

Abstract
Oxidative stress increases endothelial mannose-binding lectin (MBL) binding and activates the lectin complement pathway (LCP). However, the molecular mechanism of MBL binding to the endothelium after oxidative stress is unknown. Intermediate filaments have been previously reported to activate the classical complement pathway in an antibody-independent manner. We investigated whether oxidative stress increases human umbilical vein endothelial cell (HUVEC) cytokeratin 1 (CK1) expression and activates the LCP via MBL binding to CK1. Reoxygenation (3 hours, 21% O2) of hypoxic HUVECs (24 hours, 1% O2) significantly increased CK1 mRNA (in situ hybridization) and membrane protein expression [enzyme-linked immunosorbent assay (ELISA)/confocal microscopy]. Incubating human serum (HS) with N-acetyl-d-glucosamine or anti-human MBL monoclonal antibody attenuated MBL and C3 deposition on purified CK1 (ELISA). CK1 and MBL were co-immunoprecipitated from hypoxic HUVECs reoxygenated in HS. Treatment with anti-human cytokeratin Fab fragments attenuated endothelial MBL and C3 deposition after oxidative stress (ELISA/confocal microscopy). We conclude that: 1) endothelial oxidative stress increases CK1 expression, MBL binding, and C3 deposition; 2) inhibition of MBL attenuates purified CK1-induced complement activation; and 3) anti-human cytokeratin Fab fragments attenuate endothelial MBL and C3 deposition after oxidative stress. These results suggest that MBL binding to endothelial cytokeratins may mediate LCP activation after oxidative stress.
The human complement system is comprised of three different pathways: the classical, alternative, and recently described lectin complement pathway (LCP). 1,2 The LCP is an antibody-independent cascade that is normally initiated by binding of mannose-binding lectin (MBL) to cell surface carbohydrates on foreign bacteria, protozoa, or parasites. 3 Associated with MBL (also known as mannose-binding protein) are two serine proteases, MBL-associated serine protease-1 and MBL-associated serine protease-2, which cleave C2 and C4 to form the classical pathway C3 convertase. 1 Although there is experimental evidence linking the classical and alternative complement pathways to human disease, the role of the LCP is just beginning to be evaluated. 4
We recently demonstrated that human endothelial oxidative stress activates complement via the antibody-independent LCP. 5 Further, treatment of human sera with novel, functionally inhibitory anti-human MBL monoclonal antibodies (mAbs) significantly attenuated MBL and C3 deposition after endothelial oxidative stress. 5 These findings were extended to a rat model of myocardial ischemia-reperfusion in which rat C3 and MBL immunostaining were observed to be significantly increased throughout the ischemic area at risk, further suggesting that the LCP may mediate tissue injury in vivo after oxidative stress. 5 However, the molecular mechanism of MBL binding to the vascular endothelium after oxidative stress is unknown.
Our understanding of the role of cytoskeletal filaments in cellular function has significantly advanced in recent years. In addition to providing structural support, it is now clear that intermediate filaments play a key role in a variety of cellular functions, including cell-cell and cell-extracellular matrix interactions, cell motility, receptor-ligand interactions, and receptor internalization. 6,7 Although various intermediate filaments exist in human endothelial cells, their nonstructural roles have not been fully elucidated. Recently, we demonstrated that the plant lectin Ulex europaeus agglutinin II, which has a similar binding profile as MBL, competitively inhibits MBL deposition and subsequent activation of the LCP after human endothelial cell oxidative stress. 8 Further, in preliminary experiments performed in our laboratory, immunoprecipitation and protein sequencing of oxidatively stressed human endothelial cells with U. europaeus agglutinin II revealed the intermediate filament, cytokeratin 1 (CK1). Interestingly, CK1 was recently cloned from a human endothelial cell library and identified as a kininogen-binding protein, 9-13 suggesting that endothelial cytokeratins may function as extracellular binding proteins. Additionally, exons 1 and 9 of CK1 contain sequences highly homologous to a peptide sequence (SFGSGFGGGY) known to mimic the MBL and U. europaeus agglutinin-II ligand, N-acetyl-d-glucosamine (GlcNAc). 14,15 Recently, MBL was shown to bind specifically to this keratin peptide sequence, and pretreatment of human sera with this peptide significantly attenuated MBL and complement deposition on oxidatively stressed human endothelial cells. 16 As intermediate filaments have been previously reported to activate the classical complement pathway in an antibody-independent manner, 17,18 we investigated whether oxidative stress increases endothelial CK1 expression and activates the LCP. Our data suggest that activation of the LCP after oxidative stress may be mediated in part by MBL binding to vascular endothelial cytokeratins.
Materials and Methods
Human Umbilical Vein Endothelial Cells (HUVECs)
HUVECs were isolated and cultured as previously described. 19 Cells were used during passage 1 through 3. Separate cell isolates were used for all repeat experiments.
Isolation and Purification of Human MBL
MBL and MBL-associated serine proteases were purified from fresh frozen human plasma as previously described. 5,20 Western blot analysis confirmed the absence of IgG and/or IgM.
Isolation and Purification of Human CK1
Human CK1 was purified from human dermal keratin (Sigma, St. Louis, MO) using a monospecific rabbit anti-human CK1 polyclonal antibody (pAb) (Convance/BAbCO, Richmond, CA) conjugated to protein G Sepharose (ImmunoPure Protein G IgG Plus orientation kit; Pierce, Rockford IL). After equilibrating the protein G column with binding buffer (10 mmol/L Tris, pH 7.5), human keratin was loaded onto the column. The column was then washed extensively with binding buffer and eluted with 0.1 mol/L glycine-HCl buffer, pH 2.8, with the eluent being collected in 0.5-ml fractions containing 1 mol/L Tris-HCl buffer (1:10, v:v), pH 9.5. The protein-containing fractions were pooled, dialyzed overnight in 10 mmol/L Tris buffer, pH 7.5, and the protein concentration determined. CK1 purification was confirmed by Western blot analysis.
Generation of Anti-Human Keratin Antibodies and Fab Fragments
Male New Zealand White rabbits (Harlan, Indianapolis, IN) were immunized initially with human keratin (100 μg, s.c.) in TiterMax (Sigma) and then with human keratin (50 μg, s.c.) in phosphate-buffered saline (PBS) on a biweekly basis for 6 weeks. Two weeks after the last immunization, the animals were bled and the resultant pAb purified by protein G affinity chromatography. All pAb were dialyzed against PBS, concentrated, and sterile filtered.
