

Abstract
Alport syndrome is an inherited disorder of type IV collagen with progressive nephropathy, ocular abnormalities, and high-tone sensorineural deafness. In X-linked Alport syndrome, mutations in the COL4A5 gene encoding the α5 chain of type IV collagen lead to loss of the α3/α4/α5 network and increased susceptibility of the glomerular basement membrane to long-term damage. The molecular defects that underlie the otopathology in this disease remain poorly understood. We used a canine model of X-linked Alport syndrome to determine the expression of type IV collagen α-chains in the inner ear. By 1 month in normal adult dogs, the α3, α4, and α5 chains were co-expressed in a thin continuous line extending along the basilar membrane and the internal and external sulci, with the strongest expression along the lateral aspect of the spiral ligament in the basal turn of the cochlea. Affected dogs showed complete absence of the α3/α4/α5 network. The lateral aspect of the spiral ligament is populated by tension fibroblasts that express α-smooth muscle actin and nonmuscle myosin and are postulated to generate radial tension on the basilar membrane via the extracellular matrix for reception of high frequency sound. We propose that in Alport syndrome, the loss of the α3/α4/α5 network eventually weakens the interaction of these cells with their extracellular matrix, resulting in reduced tension on the basilar membrane and the inability to respond to high frequency sounds.
Alport syndrome is a hereditary disorder of type IV collagen characterized by progressive nephropathy, ocular abnormalities, and high-tone sensorineural deafness. 1-4 Approximately 80% of affected families show an X-linked inheritance pattern and the remainder are autosomal-dominant or -recessive. 5,6 Patients have near normal kidney function at birth, which deteriorates throughout time leading to end-stage renal disease by the end of the third decade, especially in male patients. 1-4 The pathogenesis of Alport syndrome has been linked to mutations in genes for type IV collagen. Type IV collagen is assembled from a family of distinct α-chains designated α1 to α6, which are encoded by six different genes, COL4A1 to COL4A6, respectively. 7 The glomerular basement membrane (GBM) is composed of the α1 to α5 chains, 8,9 which are assembled in triple helices that in turn self-assemble into supramolecular networks. Two distinct α1/α2-containing and α3/α4/α5-containing networks have been identified at the biochemical level. 10
Over 300 mutations have been found in the COL4A5 gene in the X-linked form of Alport syndrome 4,11 and in the autosomal recessive forms, mutations have been identified in the COL4A3 and COL4A4 genes. 12,13 These mutations lead to the assembly of a GBM that is abnormal with respect to morphology and composition of type IV collagen α-chains. In most patients with Alport syndrome, the GBM contains the α1 and α2 chains and lacks the α3, α4, and α5 chains. 8,9,14,15 The altered α-chain composition seems to reflect an arrest of an early developmental switch, wherein an immature α1/α2 network persists and is not replaced by the α3/α4/α5 network. 10,16 The existence of an α3/α4/α5 network provides a plausible explanation for the absence of these chains in Alport syndrome in the setting of COL4A3, COL4A4, or COL4A5 mutations, in that all three chains are required for the assembly of this network. 10,17
In contrast to the many advances made toward the understanding of the pathogenesis of glomerular disease in Alport syndrome throughout the past few years, relatively little is known about the basis for hearing loss in this disease. The hearing deficit in Alport syndrome consists of a bilateral sensorineural hearing loss, involving initially the high frequency range, but later spreading to other ranges. 1-4 This may progress to deafness after adulthood is reached. Hearing loss is the most common extra-renal manifestation of Alport syndrome, with a frequency as high as 79% in male patients. 4 Males are affected more frequently than females, with the onset of symptoms usually occurring before 10 years of age and following a more progressive course. In male Alport syndrome patients, the presence of hearing loss is associated with a poor renal prognosis, but patients with normal hearing may still progress to end-stage renal disease.
Pathological studies on the inner ears of Alport syndrome patients have yielded conflicting results, reporting changes in a variety of anatomical sites. These observations include no abnormality, degeneration of the stria vascularis, degeneration or loss of the organ of Corti, hyalinization or loss of the tectorial membrane, neuronal depletion in the spiral ganglion, and cystic changes of the spiral ligament. 18-22 The few studies in which the basilar membrane has been examined report no changes. 20,22 None of these studies used immunohistochemistry and only one used electron microscopy. 18
Because the pathogenesis of renal disease in Alport syndrome involves the loss of the α3/α4/α5 network from the GBM, the localization of type IV collagen α-chains in the inner ear is of particular relevance to the pathogenesis of the hearing loss in this disease. This has been investigated in normal mouse, guinea pig, and chinchilla using various antibodies. 23-26 The results obtained are not entirely consistent between studies and localize the α3, α4, and α5 chains to one or more of the organ of Corti, tectorial membrane, basilar membrane, spiral ligament, and stria vascularis. Although there are several animal models available for the renal disease of Alport syndrome, 27 none of these display a hearing impairment comparable to human Alport syndrome patients. The only animal models in which the inner ear has been examined are the COL4A3 knockout mouse 28 and the Samoyed dog model for X-linked Alport syndrome. 29 The latter model results from a nonsense mutation in the COL4A5 gene 30 and closely mimics human X-linked Alport syndrome. 31-33 Studies on the COL4A3 knockout mouse suggested changes in the stria vascularis as the possible cause of hearing loss in Alport syndrome, whereas previous studies in the dog model suggested the basilar membrane might be involved. The canine studies were performed before antibodies specific for all six α-chains were available. Therefore, in light of the present knowledge of the pathogenesis of renal disease in Alport syndrome, we decided to revisit the canine model to investigate the distribution of the α1/α2 and α3/α4/α5 networks in the inner ear.
Materials and Methods
Audiometry
Auditory thresholds were objectively assessed in dogs by measuring auditory brainstem-evoked responses (ABR). Dogs were anesthetized and maintained at a surgical plane of anesthesia using sodium pentobarbital intravenously. Acoustic stimuli were pure tone pips of 5 ms (1 ms rise/fall) at frequencies ranging from 0.5 to 32 kHz. Stimuli were delivered by an earphone held in place over the external auditory canal. Responses were recorded by means of scalp needle electrodes positioned at the mastoid process and vertex of the head. A ground electrode was inserted at the nontest side mastoid. Responses were recorded for 12 ms after the stimulus, amplified, filtered (150 to 3000 Hz bandpass), and averaged over 3 × 300 sweeps using a Tucker-Davis system (Tucker-Davis Technologies, Miami, FL). For each stimulus, condition responses were recorded for a range of stimulus intensities in 10 dB steps. Thresholds were determined as the lowest level for which there was a repeatable ABR. Single time point audiometric analysis was performed on dogs of the following ages: 11 days (two affected and two normal males), 31 days (two normal males), 56 days (two carrier and one normal females), 59 days (two affected males), 104 days (one normal and one affected male), 154 days (two affected and one normal males). Serial audiometric evaluations were performed on one affected male (31, 77, and 192 days).
