

Abstract
A common polymorphism in the cystatin C gene is associated with increased risk of developing Alzheimer’s disease (AD). To explore possible neuropathological consequences of this genetic association, we examined expression of cystatin C in brains from 22 AD and 11 control patients by immunohistochemistry. In the temporal cortex of all AD brains, there was strong cystatin C immunostaining of neurons and activated glia, whereas staining was absent or minimal in 7 of the 11 control brains. Neuronal staining of cystatin C in AD brains was primarily limited to pyramidal neurons in cortical layers III and V, which are the neurons most susceptible to cell death in AD. The increase in cystatin C staining in AD was independent of cystatin C genotype. Immunostaining of cystatin C within neurons showed a punctate distribution, which co-localized with the endosomal/lysosomal proteinase, cathepsin B. A primarily glial source for cystatin C was suggested by parallel studies using in situ hybridization of mouse brain. In human AD brain, there was little co-localization of cystatin C with parenchymal Aβ deposits, although a small fraction of cerebral blood vessels and neurofibrillary tangles were cystatin C-positive. The regional distribution of cystatin C neuronal immunostaining also duplicated the pattern of neuronal susceptibility in AD brains: the strongest staining was found in the entorhinal cortex, in the hippocampus, and in the temporal cortex; fewer pyramidal neurons were stained in frontal, parietal, and occipital lobes. These neuropathological observations reinforce the association between cystatin C and AD, and support a model of cystatin C involvement in the process of neuronal death in AD.
Cystatin C is a cysteine protease inhibitor that inhibits the cathepsin family of lysosomal proteases (cathepsin B, H, L, and S). 1 It is a 14-kd secreted protein (120 amino acids), 2 present in cerebrospinal fluid at high concentrations. 3 A mutant form of cystatin C forms the cerebrovascular amyloid in hereditary cerebral hemorrhage with amyloidosis, Icelandic-type, because of a leucine-to-glutamine substitution at position 68. 4 However, cystatin C is only found in a subset of cerebral amyloid deposits in sporadic cases of cerebral amyloid angiopathy. 5-8 In these cases, amyloid is primarily composed of the 4-kd Aβ peptide, which is also found in the parenchymal amyloid deposits in the brain of Alzheimer’s disease (AD) patients.
In addition to the rare Icelandic mutation of the cystatin C gene (CST3), there are two common haplotypes of CST3 (A and B) that differ from each other at three sites: two single base pair changes in the promoter region and one in the signal peptide domain that causes an amino acid substitution (alanine to threonine). 9 Recently, case-control studies found associations of CST3 with increased risk of late onset AD. 10,11 In the larger series, Finckh and colleagues 10 found the BB genotype in <2% of control individuals but in 5 to 9% of AD patients, and in even higher percentages in those patients with the latest ages of onset. These data suggest that inheritance of CST3 BB increases the risk of AD, particularly with onset after age 75. To determine how changes in the CST3 gene could affect the neuropathological processes of AD, we examined the distribution of the cystatin C protein in AD brain tissue.
Materials and Methods
Antibodies
For cystatin C immunohistochemistry, two rabbit polyclonal antibodies were used (Upstate Biotechnology, Lake Placid, NY, and Biogenesis, Kingston, NH). Western blots of human brain extracts with cystatin C antibodies revealed a strong band of ∼14 kd, corresponding in size to purified cystatin C (Figure 1) ▶ . Cathepsin B was detected with a sheep polyclonal antibody (ICN Biochemicals, Irvine, CA). Aβ was detected with the mouse monoclonal antibody 10D5 (courtesy of Dr. Peter Seubert, Elan Pharmaceuticals). 12 Phospho-tau was detected with the mouse monoclonal antibody PHF-1 (courtesy of Dr. Peter Davies, Albert Einstein University, New York, NY). 13 )
Figure 1.