Polyclonal anti-human keratin Fab fragments were generated by digesting anti-human keratin pAb with papain (Sigma) for 16 hours at 37°C. The reaction was terminated with iodoacetamide (Sigma). The resulting mixture was then dialyzed in PBS, pH 8.0, overnight at 4°C. Any remaining whole IgG and the Fc portion of the anti-human keratin pAb were removed from the mixture by protein G affinity chromatography. Fab fragment generation was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
HUVEC CK1 Enzyme-Linked Immunosorbent Assay (ELISA)
Confluent HUVECs were subjected to 0 or 24 hours of hypoxia (1% O2). The cell media were aspirated and 100 μl of gelatin-veronal buffer (GVB) containing Ca2+/Mg2+ was added to each well. The cells were then reoxygenated for 3 hours at 37°C, washed, and fixed with 1% paraformaldehyde (Sigma) for 30 minutes. After washing, the cells were incubated with 50 μl of rabbit anti-human CK1 pAb (1:500 dilution, Convance/BAbCO) or anti-porcine C7 pAb (20 μg/ml, isotype control) for 1 hour at 4°C. After washing, 50 μl of peroxidase-conjugated goat anti-rabbit pAb was added to each well and incubated for 1 hour at 4°C. The plates were washed, developed with 50 μl of 2,2′-azino-bis(3-ethylbenzthiazoline)-6-sulfonic acid and read (Molecular Devices, Sunnyvale, CA) at 405 nm. Background optical density was determined from cells to which only the secondary antibody was added, and was subtracted from all groups. This experiment was performed three times using three wells per experimental group (n = 3).
In Situ Hybridization of HUVEC CK1 mRNA
The vector containing the rCK-131 cDNA (nucleotide 463 to 1434, accession NM 006121) was generously provided to us by Dr. Alvin Schmaier. 11 The 971-bp EcoRI/SalI digested fragment was gel purified using a Qiagen gel extraction kit according the manufacture’s instructions (Qiagen, Valencia, CA). The cDNA fragment was labeled with biotin-16-dUTP, and the probes separated with spin columns.
Confluent HUVECs grown on LabTech tissue culture microscope slides (Nalge Nunc International, Rochester, NY) were subjected to 0 or 24 hours of hypoxia. The cell media was aspirated, 1 ml of GVB added to each slide, and the cells reoxygenated for 3 hours at 37°C. The cells were then fixed immediately with 4% paraformaldehyde-PBS (10 minutes) and washed in PBS-MgCl2 (5 mmol/L). All materials were kept RNase-free throughout the procedure. Before hybridization, the cells were hydrated in 0.2 mol/L Tris-HCl, pH 7.4, and 0.1 mol/L glycine for 10 minutes, and then changed to 50% formamide-2× standard saline citrate (SSC) (SSC contains 0.15 mol/L NaCl, 0.015 mol/L sodium citrate, pH 7.0) at 65°C for 15 minutes. While the HUVECs were being hydrated, the probe (80- to 100-μl probe), 4 μl of Escherichia coli tRNA (Sigma), and 4 μl of salmon sperm DNA (Sigma) were melted in 10 to 30 μl of 100% formamide (Sigma) at 90°C for 10 minutes. An equal volume of hybridization mix was added for a final concentration of 50% formamide, 2× SSC, 0.2% bovine serum albumin, 10 mmol/L vanadyl sulfate-ribonucleoside complex (Bethesda Research Laboratories, Bethesda, MD), 10% dextran sulfate, and 1 μg/ml each of E. coli tRNA and salmon sperm DNA. The final concentration of the probe was 80 to 100 ng/30 μl hybridization. The probe and hybridization mix were added to the tissue culture slides, the covers replaced, and the mixture incubated at 37°C (4 to 16 hours) in a closed, 2× SSC-saturated chamber. After hybridization, the cells were washed with 2× SSC-50% formamide for 30 minutes at 37°C, then in 1× SSC-50% formamide for 30 minutes at 37°C, and twice in 1× SSC at room temperature for 30 minutes.
The cells were incubated in 4× SSC-1% bovine serum albumin with avidin-fluorescein isothiocyanate (FITC) (2 μg/ml) for 30 minutes, then washed three times in 2× SSC at room temperature on a rotating shaker. The cells were then mounted in antifade mounting medium, covered, and viewed on a Leica confocal scanning microscope (Leica Exton, PA). Control hypoxic HUVECs were incubated in RNase A (100 μg/ml in 2× SSC for 1 hour at 37°C) to determine specificity of the probe for RNA. After incubation in RNase A, the cells were hybridized as described above and incubated with avidin-FITC, washed, and viewed by confocal microscopy. A second negative control preparation consisted of hypoxic HUVECs hybridized with a porcine MBL cDNA probe, washed, then reacted with FITC-avidin and viewed on a confocal microscope. All in situ hybridization studies were done in triplicate.
Immunoprecipitation and Sequencing of HUVEC CK1
To confirm the specificity of the anti-human CK1 pAb used in these experiments, HUVEC CK1 was immunoprecipitated and sequenced. Confluent HUVEC cultures grown in 100-mm Petri dishes were subjected to 24 hours of hypoxia followed by 3 hours of reoxygenation in the presence of GVB. The cells were then washed with ice cold GVB and incubated with lysing buffer (150 mmol/L NaCl, 25 mmol/L Tris, 1 mmol/L MgCL2, 1% Triton X-100, 1% Nonidet P-40, 5 mmol/L ethylenediaminetetraacetic acid, 5 μg/ml chymostatin, 2 μg/ml aprotinin, and 1.25 mmol/L phenylmethyl sulfonyl fluoride, pH 7.4, all from Sigma). Cell debris was removed by centrifugation (10,000 × g, 5 minutes). Cell lysates were precleared with 50 μl of pre-equilibrated protein-G Sepharose (Pharmacia, Uppsala, Sweden) overnight at 4°C. CK1 immunoprecipitation was performed by addition of rabbit anti-human CK1 pAb (4 μg/ml, Convance/BAbCO). After centrifugation (10,000 × g, 5 minutes) and washing, the immunoprecipitates were boiled in reducing sample buffer and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After staining with Coomassie blue, the resultant protein band was cut from the gel and sent to the Harvard University Core Microchemistry Facility for microsequencing.