Otoacoustic Emission Recording
Transient-evoked otoacoustic emission (OAE) and distortion product OAE measurements were made with the ILO 88/92 device (Otodynamics, London, UK). In each animal, OAEs were recorded from both ears in a sound-attenuating booth. The seal and position of the ear probe were checked by monitoring the amplitudes of calibration signals within the ear canal. For transient-evoked OAE recordings, the stimulus was an 80-μs unfiltered click at 80-dB sound pressure level, presented in the nonlinear mode (3 clicks of one polarity and amplitude x are followed by one click of opposite polarity and amplitude 3x) to reduce the linear stimulus artifact but not the nonlinear transient-evoked OAE signal. Analysis time was 20 ms, excluding the first 2.5 ms to further eliminate ringing components of the stimulus artifact. Data were collected by averaging responses from 260 stimulus presentations collected above a noise rejection level of 47-dB sound pressure level. Distortion product OAEs were generated by presentation of equilevel primary tones at 50-dB sound pressure level, with an f2/f1 ratio of 1.21. Distortion product OAEs were recorded with f2 set between 1 kHz and 6 kHz at 1/3 octave intervals. The amplitude of the 2f1–f2 component was measured (in dB sound pressure level) and the average of three such recordings was used for analysis.
Immunohistochemistry
Temporal bones were removed at the time of sacrifice and perfusion-fixed with 50% acetone in phosphate-buffered saline (PBS) for 15 minutes, then rinsed in PBS, and decalcified in 10% ethylenediaminetetraacetic acid. Cochleae were snap-frozen in OCT and 5-μm cryosections were stained using the following antibodies: monoclonal mouse anti-human α-smooth muscle actin (1:50 dilution; DAKO, Carpinteria, CA), polyclonal rabbit anti-human nonmuscle myosin (1:50 dilution; Biomedical Technologies, Stoughton, MA), and monoclonal rat antibodies to each of the six α-chains of human type IV collagen (1:100 dilution). 9,34 For collagen immunostaining only, sections were pretreated for 10 minutes with a 100 mmol/L acid-KCL solution (pH 1.5) to expose epitopes and then blocked with 1.5% rabbit serum (all reagents from Vector Laboratories, Burlingame, CA). Primary antibodies were applied for 90 minutes followed by a biotinylated rabbit anti-rat antibody (1:200 dilution) for 60 minutes, then a peroxidase-conjugated avidin-biotin complex for 30 minutes. Diaminobenzidine was used as a chromagen and sections were counterstained with hematoxylin. The above protocol was modified for mouse and rabbit primary antibodies in which sections were blocked with 1.5% horse or goat serum, respectively, followed by primary antibodies for 60 minutes, then a biotinylated horse anti-mouse or goat anti-rabbit antibody, respectively (both at 1:200 dilution) for 30 minutes.
Results
Audiometry
No ABR or OAE responses were detected in 11-day-old dogs. This was because of the impaired conductive properties of the poorly developed middle ear at this age, a finding consistent with those of previously published studies on the postnatal development of the ABR response in dogs. 35,36 At age 31 days, ABR and OAE responses were indicative of mature auditory function in that the thresholds and latencies of ABR responses and the amplitude of OAE were within a normal range for the adult dog. No significant differences in ABR and OAE responses were detected between age-matched normal and affected dogs up to 5 months of age. One affected dog was serially tested up to 192 days of age. At the last time point, there were no OAE responses bilaterally and ABR thresholds were elevated 25 to 40 dB at all test frequencies bilaterally. At the time of testing this animal was markedly hypocalcemic and uremic; either condition could account for the measured hearing loss.
Immunohistochemistry
The anatomy of the canine inner ear is illustrated in Figure 1 ▶ . In the inner ears of normal dogs at all ages examined, the α1 and α2 chains of type IV collagen were co-localized to all vascular and perineural basement membranes (Figures 2A and 3A) ▶ ▶ . The α1 and α2 chains were also expressed in a linear manner from the internal sulcus, along the edge of basilar membrane to the external sulcus where the staining diffused into a fine network pattern among the root cells of the spiral ligament in the zone of attachment of the basilar membrane. These chains were notably absent from the lateral aspect of the spiral ligament adjacent to the temporal bone. This pattern of staining was consistent from the apex to the base of the cochlea. The expression of the α1 and α2 chains in affected dog ears was identical to that in the normal dog and no increased staining for these chains was noted in any structure (Figures 2B and 3B) ▶ ▶ .
Figure 1.
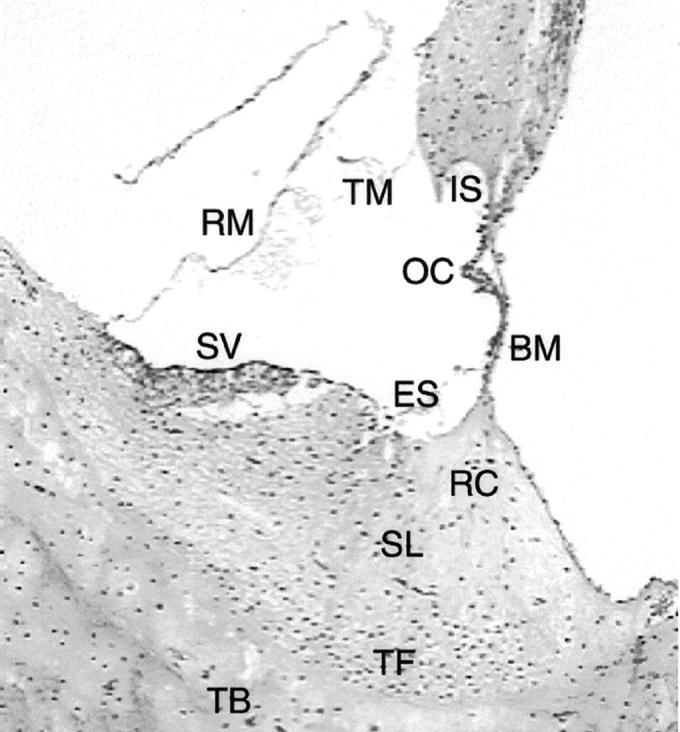
Normal anatomy of the canine inner ear. The organ of Corti (OC) sits on the basilar membrane (BM). A continuous basement membrane runs from the internal sulcus (IS) medial to the organ of Corti, along the basilar membrane and over to the external sulcus (ES) on the medial side of the spiral ligament (SL). The tectorial membrane (TM) and Reissner’s membrane (RM) are labeled as well. The stria vascularis (SV) is attached to the medial side of the spiral ligament (artifactually detached in this image). The root cells (RC) are located in the medial aspect of the spiral ligament in the region of attachment of the basilar membrane. The tension fibroblasts (TF) are situated in the lateral aspect of the spiral ligament, identifiable by increased nuclear density. The inner ear is contained with the temporal bone (TB). H&E; original magnification, ×135.
