

Abstract
The α2β1 integrin supports cell-cycle progression of mammary epithelial cells adherent to type I collagen matrices. Integrin collagen receptors containing the α2 cytoplasmic domain stimulated expression of cyclin E and cyclin-dependent kinase (cdk)2, resulting in cyclin E/cdk2 activation in the absence of growth factors other than insulin. Integrin collagen receptors in which the α2 cytoplasmic domain was replaced by the α1 cytoplasmic domain or an α2 subunit cytoplasmic domain truncated after the GFFKR sequence failed to stimulate cyclin E/cdk2 activation or entry into S phase in the absence of growth factors. Although overexpression of cyclins D or E or cdk2 in cells expressing the integrin collagen receptor with the α1-integrin cytoplasmic domain did not restore G1 progression when mammary epithelial cells adhered to type I collagen, co-expression of cyclin E and cdk2 did rescue the ability of the transfectants to enter S phase. Activation of cyclin E/cdk2 complex by mammary epithelial cells required synergy between adhesion mediated by an integrin collagen receptor containing the α2-integrin subunit cytoplasmic domain and the insulin receptor.
Integrins are heterodimeric cell-surface receptors for extracellular matrix molecules and they play a vital role in regulating cell adhesion, spreading, migration, proliferation, survival, and differentiation. 1 Signals from both integrins and growth factor receptors act cooperatively to control many functions including migration, proliferation, and survival. 2 Integrin-mediated control of cell proliferation has been shown to occur at different points throughout the G1 phase of the cell cycle by regulating both the expression of cyclin D1 and E and the activity of the cyclin D/cyclin-dependent kinase (cdk)4/6 and cyclin E/cdk2 kinase complexes. 3-6 Therefore, integrins play a vital role in controlling proliferation by relaying signals regarding the composition of the extracellular matrix from the cell surface to the nucleus where the signals influence the cell-cycle machinery and control G1 progression.
Work from our laboratory has focused primarily on the α2β1 integrin, one of four integrins including α1β1, α2β1, α10β1, and α11β1 that have been shown to act as receptors for native collagens. 7-14 Studies by several groups including our own have shown that although the α1β1 and α2β1 integrins have similarities in ligand binding they mediate different functions and are not simply redundant adhesive receptors. 15-19 Re-expression of the α2β1 integrin in a tumorigenic cell line restored the ability of the cells to form glandular structures and diminished their malignant potential despite the fact that the cells expressed high levels of endogenous α1β1 integrin. 16 In primary endothelial cells plated on laminin matrices, the α1β1 integrin signals through Shc in a caveolin-dependent manner that requires the transmembrane domain of the α1-integrin subunit to regulate cell-cycle progression and survival, whereas ligand binding to the α2β1 integrin was associated with cell-cycle arrest and apoptosis. 20 In contrast, we have shown that the α2β1 integrin supports proliferation of mammary epithelial cells when plated on monolayer type I collagen or embedded in three-dimensional collagen gels. 17
To examine the different roles that the α1β1 and α2β1 integrins play in regulating cell phenotype, we evaluated primary mammary epithelial cells and an immortalized, but nontransformed, mammary epithelial cell model lacking endogenous α1β1 or α2β1 collagen receptors but expressing either the full-length human α2-integrin subunit cDNA (X2C2), a chimeric cDNA consisting of the α2 extracellular and transmembrane domains fused to the α1 cytoplasmic domain (X2C1), or a cDNA encoding the α2 subunit with a cytoplasmic domain truncation after the conserved GFFKR motif (X2C0). 17 Primary mammary epithelial cells, as well as the X2C2 and X2C1 transfectants effectively adhered, spread, formed focal adhesion complexes, and proliferated on type I collagen matrices. We now report that although both the X2C2- and the X2C1-expressing cells enter S phase in growth factor-enriched media, the cytoplasmic domain of the α2-integrin subunit uniquely supports S-phase entry in the presence of insulin alone. The α2, but not the α1, cytoplasmic domain mediates activation of the cyclin E/cdk2 complex and entry into S phase in the absence of growth factors other than insulin. These results indicate that the α2-integrin cytoplasmic domain and the insulin receptor synergize to regulate cell-cycle progression.
