Supported by National Institutes of Health grants AI44458 (to J. A. C.) and AI42394 (R. J. K.).
References
- 1.Gepts W: Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965, 14:619-633 [DOI] [PubMed] [Google Scholar]
- 2.Corbett JA, Kwon G, Hill JR, McDaniel ML: Nitric oxide mediates cytokine-induced islet destruction. Diabetes Annual, vol 9. Edited by SM Marshall, PD Home, RA Rizza. Amsterdam, Elsevier Science, 1995
- 3.Eizirik DL, Flodstrom M, Karlsen AE, Welsh N: The harmony of the spheres: inducible nitric oxide synthase and related genes in pancreatic beta cells. Diabetologia 1996, 39:875-890 [DOI] [PubMed] [Google Scholar]
- 4.Mandrup-Poulsen T: The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 1996, 39:1005-1029 [DOI] [PubMed] [Google Scholar]
- 5.Corbett JA, Lancaster JR, Jr, Sweetland MA, McDaniel ML: Interleukin-1 beta-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 beta-induced inhibition of insulin secretion. J Biol Chem 1991, 266:21351-21354 [PubMed] [Google Scholar]
- 6.Southern C, Schulster D, Green IC: Inhibition of insulin secretion by interleukin-1 beta and tumour necrosis factor-alpha via an l-arginine-dependent nitric oxide generating mechanism. FEBS Lett 1990, 276:42-44 [DOI] [PubMed] [Google Scholar]
- 7.Welsh N, Eizirik DL, Bendtzen K, Sandler S: Interleukin-1 beta-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology 1991, 129:3167-3173 [DOI] [PubMed] [Google Scholar]
- 8.Heitmeier MR, Scarim AL, Corbett JA: Interferon-gamma increases the sensitivity of islets of Langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J Biol Chem 1997, 272:13697-13704 [DOI] [PubMed] [Google Scholar]
- 9.Corbett JA, Wang JL, Hughes JH, Wolf BA, Sweetland MA, Lancaster JR, Jr, McDaniel ML: Nitric oxide and cyclic GMP formation induced by interleukin 1 beta in islets of Langerhans. Evidence for an effector role of nitric oxide in islet dysfunction. Biochem J 1992, 287:229-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunningham JM, Green IC: Cytokines, nitric oxide and insulin secreting cells. Growth Regul 1994, 4:173-180 [PubMed] [Google Scholar]
- 11.Corbett JA, Wang JL, Sweetland MA, Lancaster JR, Jr, McDaniel ML: Interleukin 1 beta induces the formation of nitric oxide by beta-cells purified from rodent islets of Langerhans. Evidence for the beta-cell as a source and site of action of nitric oxide. J Clin Invest 1992, 90:2384-2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scarim AL, Heitmeier MR, Corbett JA: Irreversible inhibition of metabolic function and islet destruction after a 36-hour exposure to interleukin-1beta. Endocrinology 1997, 138:5301-5307 [DOI] [PubMed] [Google Scholar]
- 13.Delaney CA, Green MH, Lowe JE, Green IC: Endogenous nitric oxide induced by interleukin-1 beta in rat islets of Langerhans and HIT-T15 cells causes significant DNA damage as measured by the ‘comet’ assay. FEBS Lett 1993, 333:291-295 [DOI] [PubMed] [Google Scholar]
- 14.Fehsel K, Jalowy A, Qi S, Burkart V, Hartmann B, Kolb H: Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 1993, 42:496-500 [DOI] [PubMed] [Google Scholar]
- 15.Eizirik DL, Delaney CA, Green MH, Cunningham JM, Thorpe JR, Pipeleers G, Hellerstrom C, Green JC: Nitric oxide donors decrease the function and survival of human pancreatic islets. Mol Cell Endocrinol 1996, 118:71-83 [DOI] [PubMed] [Google Scholar]
- 16.Hadjivassiliou V, Green MH, James RF, Swift SM, Clayton HA, Green IC: Insulin secretion, DNA damage, and apoptosis in human and rat islets of Langerhans following exposure to nitric oxide, peroxynitrite, and cytokines. Nitric Oxide 1998, 2:429-441 [DOI] [PubMed] [Google Scholar]
- 17.Dunger A, Augstein P, Schmidt S, Fischer U: Identification of interleukin 1-induced apoptosis in rat islets using in situ specific labelling of fragmented DNA. J Autoimmun 1996, 9:309-313 [DOI] [PubMed] [Google Scholar]