C3 and MBL Deposition (ELISA) on Purified CK1
Purified human CK1 (50 μl; 2 μg/ml in 15 mmol/L sodium carbonate, pH 9.6) was added to 96-well microtiter plates for 12 to 16 hours at 4°C. After washing, the plates were blocked for 2 hours at room temperature with 3% bovine serum albumin and washed again. HS (2% final concentration) was incubated with 1) 100 mmol/L of GlcNAc; 2) 20 μg/ml of anti-human MBL mAb, 3F8; 5 or 3) vehicle (veronal-buffered saline containing Ca2+/Mg2+) for 30 minutes at room temperature. The plates were then inoculated with 100 μl of treated or untreated HS, and incubated for 30 minutes at 37°C. The plates were washed and 50 μl of horseradish peroxidase-conjugated goat anti-human C3 pAb (1:2000 dilution; ICN, Aurora, OH) or rabbit anti-human MBL pAb (R2.2, 1:500 dilution) added for 1 hour at room temperature. The plates were then washed and developed as described above. Background optical density was determined from wells coated with bovine serum albumin only and was subtracted from all groups. This experiment was performed three to four times using three wells per experimental group (n = 3–4).
Immunoprecipitation and Western Blot of Human CK1 and MBL
Confluent HUVEC cultures grown on 100-mm Petri dishes were subjected to 0 or 24 hours of hypoxia followed by 3 hours of reoxygenation in the presence of GVB (for CK1 analysis) or 30% HS (for MBL analysis). The cells were then washed with ice-cold GVB and incubated with lysing buffer. Cell debris was removed by centrifugation (10,000 × g, 5 minutes). Cell lysates were precleared with 50 μl of pre-equilibrated protein-G Sepharose. The lysates were then immunoprecipitated by addition of human MBL (90 μg) and 50 μl of anti-human MBL mAb (1C10) 5 or anti-human CK1 pAb (Convance/BAbCO) conjugated to protein-G Sepharose (ImmunoPure Protein G IgG Plus orientation kit, Pierce). After centrifugation (10,000 × g, 5 minutes) and washing, the immunoprecipitates were boiled in reducing sample buffer and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The gel was electroblotted to nitrocellulose and blocked with 10% nonfat dry milk overnight at 4°C.
For CK1 analysis, anti-human CK1 pAb (1:500 dilution) was incubated with the nitrocellulose in 3% nonfat dry milk for 1 hour at 4°C. The nitrocellulose was then washed and incubated with horseradish peroxidase-conjugated goat anti-rabbit pAb (1:1000 dilution, ICN) for 1 hour at 4°C. For MBL analysis, horseradish peroxidase-conjugated anti-human MBL mAb (2A9, 1:2000 dilution) was incubated with the nitrocellulose in 3% nonfat dry milk for 1 hour at 4°C. The membranes were then washed and developed with the enhanced chemiluminescence system (Amersham) and X-ray films (Eastman-Kodak, Rochester, NY). In a separate experiment, Western blotting of normoxic and hypoxic HUVEC lysates were performed under reducing conditions as described above.
HUVEC C3 and MBL ELISA
HUVEC C3 and MBL deposition after oxidative stress was measured by ELISA as previously described. 5 HUVECs were grown to confluence and then subjected to 0 (normoxia) or 24 hours of hypoxia (1% O2). The cell media were aspirated and 100 μl of one of the following was added to each well: 1) 30% HS, 2) GVB, 3) 30% HS plus 100 mmol/L of GlcNAc, 4) 30% HS plus 50 μg/ml of anti-human keratin pAb, or 5) 30% HS plus 20 μg/ml of anti-human keratin Fab fragments. The cells were then reoxygenated for 3 hours at 37°C, washed, and fixed with 1% paraformaldehyde for 30 minutes. After washing, the cells were incubated with horseradish peroxidase-conjugated goat anti-human C3 pAb (1:1000 dilution; Cappel, West Chester, PA) or the anti-human MBL mAb, 1C10 (1:1000 dilution) for 1 hour at 4°C. The plates were then washed and developed as described above. Background optical density was determined from cells to which only the anti-human C3 or MBL antibody was added, and was subtracted from all groups. As a separate control, cells were also incubated with HS treated with species-matched, isotype irrelevant control pAb (50 μg/ml) or Fab fragments (20 μg/ml) (Jackson Immunoresearch, West Grove, PA). These experiments were performed three times using four to six wells per experimental group (n = 3).
Immunofluorescent Confocal Microscopy
HUVECs grown on LabTech tissue culture microscope slides were subjected to 0 or 24 hours of hypoxia and then reoxygenated for 3 hours in GVB or 30% HS treated with GVB (vehicle), anti-human keratin Fab fragments (20 μg/ml), or GlcNAc (100 mmol/L). The slides were then washed in PBS containing calcium and magnesium and fixed in 4% paraformaldehyde for 15 minutes, washed again, and blocked with 10% goat serum. Human MBL deposition (green) was identified using biotinylated 1C10 and streptavidin-conjugated FITC (Jackson Immunoresearch). Human C3 deposition (green) was evaluated with a FITC-conjugated goat anti-human C3 F(ab′)2 antibody (ICN). HUVEC CK1 expression was evaluated with anti-human CK1 pAb (Convance/BABCO) and FITC-conjugated goat anti-rabbit IgG (Jackson Immunoresearch). After incubation with the appropriate antibodies, the slides were washed (3 times, 10 minutes each) and incubated with propidium iodide (10 μg/ml, Sigma). The slides were, then coated with anti-fade mounting media (Molecular Probes, Eugene, OR), covered, and analyzed with a Zeiss confocal microscope as previously described. 21 Controls with streptavidin-conjugated FITC only were processed as above, omitting the primary antibody to determine nonspecific binding. All analyses were conducted at the same pinhole, voltage, and laser settings. This experiment was performed three times (n = 3).
Statistical Analysis
All data presented represent the mean and SEM for number of determinations. Data analyses were performed using Sigma Stat (Jandel Scientific, San Rafael, CA). A P value of <0.05 was considered significant. Endothelial CK1 expression and MBL/C3 deposition on purified CK1-coated plates were analyzed by one-way analysis of variance. Endothelial C3 and MBL deposition on normoxic versus hypoxic HUVECs were analyzed by two-way analysis of variance. All pairwise multiple comparisons were made using the Student-Newman-Keuls test. MBL and C3 deposition on purified CK1-coated plates (ELISA; see Figure 4 ▶ ) were normalized to untreated 2% HS. Endothelial MBL and C3 deposition (ELISA; see Figure 6 ▶ ) after oxidative stress were normalized to hypoxic HUVECs reoxygenated in untreated 30% HS. Means ± SEM of the raw data used for normalization are presented in the Results and/or figure legends.
Figure 4.