Figure 2.

Distribution of the α1 to α5 chains, respectively, in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for the α3, α4, and α5 chains only. In normal dogs, the lateral aspect of the spiral ligament is the predominant location of the α3, α4, and α5 chains, whereas this region is negative for the α1 and α2 chains. This region is negative for the α3, α4, and α5 chains in normal dogs before 1 month of age. The stria vascularis contains the α1 and α2 chains only. The basilar membrane is positive for the α1 to α5 chains. The tectorial membrane is negative for the α1 to α5 chains. In affected dogs, the α1 and α2 chains have the same distribution, and the α3, α4, and α5 chains are absent. Original magnifications, ×175.
Figure 3.

Distribution of the α1 to α5 chains, respectively, in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for the α3, α4, and α5 chains only. In normal dogs, the lateral aspect of the spiral ligament is the predominant location of the α3, α4, and α5 chains, whereas this region is negative for the α1 and α2 chains. This region is negative for the α3, α4, and α5 chains in normal dogs before 1 month of age. The stria vascularis contains the α1 and α2 chains only. The basilar membrane is positive for the α1 to α5 chains. The tectorial membrane is negative for the α1 to α5 chains. In affected dogs, the α1 and α2 chains have the same distribution, and the α3, α4, and α5 chains are absent. Original magnifications, ×175.
In contrast, the expression of the α3, α4, and α5 chains of type IV collagen varied with the age of the dog, the disease status (normal versus affected), and the level (basal versus apical) of the cochlea. In normal dogs by 1 month of age, the α3, α4, and α5 chains co-localized with the α1 and α2 chains along the basilar membrane and the internal and external sulci. The stria vascularis was negative for the α3, α4, and α5 chains, as were perineural basement membranes. In the spiral ligament an almost reverse pattern of staining was noted compared to the α1 and α2 chains. In the zone of attachment of the basilar membrane that is occupied by the root cells of the spiral ligament, there was sparse staining for the α3, α4, and α5 chains, whereas the region adjacent to the temporal bone was intensely stained in a fine network pattern that appeared to envelop individual cells in this region (Figures 4A, 5A, 6A) ▶ ▶ ▶ . Of note, this area showed the strongest expression of the α3, α4, and α5 chains of any structure in the inner ear. The intensity of staining in this region varied from apex to base of the cochlea, with the most intense staining in the basal turn and the weakest in the apical turn. The few capillaries present in the spiral ligament showed positive staining for the α1 to α5 chains; capillaries elsewhere were positive for only the α1 and α2 chains. In affected adult dogs, no staining for the α3/α4/α5 chains was detected (Figures 4B, 5B, 6B) ▶ ▶ ▶ . Staining for the α6 chain was confined to the basement membranes ensheathing smooth muscle cells of arterioles in the temporal bone where it co-localized with the α5 chain (results not shown). The tectorial membrane did not stain positively for any of the type IV collagen α-chains. In 11-day-old normal dogs, the α3, α4, and α5 chains of type IV collagen were weakly expressed only along the basilar membrane and the external sulcus and there was no staining in the lateral aspect of the spiral ligament (Figures 4C, 5C, 6C) ▶ ▶ ▶ . Inner ears of affected dogs of the same age lacked any staining for the α3/α4/α5 network (Figures 4D, 5D, 6D) ▶ ▶ ▶ .
Figure 4.
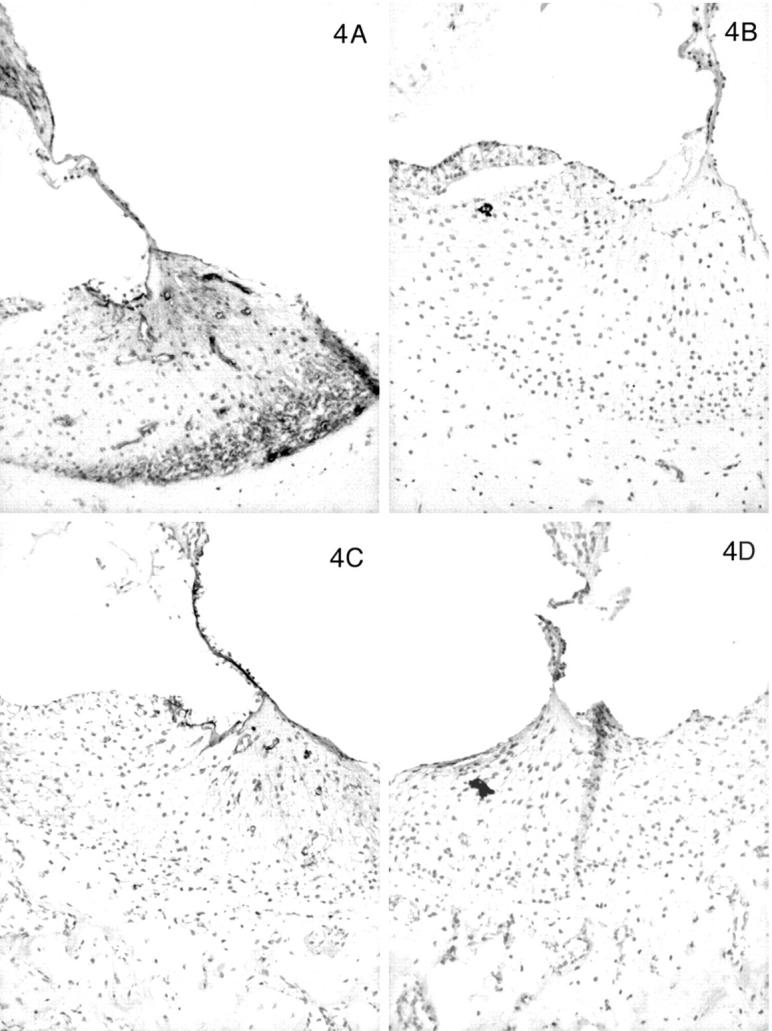
Distribution of the α1 to α5 chains, respectively, in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for the α3, α4, and α5 chains only. In normal dogs, the lateral aspect of the spiral ligament is the predominant location of the α3, α4, and α5 chains, whereas this region is negative for the α1 and α2 chains. This region is negative for the α3, α4, and α5 chains in normal dogs before 1 month of age. The stria vascularis contains the α1 and α2 chains only. The basilar membrane is positive for the α1 to α5 chains. The tectorial membrane is negative for the α1 to α5 chains. In affected dogs, the α1 and α2 chains have the same distribution, and the α3, α4, and α5 chains are absent. Original magnifications, ×175.
Figure 5.