Materials and Methods
Antibodies and Reagents
Anti-α2-integrin subunit antibody (P1E6) was purchased from Life Technologies (Gaithersburg, MD). Monoclonal anti-cyclin D1 (R-124) and D3 (D-7), as well as polyclonal anti-cyclin D2 (M-20), E (M-20), cdk2 (M2), cdk4 (C-22), cdk6 (C-21), and actin (C-11)antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-bromodeoxyuridine (BrdU) antibody fluorescein isothiocyanate (B44) was purchased from Becton-Dickinson (Mountain View, CA). Monoclonal anti-α5- (5H10–27) and anti-α6- (G0H3) integrin subunit antibodies were purchased from Pharmingen (La Jolla, CA). Monoclonal anti-human cyclin E antibody (HE-12), hygromycin B, and geneticin were from Calbiochem (La Jolla, CA). Fibronectin, BrdU, and propidium iodide were from Sigma Chemical Co. (St. Louis, MO). Polyclonal anti-phospho-Akt and anti-Akt antibodies were obtained from New England Biolabs Inc (Beverly, MA). Type I collagen was from Collaborative Biomedical Products. Anti-α1-subunit antibody (FB12) was purchased from Chemicon International, Inc. (Temecula, CA).
Cell Culture and Transfection
Normal human mammary epithelial cells were obtained from Clonetics (San Diego, CA) and maintained in mammary epithelial basal medium (MEBM) according to the supplier’s instructions. The subclones of the murine NMuMG cell line were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum and insulin (5 μg/ml; Eli Lilly and Co., Indianapolis, IN). The transfected cell lines (X2C2, X2C1, X2C0, and control) were developed as previously described and maintained in DMEM with geneticin (850 μg/ml). 17 X2C1-expressing cells were co-transfected with human cyclin D1, D3, or E cDNA subcloned into pRC/CMV vector (Invitrogen, Carlsbad, CA) or murine cyclin D2 cDNA subcloned into pcDNA3.1 vector (Invitrogen, Carlsbad, CA) (gifts from Dr. D. Dean, Washington University, St. Louis, MO) or human cdk2 cDNA subcloned into pcDNA3.1 vector (a gift from Dr. J. Baldassare, St. Louis University, St. Louis, MO) along with the Selecta Vecta-Hyg cDNA construct (Novagen, Inc., Madison, WI) using Lipofectin (Life Technologies) per the manufacturer’s instructions. Nonclonal cell lines were selected in DMEM containing hygromycin (448 μg/ml) and geneticin (850 μg/ml). The expression levels of cyclin D1, D2, D3, E, or cdk2 were evaluated by immunoblot analysis.
Flow Cytometric Analysis
Flow cytometric analysis was performed on adherent cells harvested with 2 mmol/L ethylenediaminetetraacetic acid (EDTA) in phosphate-buffered saline (PBS), pH 7.5. Single cells (1 × 106) in PBS containing 1.5% horse serum were incubated with the appropriate monoclonal antibodies at either 5 μg/ml or at the saturating concentration recommended by the manufacturer for 45 minutes at 4°C. Cells were washed three times and incubated with 2 μg/ml of a secondary goat anti-mouse antibody coupled to fluorescein (Tago Inc., Camarillo, CA) for 45 minutes at 4°C, washed twice, and resuspended in PBS. Fluorescein-labeled cells were analyzed using a Coulter XL flow cytometer (Coulter Electronics, Hialeah, FL).
Adhesion Assay
Adhesion assays were performed as described previously. 21 Briefly, 2 × 10 4 cells were allowed to adhere to type I collagen (25 μg/ml) in the absence or presence of the indicated antibody at a concentration demonstrated by the manufacturer to block adhesion for 1 hour at 37°C. Nonadherent cells were removed by washing three times.