Immunoblot of cerebrospinal fluid and brain for cystatin C. The specificity of anti-cystatin C antibody was demonstrated using standard cystatin C (lane 1), human cerebrospinal fluid (lane 2), and human brain tissue (lanes 3 and 4). Antibodies recognized monomeric cystatin C at 14 kd; dimeric cystatin C is seen with the standard cystatin C. A second band of reactivity is seen at >50 kd in cerebrospinal fluid and brain, possibly reflecting the presence of albumin.
Brain Tissue
Temporal cortex of 22 neuropathologically confirmed AD cases and 11 control brains were collected from the Massachusetts Alzheimer Disease Research Center brain bank (Dr. E. T. Hedley-Whyte, director). The AD group consisted of 9 males and 13 females (average age, 80 years; SD, 7); the control group consisted of five males and five females (average age, 73 years; SD, 20). In addition, frontal cortex, occipital cortex, parietal cortex, and cerebellum were obtained from five AD cases. Postmortem intervals averaged 19 hours. Tissue was fixed in paraformaldehyde-lysine-metaperiodate for 24 hours at 4°C, and cryoprotected in 15% glycerol in 0.1 mol/L Tris-buffered saline (pH 7.4). Fifty-μm sections were prepared using a freezing-sledge microtome. The control brain tissues were screened for AD neuropathological changes by immunostaining for amyloid deposits and neurofibrillary tangles (Table 1) ▶ . Braak and Braak 14 staging was determined by the distribution of PHF-1-stained neurofibrillary tangles.
Table 1.
Control Brain Tissue
| Control no. | Age, year | Neuronal cystatin C | Aβ plaques | Braak and Braak stage |
|---|---|---|---|---|
| 1 | 86 | + | −− | 0 |
| 2 | 82 | +++ | −− | 2 |
| 3 | 71 | + | −− | 0 |
| 4 | 72 | + | −− | 0 |
| 5 | 53 | −− | −− | 0 |
| 6 | 72 | −− | −− | 0 |
| 7 | 68 | +++ | +++ | 1 |
| 8 | 64 | +++ | ++ | 2 |
| 9 | 77 | −− | −− | 3 |
| 10 | 66 | −− | −− | 3 |
| 11 | 93 | +++ | −− | 3 |
−−, no immunoreactivity throughout the temporal cortex; +, minimal immunoreactivity only in some fields; ++, minimal immunoreactivity in all fields; +++, strong immunoreactivity in all fields.
DNA was isolated from 50 mg of frozen brain tissue using Qiagen DNA extraction kits (Qiagen, Valencia, CA). The three linked polymorphisms in the promoter and signal peptide of CST3 were analyzed by a polymerase chain reaction-restriction enzyme assay as described. 10
Immunoblotting
Standard cystatin C (50 ng; Calbiochem, La Jolla, CA), human cerebrospinal fluid (15 μl), and human brain extract (10 μg protein in 50 mmol/L Tris, pH 8.0, 0.5 mol/L NaCl, 0.1% Triton X-100) were separated by 4 to 20% polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membrane, probed with anti-cystatin C antibody, and visualized by chemiluminescence.
Immunohistochemistry
Brain tissue was immunostained using anti-cystatin C (1:500 to 1:1000), 10D5 (1:1000), PHF-1 (1:200), or anti-cathepsin B (1:250). Antibodies were detected using horseradish peroxidase-linked secondary antibodies (anti-rabbit for cystatin C, anti-mouse for Aβ, and phospho-tau; Jackson Immunoresearch, West Grove, PA) and visualized with 3,3′-diaminobenzidine. For fluorescent double labeling, tissue was initially co-incubated with two primary antibodies. Cystatin C antibodies were detected with Cy3-linked or (4,4-difluoro-5,7-dimethyl-4-bova-3a,4a-diaza-5-indacene) (BODIPY)-linked anti-rabbit secondary antibodies; Aβ and phospho-tau antibodies were detected with BODIPY-linked anti-mouse secondary antibodies; cathepsin B antibodies were detected with Cy3-linked secondary antibodies. Fluorescence was observed using the BioRad MRC-1024 confocal microscope (Cy3: excitation at 568 nm, emission at 605 nm, BODIPY: excitation at 488 nm, emission at 522 nm) (Bio-Rad, Richmond, CA). Controls without primary antibodies were included for each experiment.