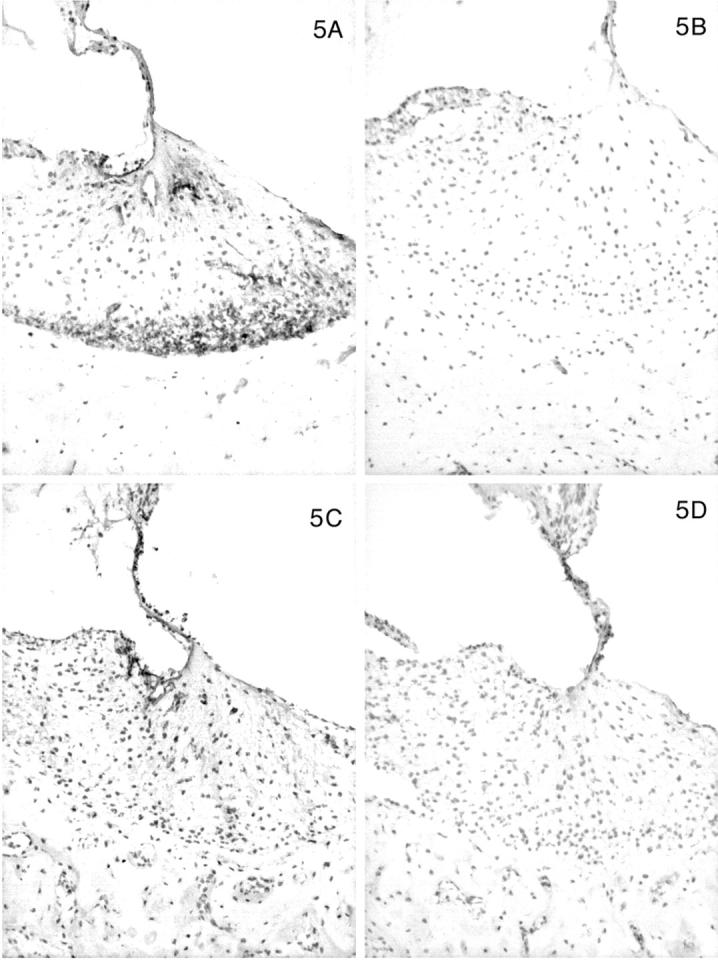
Distribution of the α1 to α5 chains, respectively, in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for the α3, α4, and α5 chains only. In normal dogs, the lateral aspect of the spiral ligament is the predominant location of the α3, α4, and α5 chains, whereas this region is negative for the α1 and α2 chains. This region is negative for the α3, α4, and α5 chains in normal dogs before 1 month of age. The stria vascularis contains the α1 and α2 chains only. The basilar membrane is positive for the α1 to α5 chains. The tectorial membrane is negative for the α1 to α5 chains. In affected dogs, the α1 and α2 chains have the same distribution, and the α3, α4, and α5 chains are absent. Original magnifications, ×175.
Figure 6.
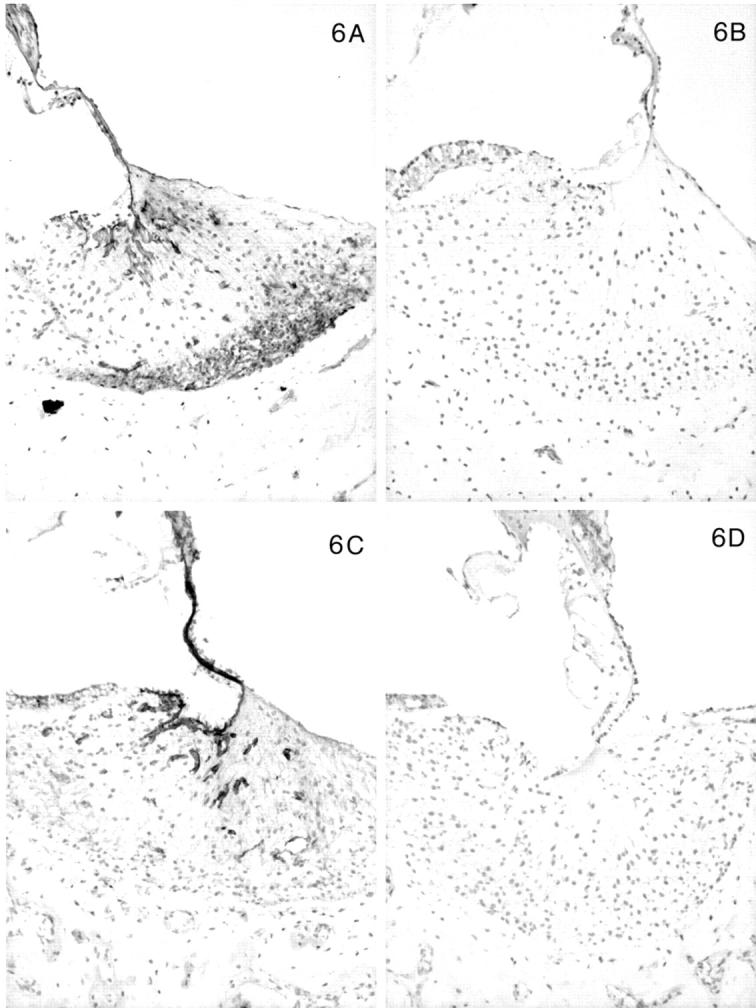
Distribution of the α1 to α5 chains, respectively, in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for the α3, α4, and α5 chains only. In normal dogs, the lateral aspect of the spiral ligament is the predominant location of the α3, α4, and α5 chains, whereas this region is negative for the α1 and α2 chains. This region is negative for the α3, α4, and α5 chains in normal dogs before 1 month of age. The stria vascularis contains the α1 and α2 chains only. The basilar membrane is positive for the α1 to α5 chains. The tectorial membrane is negative for the α1 to α5 chains. In affected dogs, the α1 and α2 chains have the same distribution, and the α3, α4, and α5 chains are absent. Original magnifications, ×175.
In normal dogs, α-smooth muscle actin was expressed by smooth muscle cells lining arteriolar vessels in the temporal bone. Strong expression of α-smooth muscle actin was also detected among the cells that populated the lateral aspect of the spiral ligament (Figure 7A) ▶ . The distribution of α-smooth muscle actin-positive cells in this site closely reflected the region of the spiral ligament that stained positively for the α3, α4, and α5 chains, except the expression of actin was limited to the most basal aspect of the cochlea near the round window. The same region in affected adult dogs was equally positive for α-smooth muscle actin (Figure 7B) ▶ . No staining for actin was observed in normal or affected dogs at 11 days of age (Figure 7, C and D) ▶ ▶ . Nonmuscle myosin was also expressed in the lateral aspect of the spiral ligament in a pattern that co-localized with that of α-smooth muscle actin and the α3/α4/α5 network. No difference in staining was detected between adult normal and affected dogs (Figure 8, A and B) ▶ ▶ , but the expression of this protein was markedly diminished at age 11 days in both normal and affected dogs (results not shown).