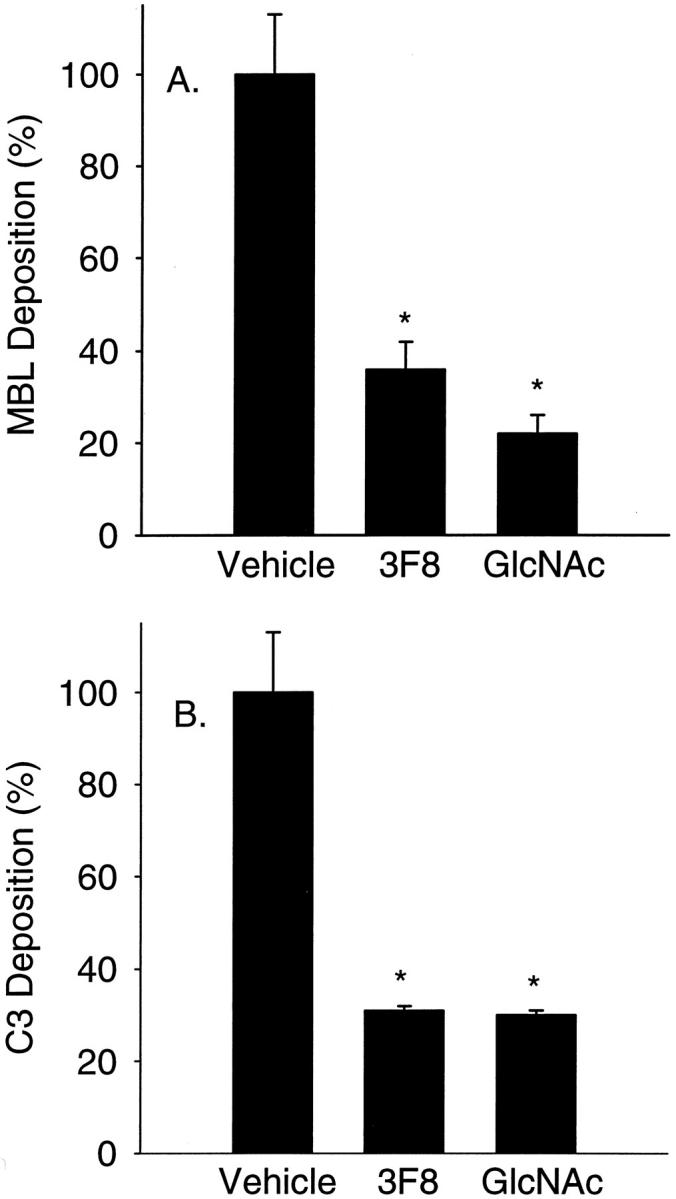
MBL and C3 deposition on purified human CK1. Human MBL (A) and C3 (B) deposition on purified human dermal CK1 was determined by ELISA. Treatment of 2% HS with GlcNAc (100 mmol/L) or the functionally inhibitory anti-human MBL mAb, 3F8 (10 μg/ml), significantly attenuated C3 and MBL deposition compared to untreated HS (vehicle) (n = 3; data normalized to vehicle; error bars = SEM; *, P < 0.05 compared to vehicle).
Figure 6.
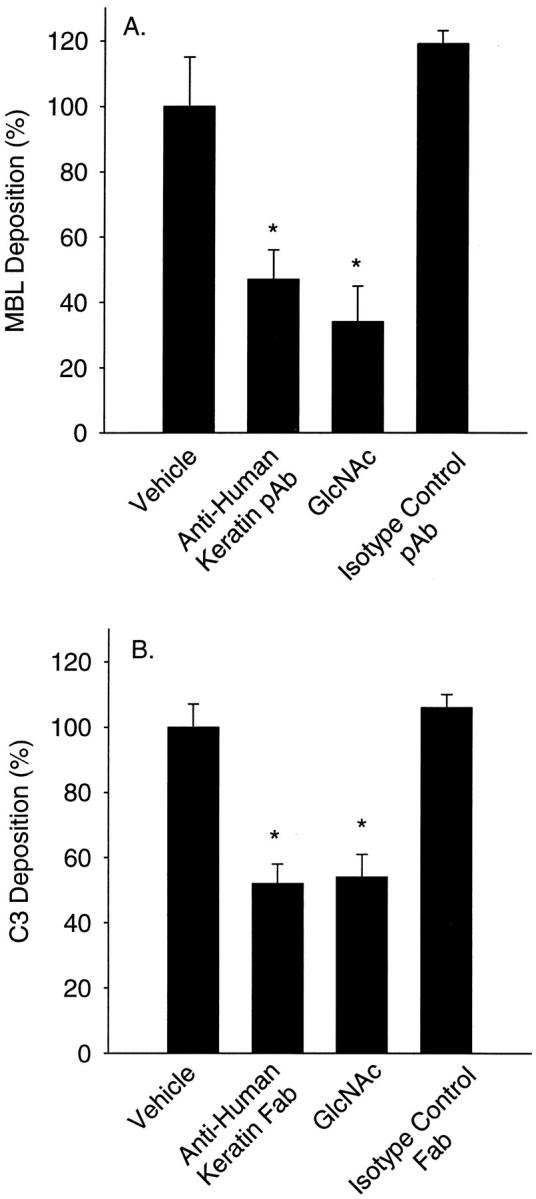
Anti-human keratin antibodies attenuate endothelial MBL and C3 deposition after oxidative stress. HUVEC MBL (A) and C3 (B) deposition after oxidative stress were measured by ELISA. MBL and C3 deposition after oxidative stress was significantly increased compared to normoxic HUVECs. Incubation of HS (vehicle) with the MBL inhibitory sugar, GlcNAc (100 mmol/L) or anti-human keratin pAb (50 μg/ml) significantly attenuated MBL deposition (A). Incubation of HS (vehicle) with GlcNAc (100 mmol/L) or anti-human keratin Fab fragments (20 μg/ml) significantly attenuated C3 deposition (B). Incubation of HS with species-matched, isotype-irrelevant control pAb (50 μg/ml) or Fab fragments (20 μg/ml) did not significantly attenuate MBL (A) or C3 deposition (B), respectively (n = 3; data normalized to vehicle; error bars = SEM; *, P < 0.05 compared to vehicle).