In Situ Hybridization
Cystatin C in situ hybridization was performed according to published protocols on 14-μm sagittal sections from five APP transgenic and three nontransgenic mice (three 12-month-old PDAPP homozygotes, 15 three 2- to 15-month-old nontransgenic mice, and two 15-month-old Tg2576 heterozygotes 16 ) using mouse cystatin C (GenBank accession no. M59470) bases 181 to 226 45-mer sense and antisense oligonucleotide probes. 17,18 Hybridized slides were dipped in Amersham LM-1 emulsion (Amersham, Arlington Heights, IL), stored at 4°C for 4 weeks, developed in Kodak D-19 (Eastman-Kodak, Rochester, NY), and counterstained with thionin and thioflavin S.
Results
We immunostained the temporal cortex from 11 control and 22 AD brains for cystatin C. In 7 of the 11 control brains, there were no cystatin C-positive cells visible (Figure 2A) ▶ , whereas there was strong cystatin C immunostaining in all AD brains (Figure 2B) ▶ . Cystatin C immunoreactivity was confined primarily to pyramidal neurons, of cortical layers III and V, although neurons in layer II were also often positive. This distribution of neuronal staining demarcated those neurons most susceptible to loss in AD cortex. To determine whether other susceptible neurons were also cystatin C-positive in the AD brain, we examined the entorhinal cortex and the hippocampus. Neurons in all hippocampal subfields were positive, with the strongest staining observed in CA1 (Figure 2C) ▶ . Similarly, entorhinal cortex neurons were also strongly positive for cystatin C (Figure 2D) ▶ . Of the 22 AD brains, 15 were the CST3 AA genotype, 6 were AB, and 1 was BB; strong neuronal staining was observed for the brains within each genotype subgroup. Thus, regardless of CST3 genotype, cystatin C is found in neurons throughout the temporal lobe that are susceptible to loss in AD.
Figure 2.

Cystatin C is increased in AD brain. Temporal lobes of control (A) and AD (B–D) were immunostained for cystatin C. Cortex showed no staining in the control case (A), but strong staining of layer III and V pyramidal neurons in AD brain (B). Cystatin C was also strongly present in neurons of CAI (C) and entorhinal cortex (D). Original magnifications: ×31 (A and B), ×80 (C and D).
Four of the 11 control brains showed a distribution of cystatin C that was similar to that seen in AD, with prominent staining of pyramidal neurons in cortical layers III and V, and glial staining throughout the cortex. There was no correlation of the cystatin C-positive controls with age. Two of the four cystatin C-positive controls showed strong AB immunostaining (Table 1) ▶ . In contrast, none of the seven cystatin C-negative controls showed strong Aβ immunostaining.
We examined the cellular distribution of cystatin C within the AD brain. The cystatin C in pyramidal neurons was present in a vesicular pattern (Figure 3A) ▶ . Cystatin C was also present in glia throughout the cortex (Figure 3B) ▶ . The morphology of the cystatin C-positive glia suggested that they were strongly activated. Cystatin C-positive glia and neurons were observed together in some fields, and separately in others.
Figure 3.