Figure 7.
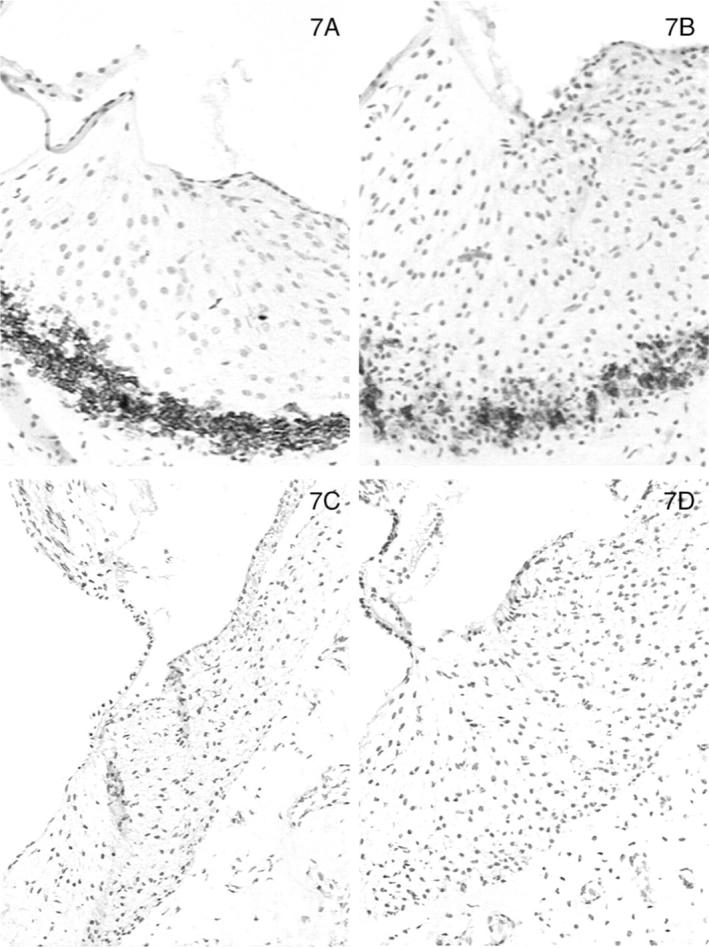
Distribution of α-smooth muscle actin (Figure 7) and nonmuscle myosin (Figure 8 ▶ ) in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for α-smooth muscle actin only. Original magnifications, ×175.
Figure 8.

Distribution of α-smooth muscle actin (Figure 7 ▶ ) and nonmuscle myosin (Figure 8) in the basal turn of the cochlea for normal (A) and affected (B) dogs at age 5 months. Normal (C) and affected (D) dogs at 1.5 weeks of age are shown for α-smooth muscle actin only. Original magnifications, ×175.
Discussion
The pathogenesis of high-tone sensorineural deafness in Alport syndrome has remained poorly understood, at least in part because of the general unavailability of lesional tissue to study. Almost all of the histological studies of the inner ear of patients with Alport syndrome have been conducted at the light microscopic level, and a variety of structures have been implicated as the basis of the hearing loss. 18-22 To circumvent this problem, two approaches have been taken. The first approach has been to study the localization of the α3, α4, and α5 chains in the normal inner ear. The hypothesis is that the pathogenesis of the hearing loss in Alport syndrome might be related to the loss of the α3/α4/α5 network, as is the case with the renal disease in this condition. Structures of the inner ear that contain this network would then be candidates for the source of the hearing loss. The second approach has been to examine the inner ears of animal models of Alport syndrome.
Most studies on the localization of the various chains of type IV collagen have been performed in mammals other than man, and include mouse, guinea pig, and chinchilla. 23-26,37 Our studies in the canine ear show similar results to those reported for other species, although there is some inconsistency for specific structures. The vascular and perineural basement membranes are consistently positive for the α1 and α2 chains only and the basilar membrane is positive for the α1 to α5 chains. Canine stria vascularis was positive for the α1 and α2 chains only, whereas a study of murine ear found positive staining for the α5 chain and weak staining for the α3 and α4 chains. 26 The tectorial membrane was negative for the α1 to α5 chains in our study as has been noted by some 23,26 but not by others. 25 Studies have shown that the tectorial membrane is mainly composed of type II collagen with some type V and IX collagen as well, rather than type IV collagen. 38,39
The spiral ligament in the normal canine ear showed a compartmentalization of type IV collagen expression with the α1 and α2 chains in the region of attachment of the basilar membrane (among root cells of the spiral ligament) and strong expression of the α3, α4, and α5 chains in a separate region along the lateral edge of the spiral ligament, which is populated by cells known as tension fibroblasts. 40,41 This region was notably devoid of the α1 and α2 chains. Results of other studies have varied for this region of the inner ear, with a more diffuse staining noted for the α1 chain 24 and no staining for the α3, α4, and α5 chains. 24,25 Somewhat similar results have been obtained for mouse inner ear 26 except the strongest expression for the α3, α4, and α5 chains was in the medial aspect of the spiral ligament, corresponding to the region positive for the α1 and α2 chains in the canine inner ear. In addition, the staining for the α3/α4/α5 network was strong in the apical turn of the mouse cochlea, whereas in the canine inner ear, the strongest staining was in the basal turn and the apical turn was poorly stained.
The reasons for these differences may be related to methods of tissue preparation, the particular antibodies used and the different species being examined. The issue becomes important when the localization of the α3/α4/α5 chains in the normal inner ear is used to explain what structure underlies the hearing loss in Alport syndrome, given that these chains are expected to be absent in this disease. Animal models of Alport syndrome permit a study of the inner ears in this condition, to circumvent the lack of available tissue from human patients. The present study advances our earlier work in the Samoyed model of X-linked Alport syndrome. 29 The inner ear has also been studied in the mouse model of autosomal recessive Alport syndrome. 28 In neither of these models is there a hearing deficit comparable to human Alport patients, although there was a slight increase in auditory thresholds throughout time for individual mice. 28 No hearing loss in the canine model was detectable by audiometry; one dog by age 7 months showed a mild decrease in hearing over all frequencies, a finding in keeping with the uremic state present at this age. Perhaps dog and mouse models for Alport syndrome have such rapidly advancing renal disease (both are essentially knockout models), that there is insufficient time for hearing loss to develop.