DNA Synthesis Assay
Transfected NMuMG cells were serum-starved in media containing 0.4% serum and insulin (5 μg/ml) for 48 hours and in DMEM without insulin or serum for 24 hours, removed from the flask with trypsin/EDTA, washed twice, and replated onto type I collagen (25 μg/ml)- or fibronectin (25 μg/ml)-coated dishes in media consisting of either MEBM containing bovine pituitary extract (13 mg/ml), hydrocortisone (0.5 μg/ml), recombinant human epidermal growth factor (10 ng/ml), and insulin (5 μg/ml) (serum-free growth factor-enriched media) or DMEM with or without insulin (5 μg/ml). After 22 hours of incubation BrdU (10 μmol/L final concentration) was added for the final 2 hours and the percentage of cells incorporating BrdU was determined, as described by Tlsty and colleagues. 22 Experiments revealed that the highest percentage of cells entered S phase between 22 and 24 hours after adhesion. No significant difference was seen by incubating the cells in BrdU for the entire 24 hours or for up to 30 hours. Normal human mammary epithelial cells were serum-starved for 72 hours in DMEM without insulin and treated as described above. After 36 hours in MEBM containing bovine pituitary extract (13 mg/ml), hydrocortisone (0.5 μg/ml), recombinant human epidermal growth factor (10 ng/ml), and insulin (5 μg/ml), BrdU was added for 12 hours and the percentage of cells incorporating BrdU was determined, as described by Tlsty and colleagues. 22 Experiments revealed that the highest percentage of cells entered S phase between 36 and 48 hours after adhesion.
Immunoblot Analysis
Cells were serum-starved as described above, plated onto type I collagen (25 μg/ml)- or fibronectin (25 μg/ml)-coated dishes and lysed after defined lengths of time in lysis buffer (50 mmol/L HEPES, pH 7.2, 250 mmol/L NaCl, 2 mmol/L EDTA, 0.1% Nonidet P-40, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 40 mmol/L NaF, 0.5 mmol/L phenylmethyl sulfonyl fluoride, 0.5 mmol/L o-vanadate, and 1 mmol/L dithiothreitol). Total protein concentration was determined by the Pierce protein assay (Fisher Scientific, Pittsburgh, PA). Equivalent amounts of protein lysate were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted onto Immobilon-P transfer membrane (Fisher Scientific). Immunoblots were incubated overnight with the appropriate dilution of primary antibody at 4°C followed by secondary horseradish peroxidase-conjugated sheep anti-mouse or anti-rabbit antibody (Amersham Pharmacia Biotech, Piscataway, NJ) for 1 hour at room temperature. Enhanced chemiluminescence system (Amersham Pharmacia Biotech) was used for visualization.
Kinase Assays
Cells were prepared as described above for the immunoblot analyses. One mg of cell lysate was precleared with rabbit anti-mouse IgG (Jackson Immunoresearch Laboratories, Inc., West Grove, PA) and immunoprecipitated with a polyclonal anti-cyclin E antibody and protein-A Sepharose beads such that equal amounts of cyclin E are immunoprecipitated (Sigma Chemical Co.). After washing, half of the beads were subjected to cyclin E/cdk2 kinase assays in kinase buffer (50 mmol/L Hepes, pH 7.5, 10 mmol/L MgCl2) with 2 μg histone H1 (Life Technologies, Inc.), 2 μmol/L ATP, 3 μCi γ-32P-ATP (ICN Biomedicals, Costa Mesa, CA). The samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis autoradiography and quantified by PhosphorImage analysis using ImageQuant (Molecular Dynamics, Sunnyvale, CA). Cyclin D/cdk4/6 kinase assays were performed by immunoprecipitating with polyclonal anti-cdk4 or cdk6 antibodies and protein-A Sepharose, as described above. The kinase reaction contained kinase buffer with 3 μg of Rb-GST fusion protein (Santa Cruz Biotechnology), 2 μmol/L ATP, 3 μCi γ-32P-ATP. The samples were analyzed and quantified as described above. The remaining 50% of the beads from the immunoprecipitation were subjected to immunoblot analysis for either cyclin E or cdk6.
Cdk2-TAT Transduction
Human cdk2 cDNA was subcloned into a TAT-HA expression vector to create a TAT-HA-cdk2 fusion protein. Overnight cultures were lysed by sonication in 20 mmol/L of Hepes, pH 7.2, 100 mmol/L NaCl, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 50 μg/ml phenylmethyl sulfonyl fluoride and purified on a Ni-NTA column (Qiagen, Inc., Valencia, CA). The eluted protein was desalted on a PD-10 column (Amersham Pharmacia Biotech) and the purity and concentration was evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by Coomassie blue staining. The cells were serum-starved as described above and removed from the flask with 0.05% EDTA, washed once in DMEM, incubated with 100 nmol/L TAT-cdk2 fusion protein for 5 minutes, and plated on type I collagen-coated dishes in DMEM plus insulin. The transduced cells were then analyzed for the incorporation of BrdU or for kinase activity, as described above.