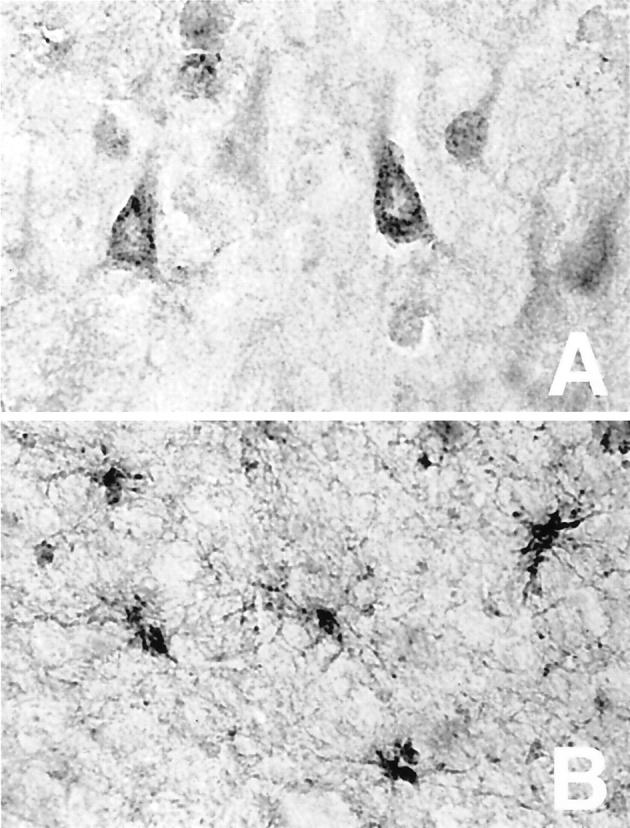
Cystatin C is present in both neurons and glia of AD brain. Temporal cortex of AD brain immunostained for cystatin C revealed a vesicular pattern in pyramidal neurons (A) as well as staining in activated glia in the white matter (B). Original magnifications, ×500.
The vesicular pattern of cystatin C immunoreactivity in neurons suggested its presence in endosomes and lysosomes. We immunostained AD brain tissue for cathepsin B, an endosomal/lysosomal protein 19 and a protease that is inhibited by cystatin C. 1 Cathepsin B and cystatin C showed similar punctate patterns of staining in neurons, with almost complete co-localization in intraneuronal vesicles (Figure 4 ▶ , in yellow). These data demonstrate the presence of cystatin C in endosomes and lysosomes in AD brain.
Figure 4.

Co-localization of cystatin C with cathepsin B. Temporal cortex of AD brain was double-immunostained for cathepsin B (in red) and cystatin C (in green), demonstrating vesicular co-localization of this protease-inhibitor pair (in yellow). Original magnifications, ×1000.
This vesicular cystatin C could be produced within neurons, or be produced in other cells and enter neurons through an endocytic pathway. To gain insight into the cellular source of cystatin C in the brain, we performed in situ hybridization of mouse brain and mouse models of Aβ deposition. Emulsion-dipped sections demonstrated predominantly glial expression of cystatin C, with emulsion granules overlying glia in white matter and interspersed between neurons in the cortex and subcortical regions (Figure 5A) ▶ . In APP transgenic mice, glia surrounding thioflavin S-labeled amyloid deposits also expressed cystatin C (Figure 5B) ▶ ; sense probes yielded no detectable signal. Microglia could not be distinguished from astrocytes, and neuronal hybridization was minimal. Thus, cystatin C seems primarily produced by glial cells in the brain, which are activated in the vicinity of amyloid deposits.
Figure 5.

In situ hybridization of cystatin C in mouse brain. Emulsion-dipped cystatin c in situ hybridization in a 15-month-old nontransgenic mouse (A) demonstrates accumulations of silver granules over glia in the dentate gyrus of the hippocampus (nissl counterstain in gray). Glia surrounding an amyloid deposit in a 12-month-old homozygous PDAPP mouse (B) also express cystatin C (nissl counterstain). Scale bars, 50 μm.
We were struck by the absence of cystatin C immunostaining of amyloid deposits in the sections of AD brain examined. Figure 6 ▶ shows the parahippocampal gyrus from one AD case, stained for cystatin C (Figure 6A) ▶ , Aβ (Figure 6B) ▶ , or phosphorylated tau (Figure 6C) ▶ . Despite the presence of large amounts of cortical Aβ, no cystatin C was observed in these parenchymal deposits (Figure 6, A and B) ▶ . A second polyclonal cystatin C antibody was used on sections from AD brains; it also identified cortical neurons without obvious staining of Aβ deposits. The layers of cystatin C-positive neurons did partially overlap with phosphorylated tau-positive neurons (Figure 6, A and C) ▶ , raising the possibility that cystatin C and neurofibrillary tangles did occasionally co-localize.