In both animal models, there is no change in the distribution of the α1 and α2 chains, and there is a generalized loss of the α3, α4, and α5 chains, except in the mouse model that retained some expression for the α5 chain in the vessels of the stria vascularis (a region negative for this chain in the normal canine ear). In the mouse model, the strial vessels showed thickened basement membranes and swollen endothelial cells, with eventual damage to the stria vascularis. 28 As the basement membrane changes bore a resemblance to those seen in the GBM of Alport syndrome, the stria vascularis was proposed to be a candidate site for the hearing loss in Alport syndrome. However, similar changes have been described in these vessels as an age-related change in gerbils. 42 Moreover, the loss of the α3/α4/α5 network is believed to be pathogenic in the glomerular disease of Alport syndrome and loss of the α3 chain usually results in loss of the α4 and α5 chains as well in Alport syndrome. 8,9,14,15 This was not the case in the Alport mouse inner ear for which selective loss of the α3 chain was implicated in the hearing loss of this disease.
In the normal canine ear, the stria vascularis lacked the α3, α4, and α5 chains. Instead, the strongest expression of the α3, α4, and α5 chains was seen in the spiral ligament, in particular the lateral aspect of this structure. This region is populated by myofibroblastic cells termed “tension fibroblasts.” 40,41 These cells connect the bony wall of the inner ear to the basilar membrane through a network of extracellular fibers and are believed to help maintain tension on the basilar membrane. Such cells contain various contractile proteins including actin, α-actinin, myosin, and tropomyosin. 43 The highest actin content is found in the basal turn of the cochlea, 40,41,44,45 which is the location of high-tone hearing, suggesting these cells may be needed to exert extra tension on the basilar membrane for reception of high frequency sounds. Our study shows that these actin-positive tension fibroblasts in the canine ear are surrounded by a distinctive extracellular matrix containing the α3, α4, and α5 chains of type IV collagen.
Developing glomeruli undergo changes in the composition of the GBM. As glomeruli mature, they undergo a developmental switch in which the α1 and α2 chains are replaced by the α3, α4, and α5 chains. 46 The renal disease in Alport syndrome can be viewed as a failure of this developmental switch, 16 an event that has been demonstrated in the Samoyed model. 33 It was also established through this model that only the α1/α2 network was essential for normal glomerular development, whereas the α3/α4/α5 network was essential for long-term stability of the GBM and maintenance of glomerular function. In other words, GBM deterioration in Alport syndrome was a postnatal process.
Parallels may be drawn with the inner ear. In many animals, there is a developmental shift in hearing such that high-tone hearing is acquired postnatally (altricious), whereas other animals are born with full hearing capabilities (precocious). Dogs, humans, and mice are altricious, with high-tone hearing acquired at around 3 weeks of age in dogs, 35,36 around 2 months of age in humans, 47 and around 8 to 12 days of age in mice. 48 No histological correlate exists in dogs and humans for this physiological shift. In the mouse ear, only the α1 and α2 chains are present at birth, with the α3, α4, and α5 chains not appearing until day 4, and not taking up their adult distribution until day 14. 49 A similar phenomenon seems to occur in the developing canine inner ear. In 11-day-old dogs, there is weak staining for the α3, α4, and α5 chains confined to the medial aspect of the spiral ligament and the lateral aspect is negative. By 1 month of age, the lateral aspect becomes the predominant location for the α3, α4, and α5 chains in the inner ear. One difference between the inner ear and the GBM is that the α1 and α2 chains do not replace the missing α3, α4, and α5 chains in affected dog inner ear, in contrast to the GBM. All other structures that contain these three chains are positive as early as 11 days, implying that either these basement membranes do not undergo a shift, or that it occurs earlier than this time point. A developmental shift in the tension fibroblasts has been documented for the gerbil inner ear. 45 Actin is first detectable after day 9 in the basal turn of the cochlea and assumes the adult distribution after day 17, a time that corresponds to the acquisition of mature auditory function. Similarly, actin was not detectable in normal or affected dog ears at 11 days of age, but was present by the time dogs had acquired high-tone hearing. The situation for the affected dog ear is then comparable to the GBM in these animals, 33 namely α3/α4/α5 network is not necessary for the development of normal structure and function, but may play a critical role instead in the long-term maintenance of function.
Based on our studies, we propose that a candidate site for the high-tone sensorineural deafness in human Alport syndrome is the spiral ligament of the inner ear, specifically in the region of the tension fibroblasts. The loss of the α3, α4, and α5 chains from this region results in a structurally abnormal matrix that connects the tension fibroblasts to the basilar membrane. This matrix normally permits the contractile fibroblasts to increase the tension on the basilar membrane to the degree needed for high-frequency sound reception. Without the α3/α4/α5 network, this type IV collagen matrix is either structurally weak or perhaps prone to proteolysis, as has been demonstrated for the GBM in Alport syndrome. 16 This could result in a gradually reduced ability of the fibroblasts in the spiral ligament to maintain sufficient tension on the basilar membrane, with loss of perception of high frequencies of sound.
If there is merit to our hypothesis, there should be evidence of pathological changes in this region of the inner ear of human patients with Alport syndrome. Although the studies are imperfect in terms of current methodologies, one of the frequently documented lesions in the inner ear of such patients is cystic and degenerative changes in the spiral ligament affecting the region of the tension fibroblasts. 19,21,50 Typically, this has involved the basal turn preferentially, ie, the region of high-tone hearing. Degeneration of the spiral ligament could be the basis for the high-tone hearing loss in human Alport syndrome, regardless of the initiating lesion. We postulate the primary lesion is the loss of the α3/α4/α5 network in the spiral ligament. An alternative explanation could be on a vascular basis: changes were reported in vessels of the stria vascularis of the COL4A3 knockout mouse 26 and some human Alport syndrome inner ears show striatal atrophy. 18,19,21 We also noted that there are a small number of vessels in the spiral ligament and these contain the α1 to α5 chains in the normal dogs but only the α1 and α2 chains in affected dogs. Such vascular changes might lead to secondary degeneration of the spiral ligament, perhaps on the basis of localized anoxia or electrolyte disturbances.
Of interest, MYH9, the gene encoding the nonmuscle myosin heavy chain IIa polypeptide was recently identified as the gene mutated in Fechtner syndrome, a rare autosomal-dominant variant of Alport syndrome characterized by nephritis, hearing loss, macrothrombocytopenia, and neutrophil inclusions. 51 This chain is part of nonmuscle myosin A, which is the only nonmuscle myosin expressed in platelets. 52 We used an antibody raised against the platelet isoform in our study and found that this protein localized to the tension fibroblasts in the basal turn of the spiral ligament in normal and affected canine inner ear. Similar findings were reported by others who used the same antibody to localize this protein in the normal rat cochlea. 53 This protein was present in tension fibroblasts of the spiral ligament, but not the stria vascularis, prompting speculation that the spiral ligament was involved in the hearing loss of Fechtner syndrome. We propose that the tension fibroblasts and their specialized extracellular matrix may constitute a functional unit for high frequency hearing, and loss of either component can lead to hearing loss. We can then put forth a unifying hypothesis that the spiral ligament may be the structure involved in hearing loss in both autosomal-dominant and X-linked forms of Alport syndrome.