Results
Endothelial CK1 Protein Expression after Oxidative Stress
Human endothelial cell-surface CK1 protein expression after oxidative stress was determined by ELISA, Western blot (Figure 1) ▶ , and confocal microscopy (Figure 2) ▶ . HUVEC CK1 expression after oxidative stress was significantly (P < 0.05) increased compared to normoxic HUVECs as demonstrated by ELISA and Western blot under reducing conditions (Figure 1) ▶ . Immunofluorescent confocal microscopy also demonstrated a significant increase in cell-surface HUVEC CK1 after oxidative stress (Figure 2B) ▶ compared to normoxic cells (Figure 2A) ▶ . Cell-surface CK1 expression after endothelial oxidative stress was also confirmed by Z-section scanning (Figure 2C) ▶ . To demonstrate the specificity of the anti-human CK1 pAb used in these experiments, HUVEC CK1 was immunoprecipitated (67-kd band, reduced 9% linear sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel; data not shown) and confirmed by protein sequencing (human keratin, type II cytoskeletal 1). Thus, these data demonstrate that oxidative stress increases human endothelial cell-surface CK1 protein expression.
Figure 1.

Endothelial CK1 protein expression after oxidative stress. HUVEC CK1 expression after oxidative stress was determined by ELISA and Western blot (inset). CK1 expression was significantly increased after oxidative stress compared to normoxic HUVECs or hypoxic/reoxygenated HUVECs incubated with an isotype control pAb (n = 3; error bars = SEM; *, P < 0.05 compared to normoxia).
Figure 2.

Endothelial CK1 protein expression after oxidative stress. HUVEC CK1 expression after oxidative stress was determined by immunofluorescent confocal microscopy. CK1 expression (green) was significantly increased after oxidative stress (B) compared to normoxic HUVECs (A). Z-section scanning further confirmed cell membrane CK1 expression after endothelial oxidative stress (C). These panels are representative of three different experiments (red = propidium iodide-stained endothelial nuclei).
Endothelial CK1 mRNA Expression after Oxidative Stress
Human endothelial cell CK1 mRNA expression after oxidative stress was determined by in situ hybridization (Figure 3) ▶ . An increase in HUVEC cytoplasmic staining and negative nuclear staining was observed after oxidative stress (Figure 3B) ▶ compared to normoxic cells (Figure 3A) ▶ . Hypoxic HUVECs hybridized with a porcine MBL cDNA probe (Figure 3D) ▶ and RNase-treated hypoxic HUVECs (Figure 3C) ▶ were used as negative controls and to demonstrate staining specificity. These data confirm the presence of HUVEC CK1 mRNA 12 and demonstrate that oxidative stress increases HUVEC CK1 mRNA expression.
Figure 3.

Endothelial CK1 mRNA expression after oxidative stress. HUVEC CK1 mRNA expression after oxidative stress was determined by in situ hybridization. HUVECs were grown under normoxic (A) or hypoxic (B) conditions as described and hybridized with a CK1 cDNA probe. Analysis by confocal microscopy revealed a diffuse cytoplasmic staining pattern, leaving the nucleus devoid of staining. Oxidative stress (B) increased the level of fluorescent staining indicating an increase in CK1 mRNA. C demonstrates that normoxic HUVECs do not express porcine MBL mRNA (normoxia control). D shows hypoxic HUVECs do not express porcine MBL mRNA (hypoxia control). No fluorescence was observed when normoxic or hypoxic HUVECs were incubated with RNase A (data not shown). This figure is representative of three experiments.
MBL and C3 Deposition on Purified Human CK1
We next investigated whether purified human CK1 activates the LCP (Figure 4) ▶ . Treatment of 2% HS with GlcNAc (100 mmol/L) or the functionally inhibitory anti-human MBL mAb, 3F8 (10 μg/ml) significantly inhibited MBL deposition (Figure 4A) ▶ on purified CK1-coated plates by 78 ± 4% and 64 ± 6%, respectively, compared to untreated HS (vehicle). Further, treatment with GlcNAc (100 mmol/L) or 3F8 (10 μg/ml) significantly inhibited C3 deposition (Figure 4B) ▶ on CK1-coated plates by 70 ± 1% and 69 ± 1%, respectively, compared to untreated HS. These data suggest that MBL binds human CK1 and activates the LCP. Further, these data demonstrate that MBL inhibition attenuates CK1-induced complement activation.
Immunoprecipitation and Western Blot of Endothelial CK1 and MBL
To determine whether human MBL binds endothelial CK1, purified MBL was used to immunoprecipitate HUVEC CK1 (Figure 5A) ▶ . Western blot of the immunoprecipitates using a monospecific anti-human CK1 antibody revealed a 67-kd band (lane 5) consistent with human CK1. Interestingly, the 67-kd band was observed after endothelial oxidative stress (lane 5), but not in normoxic HUVECs (lane 4) or in the control lanes (lanes 1 to 3). These data suggest that human MBL recognizes endothelial CK1 after oxidative stress.
Figure 5.

Immunoprecipitation of human CK1 or MBL after endothelial oxidative stress. A: Western blot analysis of human CK1. Purified human MBL was used to immunoprecipitate HUVEC CK1. Western blot of reduced immunoprecipitates revealed a 67-kd band (lane 5) consistent with human CK1. Note that the 67-kd band was observed after endothelial oxidative stress (lane 5), but not in normoxic HUVECs (lane 4) or the controls (lane 1, normoxic HUVEC lysate preclear; lane 2, hypoxic HUVEC lysate preclear; lane 3, anti-human MBL mAb [1C10]-conjugated protein G and human MBL only). B: Western blot analysis of human MBL. To further confirm that MBL binds endothelial CK1 after oxidative stress, MBL was co-immunoprecipitated with CK1 from hypoxic HUVECs reoxygenated in HS. Western blot of HUVEC lysates immunoprecipitated with a monospecific anti-human CK1 pAb revealed a 32-kd band consistent with reduced human MBL (lane 6). Note that the 32-kd band was observed after endothelial oxidative stress (lane 6), but not in normoxic HUVECs (lane 5) or the controls (lane 1, purified human MBL standard; lane 2, normoxic HUVEC lysate preclear; lane 3, hypoxic HUVEC lysate preclear; lane 4, anti-human CK1 pAb and protein G only).
To further confirm that human MBL binds endothelial CK1 after oxidative stress, MBL and CK1 were co-immunoprecipitated from hypoxic HUVECs reoxygenated in HS (Figure 5B) ▶ . Western blot of HUVEC lysates immunoprecipitated with a monospecific anti-human CK1 pAb revealed a 32-kd band consistent with reduced purified human MBL (lane 1). Further, the 32-kd band was observed after endothelial oxidative stress (lane 6), but not in normoxic HUVECs (lane 5) or in the control lanes (lanes 2 to 4). These data further suggest that human MBL binds endothelial CK1 after oxidative stress.