Figure 6.

Neuronal cystatin C in regions of AD neuropathological changes. Parahippocampal gyrus of AD patient immunostained for cystatin C (A), Aβ (B), and phospho-tau (C). The pattern of cystatin C staining is primarily independent of plaques and tangles. Original magnifications, ×80.
The pattern of co-localization of cystatin C with Aβ and neurofibrillary tangles was more closely examined by confocal microscopy using double immunofluorescence (Figure 7) ▶ . Cystatin C (in red) immunostained neurons in the vicinity of parenchymal Aβ deposits (in green) without evidence of co-localization with the plaques (Figure 7A) ▶ . In contrast to the plaques, there were some cerebrovascular deposits that immunostained strongly with both Aβ and cystatin C (Figure 7A ▶ , inset, in yellow). There was partial co-localization of cystatin C with phospho-tau in some cortical neurons (Figure 7B ▶ , arrows), but most neurofibrillary tangles were cystatin C-negative and most cystatin C-positive neurons were phospho-tau-negative. Control immunofluorescence studies without primary antibodies revealed no specific extracellular or intracellular staining.
Figure 7.

Confocal imaging of cystatin C with Aβ and phospho-tau immunostains. A: Temporal lobe of AD brain was co-labeled with antibodies against cystatin C (in red) and Aβ (in green). Cystatin C and parenchymal Aβ did not co-localize, although cystatin C and cerebrovascular amyloid did occasionally co-localize (see inset, in yellow). B: Temporal lobe of AD brain was co-labeled with antibodies against cystatin C (in red) and phospho-tau (in green). Cystatin C and neurofibrillary tangles mostly labeled separate structures, although there were regions of co-localization (in yellow, denoted by arrows). Original magnifications, ×400.
We examined the distribution of cystatin C staining in other cortical regions of five AD brains (Figure 8) ▶ . Cystatin C-positive pyramidal neurons were found in temporal (Figure 8A) ▶ , frontal (Figure 8B) ▶ , parietal (Figure 8C) ▶ ,and occipital lobes (Figure 8D) ▶ . This staining respected a laminar distribution in all brain regions, and was consistently strongest in the temporal lobe. We observed diffuse laminar immunostaining in the cerebellar cortex and cerebellar dentate nucleus, with cellular staining also apparent in the dentate nucleus. Cystatin-C-positive glia were found in regions of cortex and white matter in all regions. The strongest neuronal staining in the temporal cortex compared to other brain regions again mirrored the differential susceptibility of brain regions in AD.
Figure 8.

Regional distribution of cystatin C in AD brain. Various regions of an AD brain were immunostained for cystatin C: temporal lobe (A), frontal lobe (B), parietal lobe (C), occipital lobe (D). Neuronal staining was more pervasive in temporal lobe than in other brain regions. Original magnifications, ×80.
Discussion
The genetic associations of CST3 with AD 10,11 led us to examine the neuropathological distribution of the cystatin C protein in AD brain. We found a dramatic neuronal staining in the 22 AD brains we examined, but no or minimal staining in 7 of the 11 aged control brains examined. In addition, we found activated glia that were cystatin C-positive in the AD brains, thereby confirming a previous report regarding glial staining in AD. 20 We and others have found cystatin C message primarily in glia (Figure 5) ▶ 21 ) as well as choroid plexus. 22 We suggest that the immunostaining of glia represents cystatin C that is being secreted, and the immunostaining of neurons represents cystatin C that is endocytosed. We observed this pattern of immunostaining with two cystatin C antibodies, and found it present in AD brains regardless of CST3 genotype, suggesting that cystatin C involvement in AD pathogenesis is not limited to those individuals with the at-risk CST3 genotypes.