Footnotes
Address reprint requests to Paul Thorner, MD, Ph.D., Division of Pathology, The Hospital for Sick Children, 555 University Ave., Toronto, Ontario, Canada M5G 1X8. E-mail: thorner@sickkids.on.ca.
Supported by the Medical Research Council of Canada (grant MT-1325 to P. T. and R. J.), the National Institutes of Health (grant P01 DK 53763-01 to P. T), by the Grant-in-Aid for International Scientific Research of the Ministry of Education, Science, Sports, and Culture of Japan (Joint Research, 09044308 to Y. N.).
References
- 1.Grunfeld J-P: The clinical spectrum of hereditary nephritis. Kidney Int 1985, 27:83-92 [DOI] [PubMed] [Google Scholar]
- 2.Habib R, Gubler M-C, Hinglais N, Noël L-H, Droz D, Levy M, Mahieu P, Foidart J-M, Perrin D, Bois E, Grünfeld J-P: Alport’s syndrome: experience at Hôpital Necker. Kidney Int 1982, 21(suppl 11):S20-S28 [PubMed] [Google Scholar]
- 3.Kashtan C, Michael A: Alport syndrome. Kidney Int 1996, 50:1445-1463 [DOI] [PubMed] [Google Scholar]
- 4.Jais J, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer K-O, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz J, Schroder C, Sanak M, Krejcova S, Carvalho M, Saus J, Antignac C, Smeets H, Gubler M: X-linked Alport syndrome: natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol 2000, 11:649-657 [DOI] [PubMed] [Google Scholar]
- 5.Feingold J, Bois E, Chompret A, Broyer M, Gubler M-C, Grünfeld J-P: Genetic heterogeneity of Alport Syndrome. Kidney Int 1985, 27:672-677 [DOI] [PubMed] [Google Scholar]
- 6.Reeders S: Molecular genetics of hereditary nephritis. Kidney Int 1992, 42:783-792 [DOI] [PubMed] [Google Scholar]
- 7.Hudson B, Reeders S, Tryggvason K: Type IV collagen: structure, gene organization and role in human diseases: molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. Minireview. J Biol Chem 1993, 268:26033-26036 [PubMed] [Google Scholar]
- 8.Peissel B, Geng L, Kalluri R, Kashtan C, Rennke H, Gallo G, Yoshioka K, Sun M, Hudson B, Neilson E, Zhou J: Comparative distribution of the α1(IV), α5(IV), and α6(IV) collagen chains in normal human adult and fetal tissues and in kidneys from X-linked Alport syndrome patients. J Clin Invest 1995, 96:1948-1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ninomiya Y, Kagawa M, Iyama K, Naito I, Kishiro Y, Seyer J, Sugimoto M, Oohashi T, Sado Y: Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J Cell Biol 1995, 130:1219-1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunwar S, Ballester F, Noelken M, Sado Y, Ninomiya Y, Hudson B: Identification of a novel disulfide-cross-linked network of α3, α4 and α5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem 1998, 273:8767-8775 [DOI] [PubMed] [Google Scholar]
- 11.Knebelmann B, Breillat C, Forestier L, Arrondel C, Jacassier D, Giatras I, Drouot L, Deschênes G, Grünfeld J-P, Broyer M, Gubler M-C, Antignac C: Spectrum of mutations in the COL4A5 collagen gene in X-linked Alport syndrome. Am J Hum Genet 1996, 59:1221-1232 [PMC free article] [PubMed] [Google Scholar]
- 12.Mochizuki T, Lemmink H, Mariyama M, Antignac C, Gubler M-C, Pirson Y, Verellen-Dumoulin C, Chan B, Schröder C, Smeets H, Reeders S: Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 1994, 8:77-82 [DOI] [PubMed] [Google Scholar]
- 13.Heidet L, Arrondel C, Forestier L, Cohen-Solal L, Mollet G, Gutierrez B, Stavrou C, Gubler M, Antignac C: Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol 2001, 12:97-106 [DOI] [PubMed] [Google Scholar]
- 14.Yoshioka K, Hino S, Takemura T, Maki S, Wieslander J, Takekoshi Y, Makino H, Kagawa M, Sado Y, Kashtan C: Type IV collagen α5 chain: normal distribution and abnormalities in X-linked Alport syndrome revealed by monoclonal antibody. Am J Pathol 1994, 144:986-996 [PMC free article] [PubMed] [Google Scholar]
- 15.Kleppel M, Fan W, Cheong H, Michael A: Evidence for separate networks of classical and novel basement membrane collagen. Characterization of α3(IV)-Alport antigen heterodimer. J Biol Chem 1992, 267:4137-4142 [PubMed] [Google Scholar]
- 16.Kalluri R, Shield F, Todd P, Hudson B, Neilson E: Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest 1997, 99:2470-2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kahsai T, Enders G, Gunwar S, Brunmark C, Wieslander J, Kalluri R, Zhou J, Noelken M, Hudson B: Seminiferous tubule membrane. Composition and organization of type IV collagen chains, and the linkage of α3(IV) and α5(IV) chains. J Biol Chem 1997, 272:17023-17032 [DOI] [PubMed] [Google Scholar]
- 18.Arnold W: Uberlegungen zur pathogenese des cochleo-renalen syndroms. Acta Otolaryngol 1980, 89:330-341 [DOI] [PubMed] [Google Scholar]
- 19.Crawford MDA, Toghill P: Alport’s syndrome of hereditary nephritis and deafness. Q J Med 1968, 148:563-576 [PubMed] [Google Scholar]
- 20.Gregg J, Becker S: Concomitant progressive deafness, chronic nephritis, and ocular lens disease. Arch Ophthalmol 1963, 69:293-299 [DOI] [PubMed] [Google Scholar]
- 21.Johnsson L-G, Arenberg I: Cochlear abnormalities in Alport’s syndrome. Arch Otolaryngol 1981, 107:340-349 [DOI] [PubMed] [Google Scholar]
- 22.Myers G, Tyler H: The etiology of deafness in Alport’s syndrome. Arch Otolaryngol 1972, 96:333-340 [DOI] [PubMed] [Google Scholar]
- 23.Takahashi M, Hokunan K: Localization of type IV collagen and laminin in the guinea pig inner ear. Ann Otol Rhinol Laryngol 1992, 157:58-62 [DOI] [PubMed] [Google Scholar]
- 24.Kleppel M, Santi P, Cameron J, Wieslander J, Michael A: Human tissue distribution of novel basement membrane collagen. Am J Pathol 1989, 134:813-825 [PMC free article] [PubMed] [Google Scholar]
- 25.Kalluri R, Gattone V, Hudson B: Identification and localization of type IV collagen chains in the inner ear cochlea. Connect Tissue Res 1998, 37:143-150 [DOI] [PubMed] [Google Scholar]
- 26.Cosgrove D, Samuelson G, Pinnt J: Immunohistochemical localization of basement membrane collagens and associated proteins in the murine cochlea. Hearing Res 1996, 97:54-65 [PubMed] [Google Scholar]