Anti-Human Keratin Treatment Attenuates MBL and C3 Deposition after Endothelial Oxidative Stress
Oxidative stress increases MBL deposition on HUVECs and activates the LCP. 5 We investigated whether anti-human keratin antibodies attenuate endothelial MBL and C3 deposition. HUVEC MBL (Figure 6A) ▶ and C3 (Figure 6B) ▶ deposition after oxidative stress were measured by ELISA. Consistent with our previous findings, 5,22 a significant increase in MBL (OD405 = 0.05 ± 0.01) and C3 (OD405 = 0.21 ± 0.02) deposition was observed after oxidative stress compared to normoxic HUVECs (OD405 = 0.01 ± 0.01 and 0.07 ± 0.01, respectively; P < 0.05). Treatment with GlcNAc (100 mmol/L) or anti-human keratin pAb (50 μg/ml) significantly inhibited MBL deposition (Figure 6A) ▶ by 66 ± 11% and 53 ± 9%, respectively, compared to untreated HS (vehicle). Further, treatment with GlcNAc (100 mmol/L) or anti-human keratin Fab fragments (20 μg/ml) significantly inhibited C3 deposition (Figure 6B) ▶ by 46 ± 7% and 48 ± 6%, respectively, compared to untreated HS. Incubation of HS with species-matched, isotype irrelevant control pAb (50 μg/ml) or Fab fragments (20 μg/ml) did not significantly attenuate MBL (Figure 6A) ▶ or C3 deposition (Figure 6B) ▶ . These data demonstrate that anti-MBL or anti-keratin treatment significantly attenuates endothelial MBL and C3 deposition after oxidative stress.
To further confirm these findings, HUVEC MBL and C3 deposition after oxidative stress was determined by immunofluorescent confocal microscopy (Figure 7) ▶ . Normoxic and hypoxic HUVECs were reoxygenated in 30% HS treated with and without anti-human keratin Fab fragments (20 μg/ml). Small amounts of MBL (Figure 7A ▶ 1) and C3 (Figure 7B ▶ 1) staining were observed under normoxic conditions, confirming our previous finding of low level C3 deposition under normoxic conditions. 19,23 MBL (Figure 7A ▶ 2) and C3 (Figure 7B ▶ 2) staining after HUVEC oxidative stress was increased compared to normoxic cells. Treatment with anti-human keratin Fab fragments attenuated MBL (Figure 7A ▶ 3) and C3 (Figure 7B ▶ 3) staining. These data further demonstrate that anti-keratin treatment inhibits MBL deposition and complement activation after endothelial oxidative stress.
Figure 7.

Anti-human keratin Fab fragments attenuate endothelial C3 and MBL deposition after oxidative stress. Immunofluorescent confocal microscopical demonstration of MBL (row A; green) and C3 (row B; green) deposition on normoxic (column 1) and hypoxic (24 hours) HUVECs reoxygenated (3 hours) in 30% HS (column 2) with and without anti-human keratin Fab fragments (20 μg/ml; column 3). MBL (A2) and C3 (B2) staining after oxidative stress was significantly increased compared to normoxic HUVECs (A1 and B1, respectively). Treatment of HS with anti-human keratin Fab fragments significantly decreased MBL (A3) and C3 (B3) staining. These panels are representative of three different experiments.
Discussion
Growing evidence suggests that complement plays an important role in the pathogenesis of cardiovascular injury after oxidative stress. 24-26 However, the role of the LCP in human cardiovascular disease is unknown. Human endothelial oxidative stress in vitro increases MBL deposition and activates the LCP. 5 Further, rat myocardial MBL and C3 deposition is increased after in vivo ischemia-reperfusion. 5 Although these data suggest a novel pathophysiological role for the LCP after oxidative stress, the molecular mechanism of MBL binding to the vascular endothelium is unknown. We demonstrate in the present study that activation of the LCP after oxidative stress may be mediated by MBL binding to vascular endothelial cytokeratins. Additionally, we demonstrate that human endothelial CK1 may be a novel ligand for human MBL.
Ogawa and colleagues 27 have shown that hypoxia induces endothelial neoantigen expression, creating the possibility that de novo surface ligand expression plays a role in the hypoxic response. Although endothelial cytokeratins have been previously identified, 28-30 the effect of oxidative stress on endothelial cytokeratin expression is primarily unknown. Consistent with previous reports, 11,12 HUVEC CK1 expression was confirmed by immunoprecipitation and protein sequencing. Further, oxidative stress significantly increased HUVEC CK1 mRNA and surface protein expression. Thus, oxidative stress alters human endothelial cytokeratin expression.
Historically, CK1 is known to be an intracellular structural protein that functions to maintain cytoskeletal integrity. 7 Consistent with previous reports of endothelial CK1 surface expression, 12,13 our data also demonstrates HUVEC CK1 surface expression. However, the mechanism of cell-surface CK1 expression is unclear. Although CK1 contains an internal hydrophobic rod domain, neither a membrane-spanning region, nor an insertional signal sequence has been described. Nonetheless, proteins may be externalized in the absence of vesicular transport. For example, the phospholipid-associated protein, annexin I, localizes to the extracellular membrane despite the lack of a signal peptide and co-localizes with CK8. 31 In this context, it is possible that membrane remodeling after oxidative stress results in transport of CK1 to the cell surface. Consistent with this hypothesis, endothelial cells subjected to chemical oxidative stress have been shown to undergo cytoskeletal remodeling and membrane blebbing in the absence of apoptosis. 32 Studies are ongoing in our laboratory to address the mechanisms regulating CK1 transport and expression.
Several lines of evidence suggest that cytokeratins may mediate immune responses. First, specific cytokeratin peptide sequences have been shown to be molecular mimics of the MBL ligand, GlcNAc and to induce in vivo anti-carbohydrate antibody responses. 14,15 Second, autoantibodies to cytokeratin were recently demonstrated in cardiac transplant patients with accelerated coronary artery disease. 33 Third, inflammation associated with blistering skin disorders is observed in humans with mutant cytokeratins. 7 Thus, cytokeratins may represent a novel class of endothelial ligands that mediate immune responses. Data from our laboratory demonstrate that GlcNAc, anti-human MBL mAbs or anti-human keratin Fab fragments inhibit human MBL and C3 deposition on purified human CK1-coated plastic plates or oxidatively stressed human endothelial cells. Together, these data suggest that human MBL binds CK1 and activates the LCP. Additionally, these data demonstrate that anti-human MBL treatment attenuates CK1-induced complement activation. Although we have not yet identified the MBL-binding site on keratin, the CK1 head and tail regions contain sequences highly homologous to a CK14 decapeptide (SFGSGFGGGY) known to mimic the MBL ligand, GlcNAc. 14,15 MBL was recently shown to specifically bind this GlcNAc-mimicking peptide sequence, and pretreatment of human sera with this peptide significantly attenuated MBL and complement deposition on oxidatively stressed human endothelial cells. 16 Thus, MBL may bind the CK1 head and/or tail region(s) after endothelial oxidative stress. Epitope mapping the MBL binding site on CK1 will address this issue.