Our data extend a recent report of neuronal cystatin C immunostaining in cortical neurons 23 by demonstrating the topographical pattern of neuronal staining. Neuronal staining was most intense in the temporal lobe compared with the frontal, parietal, and occipital lobes and least intense in the cerebellum (Figure 8) ▶ . The strongest immunostaining was found in pyramidal neurons, and more specifically, in neurons of cortical layers III and V (Figure 2) ▶ . Neurons of the entorhinal cortex and hippocampal subfields were very strongly stained (Figure 3) ▶ . This pattern of cystatin C immunostaining recapitulates the hierarchical vulnerability of neurons in the AD brain. 14,24 We hypothesize that cystatin C immunoreactivity identified neurons that were damaged by AD neuropathological processes. This hypothesis is further supported by our observation that cystatin C and neurofibrillary tangles occasionally co-localize within neurons (Figure 7B) ▶ .
In both this study and that of Yasuhara and colleagues, 20 there were a minority of aged control brains that were strongly cystatin C-positive. In our study, two of the cystatin C-positive control brains showed substantial amounts of Aβ deposits whereas none of the seven cystatin C-negative brains showed as much Aβ (Table 1) ▶ . These data imply there may be some connection between the accumulation of Aβ early in AD and cystatin C accumulation in intraneuronal vesicles. This situation parallels precisely the changes in early stages of AD observed for lysosomal proteases, the class of proteases inhibited by cystatin C. Lysosomal proteases (including cathepsins B and D) are up-regulated in the AD brain 19,25 and these changes occur as the earliest markers of AD neuropathological changes. 26 We found cystatin C and cathepsin B co-localized in these neuronal vesicles (Figure 4) ▶ , which are increased in size and number in the AD brain. 19,27 These neurons have also been reported to possess vesicular accumulations of Aβ42. 28 We hypothesize that cystatin C is accumulating in endocytic vesicles to aid in inhibition of the increased levels of lysosomal proteases (eg, cathepsin B), which may be a neuronal response to intracellular Aβ.
We did not observe frequent staining of parenchymal amyloid deposits in AD brains (Figure 6) ▶ , although we did note many instances of cerebrovascular amyloid that were cystatin C-positive (Figure 7A) ▶ . In studies of AD, cystatin C has been observed in some parenchymal amyloid deposits in AD, 23 but more often in amyloid-surrounding blood vessels. 5,6 Cystatin C is also found in cerebrovascular amyloid in cases of sporadic cerebral amyloid angiopathy 5-8 in addition to the cases of cerebral amyloid angiopathy caused by the cystatin C L68Q mutation. 4 The weak staining of parenchymal amyloid deposits with cystatin C contrasts with the prominent co-localization of cystatin C and Aβ in plaques observed in transgenic APP mice. 29 These apparent disparities between parenchymal and cerebrovascular amyloid and between human and mouse brain tissues remain unexplained, but it may provide clues into the process of Aβ deposition.
One basic question is whether elevated cystatin C is specific to the chronic neurodegeneration of AD, or whether it is also present in conditions of acute neuronal injury. In models of transient global ischemia in rodents, cystatin C was found elevated in hippocampal neurons, 30 and well as glia. 31 Similarly, after facial nerve axotomy, microglial cystatin C mRNA was up-regulated in the damaged facial nucleus. 21 Cystatin C has recently been found to act as an autocrine or paracrine factor independent of its function as a protease inhibitor; its presence in the adult brain may be related to its role in promoting neurogenesis in neural stem cells. 32 Together these findings suggest that cystatin C may be a marker for damaged or degenerating neurons in conditions other than just AD.
The pattern of cystatin C immunostaining in susceptible neurons and its elevation in models of neuronal loss have focused our attention to a role for cystatin C in neuronal protection in AD. Our working model is that cystatin C is endocytosed by damaged neurons and targeted to the lysosome. These neurons would have begun to accumulate lysosomal proteases and hydrolases, and cystatin C could act as an inhibitor of some of these proteases to protect the cell from excessive lysosomal dysfunction. If the neuronal insult is too profound or too persistent, the neurons may accumulate neurofibrillary changes and degenerate.