- 27.Heikkila P, Tryggvason K, Thorner P: Animal models of Alport syndrome—advancing the prospects for effective human gene therapy. Exp Nephrol 2000, 8:1-7 [DOI] [PubMed] [Google Scholar]
- 28.Cosgrove D, Samuelson G, Meehan D, Miller C, McGee J, Walsh E, Siegel M: Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hearing Res 1998, 121:84-98 [DOI] [PubMed] [Google Scholar]
- 29.Thorner P, Jansen B, Baumal R, Harrison R, Mount R, Valli V, Spicer P, Marrano P: An immunohistochemical and electron microscopy study of extra-renal basement membranes in dogs with Samoyed hereditary glomerulopathy. Virchows Arch [A] 1988, 412:281-290 [DOI] [PubMed] [Google Scholar]
- 30.Zheng K, Thorner P, Marrano P, Baumal R, McInnes R: Canine X chromosome-linked hereditary nephritis: a genetic model for human X-linked hereditary nephritis resulting from a single base mutation in the gene encoding the α5 chain of collagen type IV. Proc Acad Natl Sci USA 1994, 91:3989-3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorner P, Zheng K, Kalluri R, Jacobs R, Hudson B: Coordinate gene expression of the α3, α4 and α5 chains of collagen type IV: evidence from a canine model of X-linked nephritis with a COL4A5 gene mutation. J Biol Chem 1996, 271:13821-13828 [DOI] [PubMed] [Google Scholar]
- 32.Jansen B, Thorner P, Baumal R, Valli V, Maxie M, Singh A: Samoyed hereditary glomerulopathy (SHG): evolution of splitting of glomerular capillary basement membranes. Am J Pathol 1986, 125:536-545 [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey S, Zheng K, Sado Y, Naito I, Ninomiya Y, Jacobs R, Hudson B, Thorner P: The role of distinct type IV collagen networks in glomerular development and function. Kidney Int 1998, 54:1857-1866 [DOI] [PubMed] [Google Scholar]
- 34.Sado Y, Kagawa M, Kishiro Y, Sugihara K, Naito I, Seyer J, Sugimoto M, Oohashi T, Ninomiya Y: Establishment by the rat lymph node method of epitope-defined monoclonal antibodies recognizing the six different α chains of human type IV collagen. Histochem Cell Biol 1995, 104:267-275 [DOI] [PubMed] [Google Scholar]
- 35.Kay R, Palmer A, Taylor P: Hearing in the dog as assessed by auditory brainstem evoked potentials. Vet Record 1984, 114:81-84 [DOI] [PubMed] [Google Scholar]
- 36.Strain G, Tedford B, Jackson R: Postnatal development of the brain stem auditory-evoked potential in dogs. Am J Vet Res 1991, 52:410-415 [PubMed] [Google Scholar]
- 37.Kleppel M, Kashtan C, Santi P, Wieslander J, Michael A: Distribution of familial nephritis antigen in normal tissue and renal basement membranes of patients with homozygous and heterozygous Alport familial nephritis. Relationship of familial nephritis and Goodpasture antigens to novel collagen chains and type IV collagen. Lab Invest 1989, 61:278-289 [PubMed] [Google Scholar]
- 38.Richardson G, Russell I, Duance V, Bailey A: Polypeptide composition of the mammalian tectorial membrane. Hearing Res 1987, 25:45-60 [DOI] [PubMed] [Google Scholar]
- 39.Slepecky N, Cefaratti L, Yoo T: Type II and type IX collagen form heterotopic fibers in the tectorial membrane of the inner ear. Matrix 1992, 12:80-86 [DOI] [PubMed] [Google Scholar]
- 40.Henson M, Henson OJ, Jenkins D: The attachment of the spiral ligament to the cochlear wall: anchoring cells and the creation of tension. Hearing Res 1984, 16:231-242 [DOI] [PubMed] [Google Scholar]
- 41.Henson M, Henson OJ: Tension fibroblasts and the connective tissue matrix of the spiral ligament. Hearing Res 1988, 35:237-258 [DOI] [PubMed] [Google Scholar]
- 42.Thomopoulos G, Spicer S, Gratton M, Schulte B: Age-related thickening of basement membrane in stria vascularis capillaries. Hearing Res 1997, 111:31-41 [DOI] [PubMed] [Google Scholar]
- 43.Henson M, Burridge K, Fitzpatrick D, Jenkins D, Pillsbury H, Henson OJ: Immunocytochemical localization of contractile and contraction associated proteins in the spiral ligament of the cochlea. Hearing Res 1985, 20:207-214 [DOI] [PubMed] [Google Scholar]
- 44.Sim D: Spiral ligament tension fibroblasts and extracellular fibres in the guinea pig. Acta Anat 1990, 138:195-199 [DOI] [PubMed] [Google Scholar]
- 45.Kuhn B, Vater M: The postnatal development of F-actin in tension fibroblasts of the spiral ligament of the gerbil cochlea. Hearing Res 1997, 108:180-190 [DOI] [PubMed] [Google Scholar]
- 46.Miner J, Sanes J: Collagen IV α3, α4, and α5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol 1994, 127:879-891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mochizuki Y, Go T, Ohkubo H, Motomura T: Development of human brainstem auditory evoked potentials and gender differences from infants to young adults. Prog Neurobiol 1983, 20:273-285 [DOI] [PubMed] [Google Scholar]
- 48.Ehret G: Development of hearing and response behavior to sound stimuli: behavioural studies. Ramond R eds. Development of Auditory and Vestibular Systems. 1983, :pp 211-237 Academic Press, New York [Google Scholar]
- 49.Cosgrove D, Kornak J, Samuelson G: Expression of basement membrane type IV collagen chains during postnatal development in the murine cochlea. Hearing Res 1996, 100:21-32 [DOI] [PubMed] [Google Scholar]
- 50.Gussen R: Scala vestibuli partition with deafness and renal disease. Ann Otol 1973, 82:871-875 [DOI] [PubMed] [Google Scholar]
- 51.Consortium M-HFS: Mutations in MYH9 result in May-Hegglin anomaly, and Fechtner and Sebastian syndromes. Nat Genet 2000, 26:103-105 [DOI] [PubMed] [Google Scholar]
- 52.Kelley M, Jawien W, Ortel T, Korczak J: Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet 2000, 26:106-108 [DOI] [PubMed] [Google Scholar]
- 53.Lalwani A, Goldstein J, Kelley M, Luxford W, Castelein C, Mhatre A: Human nonsyndromic deafness DFNA17 is due to a mutation in nonmuscle mysoin MYH9. Am J Hum Genet 2000, 67:1121-1128 [DOI] [PMC free article] [PubMed] [Google Scholar]