To determine whether endothelial cytokeratins-induce complement activation after oxidative stress, endothelial MBL and C3 deposition were measured. Treatment with anti-human dermal cytokeratin Fab fragments significantly attenuated endothelial MBL and C3 deposition, suggesting that endothelial cytokeratin epitope blockade attenuates MBL deposition and complement activation after oxidative stress. Interestingly, minimal MBL deposition was observed on untreated, normoxic cells despite the fact that CK1 has been previously cloned from and shown to be expressed by normoxic HUVECs. 11,12 Although we were able to immunoprecipitate and sequence CK1 with a monospecific anti-CK1 pAb from both normoxic and hypoxic/reoxygenated HUVECs, CK1 could only be immunoprecipitated with purified MBL from hypoxic/reoxygenated HUVECs. Additionally, MBL and CK1 could only be co-immunoprecipitated with the anti-CK1 pAb from hypoxic/reoxygenated HUVECs. Together, these data suggest that oxidative stress increases total endothelial CK1 protein expression resulting in a neo-epitope recognized by MBL. These observations are consistent with our previous finding that inhibition of de novo protein synthesis or nuclear factor-κB translocation attenuates endothelial C3 deposition after oxidative stress. 23 Cytokeratin organization and expression is known to be regulated by nuclear factor-κB, protein kinase C, activator protein-1 and cAMP-dependent phosphorylases 34-37 that, in turn, may be altered by hypoxia. 38,39 Indeed, oxidative stress in the present study increased HUVEC CK1 mRNA and cell-surface protein expression. Further studies on the effect of oxidative stress on endothelial cytokeratin protein expression are warranted.
In summary, 1) CK1 mRNA and protein expression, MBL binding, and C3 deposition are increased after human endothelial oxidative stress in vitro, 2) inhibition of MBL deposition with GlcNAc or anti-human MBL mAb attenuates MBL and C3 deposition on purified CK1, and 3) anti-human cytokeratin Fab fragments attenuate endothelial MBL and C3 deposition after oxidative stress. These results suggest that MBL deposition and activation of the LCP after oxidative stress may be mediated by endothelial cytokeratins. Further, these results identify human CK1 as a novel ligand for human MBL. Future studies with specific inhibitors of MBL and cytokeratin in animal models will help define the role of MBL and the LCP in cardiovascular disease.
Acknowledgments
We thank Margaret A. Morrissey and Meera Grover for assistance with HUVEC culture.
Footnotes
Address reprint requests to Gregory L. Stahl, Ph.D., Center for Experimental Therapeutics and Reperfusion Injury, Department of Anesthesiology, Pain, and Perioperative Medicine, Brigham and Women’s Hospital, 75 Francis St., Boston, MA 02115. E-mail: gstahl@zeus.bwh.harvard.edu.
Supported by the National Institutes of Health (HL-03854 to C. D. C., F32-HL103870 to M. C. M., and HL-52886 to G. L. S.), and an American Heart Association Established Investigator Award (to G. L. S.).
These results were presented in part at the XVIIIth International Complement Workshop (Salt Lake City, UT).
Drs. Collard and Montalto contributed equally to this manuscript.
References
- 1.Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, Reid KB, Jensenius JC: A second serine protease associated with mannan-binding lectin that activates complement. Nature 1997, 386:506-510 [DOI] [PubMed] [Google Scholar]
- 2.Turner MW: The lectin pathway of complement activation. Res Immunol 1996, 147:110-115 [DOI] [PubMed] [Google Scholar]
- 3.Turner MW: Mannose-binding lectin: the pluripotent molecule of the innate immune system. Immunol Today 1996, 17:532-540 [DOI] [PubMed] [Google Scholar]
- 4.Turner MW: Mannose-binding lectin (MBL) in health and disease. Immunobiology 1998, 199:327-339 [DOI] [PubMed] [Google Scholar]
- 5.Collard CD, Väkevä A, Morrissey MA, Agah A, Rollins SA, Reenstra WR, Buras JA, Meri S, Stahl GL: Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol 2000, 156:1549-1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pavalko FM, Otey CA: Role of adhesion molecule cytoplasmic domains in mediating interactions with the cytoskeleton. Proc Soc Exp Biol Med 1994, 205:282-293 [DOI] [PubMed] [Google Scholar]
- 7.Fuchs E, Cleveland DW: A structural scaffolding of intermediate filaments in health and disease. Science 1998, 279:514-519 [DOI] [PubMed] [Google Scholar]
- 8.Lekowski R, Collard CD, Reenstra WR, Stahl GL: Ulex europaeus agglutinin II (UEA II) is a novel and potent inhibitor of complement activation. Protein Sci 2001, 10:277-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joseph K, Shibayama Y, Nakazawa Y, Peerschke EI, Ghebrehiwet B, Kaplan AP: Interaction of factor XII and high molecular weight kininogen with cytokeratin 1 and gC1qR of vascular endothelial cells and with aggregated Abeta protein of Alzheimer’s disease. Immunopharmacology 1999, 43:203-210 [DOI] [PubMed] [Google Scholar]
- 10.Joseph K, Ghebrehiwet B, Kaplan AP: Cytokeratin 1 and gC1qR mediate high molecular weight kininogen binding to endothelial cells. Clin Immunol 1999, 92:246-255 [DOI] [PubMed] [Google Scholar]
- 11.Shariat-Madar Z, Mahdi F, Schmaier AH: Mapping binding domains of kininogens on endothelial cell cytokeratin 1. J Biol Chem 1999, 274:7137-7145 [DOI] [PubMed] [Google Scholar]
- 12.Hasan AAK, Zisman T, Schmaier AH: Identification of cytokeratin 1 as a binding protein and presentation receptor for kininogens on endothelial cells. Proc Natl Acad Sci USA 1998, 95:3615-3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahdi F, Shariat-Madar Z, Todd RF, III, Figueroa CD, Schmaier AH: Expression and colocalization of cytokeratin 1 and urokinase plasminogen activator receptor on endothelial cells. Blood 2001, 97:2342-2350 [DOI] [PubMed] [Google Scholar]