- 18.Mauricio D, Mandrup-Poulsen T: Apoptosis and the pathogenesis of IDDM: a question of life and death. Diabetes 1998, 47:1537-1543 [DOI] [PubMed] [Google Scholar]
- 19.Iwahashi H, Hanafusa T, Eguchi Y, Nakajima H, Miyagawa J, Itoh N, Tomita K, Namba M, Kuwajima M, Noguchi T, Tsujimoto Y, Matsuzawa Y: Cytokine-induced apoptotic cell death in a mouse pancreatic beta-cell line: inhibition by Bcl-2. Diabetologia 1996, 39:530-536 [DOI] [PubMed] [Google Scholar]
- 20.Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S, Nicotera P: Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res 1999, 249:396-403 [DOI] [PubMed] [Google Scholar]
- 21.Nicotera P, Leist M, Ferrando-May E: Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 1998, 103:139-142 [DOI] [PubMed] [Google Scholar]
- 22.Dimmeler S, Haendeler J, Sause A, Zeiher AM: Nitric oxide inhibits APO-1/Fas-mediated cell death. Cell Growth Differ 1998, 9:415-422 [PubMed] [Google Scholar]
- 23.Li J, Bombeck CA, Yang S, Kim YM, Billiar TR: Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J Biol Chem 1999, 274:17325-17333 [DOI] [PubMed] [Google Scholar]
- 24.Bach JF: Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev 1994, 15:516-542 [DOI] [PubMed] [Google Scholar]
- 25.Rossini AA, Mordes JP, Like AA: Immunology of insulin-dependent diabetes mellitus. Annu Rev Immunol 1985, 3:289-320 [DOI] [PubMed] [Google Scholar]
- 26.Yoon JW, Jun HS, Santamaria P: Cellular and molecular mechanisms for the initiation and progression of beta cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity 1998, 27:109-122 [DOI] [PubMed] [Google Scholar]
- 27.Yoon JW: The role of viruses and environmental factors in the induction of diabetes. Curr Top Microbiol Immunol 1990, 164:95-123 [DOI] [PubMed] [Google Scholar]
- 28.Chung YH, Jun HS, Kang Y, Hirasawa K, Lee BR, Van Rooijen N, Yoon JW: Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. J Immunol 1997, 159:466-471 [PubMed] [Google Scholar]
- 29.Baek HS, Yoon JW: Direct involvement of macrophages in destruction of beta-cells leading to development of diabetes in virus-infected mice. Diabetes 1991, 40:1586-1597 [DOI] [PubMed] [Google Scholar]
- 30.Rayfield EJ, Kelly KJ, Yoon JW: Rubella virus-induced diabetes in the hamster. Diabetes 1986, 35:1278-1281 [DOI] [PubMed] [Google Scholar]
- 31.Signore A, Procaccini E, Chianelli M, Pozzilli P: Retroviruses and diabetes in animal models: hypotheses for the induction of the disease [published erratum appears in Diabete Metab Dec;21(5):384]. Diabete Metab 1995, 21:147-155 [PubMed] [Google Scholar]
- 32.Clemens MJ, Elia A: The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res 1997, 17:503-524 [DOI] [PubMed] [Google Scholar]
- 33.Williams BRG: PKR: a sentinel kinase for cellular stress. Oncogene 1999, 18:6112-6120 [DOI] [PubMed] [Google Scholar]
- 34.Heitmeier MR, Scarim AL, Corbett JA: Double-stranded RNA inhibits beta-cell function and induces islet damage by stimulating beta-cell production of nitric oxide. J Biol Chem 1999, 274:12531-12536 [DOI] [PubMed] [Google Scholar]
- 35.Proud CG: PKR: a new name and new roles. Trends Biochem Sci 1995, 20:241-246 [DOI] [PubMed] [Google Scholar]
- 36.Gale M, Katze MG: Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther 1998, 78:29-46 [DOI] [PubMed] [Google Scholar]
- 37.Kumar KU, Srivastava SP, Kaufman RJ: Double-stranded RNA-activated protein kinase (PKR) is negatively regulated by 60S ribosomal subunit protein L18. Mol Cell Biol 1999, 19:1116-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BRG: NF-kappa B activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappa B-inducing kinase and I kappa B kinase. Mol Cell Biol 2000, 20:1278-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J, Karin M: JNK2 and IKK beta are required for activating the innate response to viral infection. Immunity 1999, 11:721-731 [DOI] [PubMed] [Google Scholar]