Acknowledgments
We thank the Massachusetts Alzheimer Disease Research Center brain bank (Dr. E. T. Hedley-Whyte, director) for access to brain tissue; Brad Hyman for helpful discussions; Efrat Levy for sharing her unpublished data; Dr. Peter Seubert for the Aβ antibody; and Dr. Peter Davies for the phopsho-tau antibody.
Footnotes
Address reprint requests to G. William Rebeck, Alzheimer Research Unit, 114 16th St., Charlestown, MA 02129. E-mail: rebeck@helix.mgh.harvard.edu.
Supported by the Alzheimer’s Association (grant IIRG-99-1755), the National Institutes of Health (grant AG 05134), and the Luttrell Foundation for Alzheimer’s Research.
References
- 1.Abrahamson M, Barrett AJ, Salvesen G, Grubb A: Isolation of six cysteine proteinase inhibitors from human urine. J Biol Chem 1986, 261:11282-11289 [PubMed] [Google Scholar]
- 2.Grubb A, Lofberg H: Human gamma-trace, a basic microprotein: amino acid sequence and presence in the adenohypophysis. Proc Natl Acad Sci USA 1982, 79:3024-3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lofberg H, Grubb AO, Olsson J-E: The cerebrospinal fluid and plasma concentrations of gamma-trace and β2-microglobulin at various ages and in neurological disorders. J Neurol 1980, 223:159-170 [DOI] [PubMed] [Google Scholar]
- 4.Ghiso J, Jensson O, Frangione B: Amyloid fibrils in hereditary cerebral hemorrhage with amyloidosis of Icelandic type is a variant of gamma-trace basic protein (cystatin C). Proc Natl Acad Sci USA 1986, 83:2974-2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinters HV, Nishimura GS, Secor DL, Pardridge WM: Immunoreactive A4 and gamma-trace peptide colocalization in amyloidotic arteriolar lesions in brains of patients with Alzheimer’s disease. Am J Pathol 1990, 137:233-240 [PMC free article] [PubMed] [Google Scholar]
- 6.Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N: Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to β protein and cystatin C. Stroke 1990, 21:397-403 [DOI] [PubMed] [Google Scholar]
- 7.Haan J, Maat-Schieman MLC, van Duinen SG, Jensson O, Thorsteinsson L, Roos RAC: Co-localization of β/A4 and cystatin C in cortical blood vessels in Dutch, but not in Icelandic hereditary cerebral hemorrhage with amyloidosis. Acta Neurol Scand 1994, 89:367-371 [DOI] [PubMed] [Google Scholar]
- 8.McCarron MO, Nicoll JAR, Stewart J, Ironside JW, Mann DMA, Love S, Graham DI, Dewar D: The apolipoprotein E e2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol 1999, 58:711-718 [DOI] [PubMed] [Google Scholar]
- 9.Balbin M, Abrahamson M: SstIl polymorphic sites in the promoter region of the human cystatin C gene. Hum Genet 1991, 87:751-752 [DOI] [PubMed] [Google Scholar]
- 10.Finckh U, von der Kammer H, Velden J, Michel T, Anderson B, Deng A, Zhang J, Mueller-Thomsen T, Zuchowski K, Menzer G: Genetic association of a cystatin C gene CST3 polymorphism with sporadic late-onset Alzheimer disease. Arch Neurol 2000, 57:1579-1583 [DOI] [PubMed] [Google Scholar]
- 11.Crawford FC, Freeman MJ, Schinka JA, Abdullah LI, Gold M, Hartman R, Krivian K, Morris MD, Richards D, Duara R, Anand R, Mullan MJ: A polymorphism in the cystatin C gene is a novel risk factor for late-onset Alzheimer’s disease. Neurology 2000, 55:763-768 [DOI] [PubMed] [Google Scholar]
- 12.Hyman BT, Tanzi RE, Marzloff KM, Barbour R, Schenk D: Kunitz protease inhibitor containing amyloid precursor protein immunoreactivity in Alzheimer’s disease: a quantitative study. J Neuropath Exp Neurol 1992, 51:76-83 [DOI] [PubMed] [Google Scholar]