- 14.Shikhman AR, Cunningham MW: Immunological mimicry between N-acetyl-beta-D-glucosamine and cytokeratin peptides. Evidence for a microbially driven anti-keratin antibody response. J Immunol 1994, 152:4375-4387 [PubMed] [Google Scholar]
- 15.Shikhman AR, Greenspan NS, Cunningham MW: Cytokeratin peptide SFGSGFGGGY mimics N-acetyl-beta-D-glucosamine in reaction with antibodies and lectins, and induces in vivo anti-carbohydrate antibody response. J Immunol 1994, 153:5593-5606 [PubMed] [Google Scholar]
- 16.Montalto MC, Collard CD, Buras JA, Reenstra WR, McClaine R, Geis DR, Rother RP, Stahl GL: A keratin peptide inhibits mannose-binding lectin. J Immunol 2001, 166:4148-4153 [DOI] [PubMed] [Google Scholar]
- 17.Linder E, Lehto VP, Stenman S: Activation of complement by cytoskeletal intermediate filaments. Nature 1979, 278:176-178 [DOI] [PubMed] [Google Scholar]
- 18.Linder E, Helin H, Rantala I: Activation of complement by intermediate filaments of glomerular epithelial cells. Clin Immunol Immunopathol 1986, 40:265-275 [DOI] [PubMed] [Google Scholar]
- 19.Collard CD, Vakeva A, Bukusoglu C, Zünd G, Sperati CJ, Colgan SP, Stahl GL: Reoxygenation of hypoxic human umbilical vein endothelial cells activates the classic complement pathway. Circulation 1997, 96:326-333 [DOI] [PubMed] [Google Scholar]
- 20.Tan SM, Chung MCM, Kon OL, Thiel S, Lee SH, Lu J: Improvements on the purification of mannan-binding lectin and demonstration of its calcium-independent association with a C1s-like serine protease. Biochem J 1996, 319:329-332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collard CD, Bukusoglu C, Agah A, Colgan SP, Reenstra WR, Morgan BP, Stahl GL: Hypoxia-induced expression of complement receptor type 1 (CR1, CD35) in human vascular endothelial cells. Am J Physiol 1999, 276:C450-C458 [DOI] [PubMed] [Google Scholar]
- 22.Collard CD, Lekowski R, Jordan JE, Agah A, Stahl GL: Complement activation following oxidative stress. Mol Immunol 1999, 36:941-948 [DOI] [PubMed] [Google Scholar]
- 23.Collard CD, Agah A, Stahl GL: Complement activation following reoxygenation of hypoxic human endothelial cells: role of intracellular reactive oxygen species, NF-kB and new protein synthesis. Immunopharmacology 1998, 39:39-50 [DOI] [PubMed] [Google Scholar]
- 24.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT: Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990, 249:146-151 [DOI] [PubMed] [Google Scholar]
- 25.Buerke M, Murohara T, Lefer AM: Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation 1995, 91:393-402 [DOI] [PubMed] [Google Scholar]
- 26.Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL: Myocardial infarction and apoptosis after myocardial ischemia and reperfusion—role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 1998, 97:2259-2267 [DOI] [PubMed] [Google Scholar]
- 27.Ogawa S, Clauss M, Kuwabara K, Shreeniwas R, Butura C, Koga S, Stern D: Hypoxia induces endothelial cell synthesis of membrane-associated proteins. Proc Natl Acad Sci USA 1991, 88:9897-9901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattey DL, Nixon N, Wynn-Jones C, Dawes PT: Demonstration of cytokeratin in endothelial cells of the synovial microvasculature in situ and in vitro. Br J Rheumatol 1993, 32:676-682 [DOI] [PubMed] [Google Scholar]
- 29.Carley WW, Szczepanski A, Gerritsen ME: Cytokeratin expression and hyaluronic acid production in cultures of human synovial microvascular endothelial cells: influence of cytokines and growth factors. Microcirculation 1996, 3:359-370 [DOI] [PubMed] [Google Scholar]
- 30.Traweek ST, Liu J, Battifora H: Keratin gene expression in non-epithelial tissues. Detection with polymerase chain reaction. Am J Pathol 1993, 142:1111-1118 [PMC free article] [PubMed] [Google Scholar]
- 31.Traverso V, Morris JF, Flower RJ, Buckingham J: Lipocortin 1 (annexin 1) in patches associated with the membrane of a lung adenocarcinoma cell line and in the cell cytoplasm. J Cell Sci 1998, 111:1405-1418 [DOI] [PubMed] [Google Scholar]
- 32.van Gorp RM, Broers JL, Reutelingsperger CP, Bronnenberg NM, Hornstra G, Dam-Mieras MC, Heemskerk JW: Peroxide-induced membrane blebbing in endothelial cells associated with glutathione oxidation but not apoptosis. Am J Physiol 1999, 277:C20-C28 [DOI] [PubMed] [Google Scholar]
- 33.Ationu A: Identification of endothelial antigens relevant to transplant coronary artery disease from a human endothelial cell cDNA expression library. Int J Mol Med 1998, 1:1007-1010 [DOI] [PubMed] [Google Scholar]
- 34.Ma S, Rao L, Freedberg IM, Blumenberg M: Transcriptional control of K5, K6, K14, and K17 keratin genes by AP-1 and NF-kappaB family members. Gene Expr 1997, 6:361-370 [PMC free article] [PubMed] [Google Scholar]
- 35.Yano T, Tokui T, Nishi Y, Nishizawa K, Shibata M, Kikuchi K, Tsuiki S, Yamauchi T, Inagaki M: Phosphorylation of keratin intermediate filaments by protein kinase C, by calmodulin-dependent protein kinase and by cAMP-dependent protein kinase. Eur J Biochem 1991, 197:281-290 [DOI] [PubMed] [Google Scholar]
- 36.Yeagle PL, Frye J, Eckert BS: Phosphorylation modulates keratin structure. Biochemistry 1990, 29:1508-1514 [DOI] [PubMed] [Google Scholar]
- 37.Eckert BS, Yeagle PL: Modulation of keratin intermediate filament distribution in vivo by induced changes in cyclic AMP-dependent phosphorylation. Cell Motil Cytoskeleton 1990, 17:291-300 [DOI] [PubMed] [Google Scholar]
- 38.Stevens T, Rodman DM: The effect of hypoxia on endothelial cell function. Endothelium 1995, 3:1-11 [Google Scholar]
- 39.Pinsky DJ, Yan SF, Lawson C, Naka Y, Chen JX, Connolly ESJ, Stern DM: Hypoxia and modification of the endothelium: implications for regulation of vascular homeostatic properties. Semin Cell Biol 1995, 6:283-294 [DOI] [PubMed] [Google Scholar]