- 40.Kaufman RJ: Double-stranded RNA-activated protein kinase mediates virus-induced apoptosis: a new role for an old actor. Proc Natl Acad Sci USA 1999, 96:11693-11695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jagus R, Joshi B, Barber GN: PKR, apoptosis and cancer. Int J Biochem Cell Biol 1999, 31:123-138 [DOI] [PubMed] [Google Scholar]
- 42.Kibler KV, Shors T, Perkins KB, Zeman CC, Banaszak MP, Biesterfeldt J, Langland JO, Jacobs BL: Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol 1997, 71:1992-2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN: Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J 1998, 17:6888-6902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srivastava SP, Kumar KU, Kaufman RJ: Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem 1998, 273:2416-2423 [DOI] [PubMed] [Google Scholar]
- 45.Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN: Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13:129-141 [DOI] [PubMed] [Google Scholar]
- 46.Yang YL, Reis LF, Pavlovic J, et al: Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J 1995, 14:6095-6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacy PE, Kostianovsky M: Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 1967, 16:35-39 [DOI] [PubMed] [Google Scholar]
- 48.McDaniel ML, Colca JR, Kotagal N, Lacy PE: A subcellular fractionation approach for studying insulin release mechanisms and calcium metabolism in islets of Langerhans. Methods Enzymol 1983, 98:182-200 [DOI] [PubMed] [Google Scholar]
- 49.Pipeleers DG, In’t Veld PA, Van de Winkel M, Maes E, Schuit FC, Gepts W: A new in vitro model for the study of pancreatic A and B cells. Endocrinology 1985, 117:806-816 [DOI] [PubMed] [Google Scholar]
- 50.Wright PH, Makulu DR, Vichick D, Sussman KE: Insulin immunoassay by back-titration. Diabetes 1971, 20:33-45 [DOI] [PubMed] [Google Scholar]
- 51.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR: Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 1982, 126:131-138 [DOI] [PubMed] [Google Scholar]
- 52.McGahon AJ, Martin SJ, Bissonnette RP, Mahboubi A, Shi Y, Mogil RJ, Nishioka WK, Green DR: The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol 1995, 46:153-185 [DOI] [PubMed] [Google Scholar]
- 53.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR: The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res 1999, 424:37-49 [DOI] [PubMed] [Google Scholar]
- 54.Jacobs BL, Langland JO: When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 1996, 219:339-349 [DOI] [PubMed] [Google Scholar]
- 55.Ewel CH, Sobel DO, Zeligs BJ, Bellanti JA: Poly IC accelerates development of diabetes mellitus in diabetes-prone BB rat. Diabetes 1992, 41:1016-1021 [DOI] [PubMed] [Google Scholar]
- 56.Sobel DO, Ewel CH, Zeligs B, Abbassi V, Rossio J, Bellanti JA: Poly IC induction of alpha-interferon in the diabetes-prone BB and normal Wistar rats. Dose-response relationships. Diabetes 1994, 43:518-522 [DOI] [PubMed] [Google Scholar]
- 57.Ellerman KE, Like AA: Susceptibility to diabetes is widely distributed in normal class IIu haplotype rats. Diabetologia 2000, 43:890-898 [DOI] [PubMed] [Google Scholar]
- 58.Thomas HE, Parker JL, Schreiber RD, Kay TW: IFN-gamma action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J Clin Invest 1998, 102:1249-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Der SD, Yang YL, Weissmann C, Williams BRG: A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA 1997, 94:3279-3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gil J, Alcami J, Esteban M: Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the a subunit of eukaryotic translation initiation factor 2 and NF-kappa B. Mol Cell Biol 1999, 19:4653-4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blair LA, Heitmeier MR, Scarim AL, Maggi LB, Jr, Corbett JA: Double-stranded RNA-dependent protein kinase is not required for double-stranded RNA-induced nitric oxide synthase expression or nuclear factor-κB activation by islets. Diabetes 2001, 50:283-290 [DOI] [PubMed] [Google Scholar]