- 13.Greenberg S, Davies P: A preparation of Alzheimer paired helical filaments that display distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 1990, 87:5827-5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braak H, Braak E: Neuropathological staging of Alzheimer related changes. Acta Neuropathol 1991, 82:239-259 [DOI] [PubMed] [Google Scholar]
- 15.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 16.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 17.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT: AB deposition is associated with neuropil changes, but not with overt neuronal loss in the PDAPP transgenic mice. J Neurosci 1997, 17:7053-7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sirinathsinghji DJS, Dunnett SB: Imaging gene expression in neural grafts. Molecular Imaging in Neuroscience: A Practical Approach. Edited by NA Sharif. 1993:pp 43-70 Oxford University Press, New York
- 19.Cataldo AM, Barnett JL, Pieroni C, Nixon RA: Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci 1997, 17:6142-6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasuhara O, Hanai K, Ohkubo I, Sasaki M, McGeer PL, Kimura H: Expression of cystatin C in rat, monkey and human brains. Brain Res 1993, 628:85-92 [DOI] [PubMed] [Google Scholar]
- 21.Miyake T, Gahara Y, Nakayama M, Yamada H, Uwabe K, Kitamura T: Up-regulation of cystatin C my microglia in the rat facial nucleus following axotomy. Mol Brain Res 1996, 37:273-282 [DOI] [PubMed] [Google Scholar]
- 22.Cole T, Dickson PW, Esnard F, Averill S, Risbridger GP, Gouthiar F, Schreiber G: The CDNA structure and expression analysis of the genes for the cysteine proteinase inhibitor cystatin C and for β2-microglobulin in rat brain. Eur J Biochem 1989, 186:35-42 [DOI] [PubMed] [Google Scholar]
- 23.Levy E, Sastre M, Kumar A, Gallo G, Piccardo P, Ghetti B, Tagliavini F: Codeposition of cystatin C with amyloid-β protein in the brain of Alzheimer’s disease patients. J Neuropathol Exp Neurol 2001, 60:94-104 [DOI] [PubMed] [Google Scholar]
- 24.Arnold SE, Hyman BT, Flory J, Dainasio AR, Van Hoesen GW: The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in cerebral cortex of patients with Alzheimer’s disease. Cerebral Cortex 1991, 1:103-116 [DOI] [PubMed] [Google Scholar]
- 25.Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Burgztajn S, Lippa C, Nixon RA: Gene expression and cellular content of cathepsin D in Alzheimer’s disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron 1995, 14:671-680 [DOI] [PubMed] [Google Scholar]
- 26.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA: Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome. Am J Pathol 2000, 157:277-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA: Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci 1996, 16:186-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR: Intraneuronal Aβ42 accumulation in human brain. Am J Pathol 2000, 156:15-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michel T, Deng A, Growdon JH, Nitsch RM: Increased cystatin C in transgenic mice overexpressing Swedish APP. Soc Neurosci 2000, 26:495A [Google Scholar]
- 30.Palm DE, Knuckley NW, Primiano MJ, Spangenberger AG, Johanson CE: Cystatin C, a protease inhibitor, in degenerating rat hippocampal neurons following transient forebrain ischemia. Brain Res 1995, 691:1-8 [DOI] [PubMed] [Google Scholar]
- 31.Ishimaru H, Ishikawa K, Ohe Y, Takahashi A, Maruyama Y: Cystatin C and apolipoprotein E immunoreactivities in CAI neurons in ischemic gerbil hippocampus. Brain Res 1995, 709:155-162 [DOI] [PubMed] [Google Scholar]
- 32.Taupin P, Ray J, Fischer VM, Suhr ST, Hakansson K, Grubb A, Gage FH: FGF-2-responsive neural stem cell proliferation requires CCg, a novel autocrine/paracrine cofactor. Neuron 2000, 28:385-397 [DOI] [PubMed] [Google Scholar]