Data Analysis
All results shown are from at least three independent experiments and data are presented as the mean ± SEM. Multiple clones of each clonal cell line were used in independent experiments. Statistical analysis and graphing was performed using GraphPad Prism V2.01 (San Diego, CA).
Results
Primary Mammary Epithelial Cells Enter S Phase on Type I Collagen
Normal human primary mammary epithelial cells express the α2 but low or undetectable levels of the α1-integrin subunit as shown in Figure 1A ▶ . These observations are consistent with previous studies from our laboratories. 23,24 To verify that normal human primary mammary epithelial cells adhere to type I collagen in an α2β1 integrin-dependent manner, adhesion assays were performed. The data in Figure 1B ▶ demonstrate that mammary epithelial cells adhere to type I collagen in a Mg2+-dependent manner. The cells failed to adhere to type I collagen in the presence of EDTA and failed to adhere to bovine serum albumin. Adhesion to type I collagen was abrogated by a function-blocking antibody against the α2-integrin subunit, but not by antibodies with specificities for the α5- or the α6-integrin subunits demonstrating that mammary epithelial cell adhesion to type I collagen is α2β1 integrin-dependent.
Figure 1.
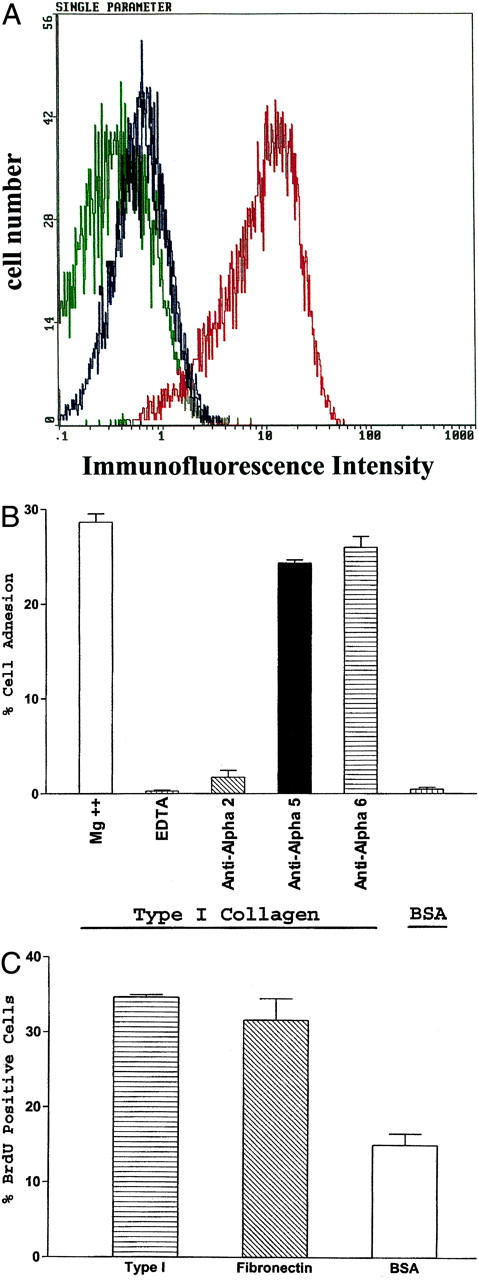
Primary mammary epithelial cell entry into S phase supported by adhesion to type I collagen. A: Expression of the α1 and α2 integrin subunits on the surface of primary mammary epithelial cells was evaluated by flow cytometric analysis (blue = anti-α1 antibody; red = anti-α2 antibody; green = isotype control antibody). B: Adhesion of primary mammary epithelial cells to type I collagen was Mg++-dependent and blocked by anti-α2-integrin subunit antibodies but not anti-α5 or -α6 integrin subunit antibodies. C: Entry into S phase by primary mammary epithelial cells was determined by BrdU incorporation 48 hours after plating on dishes coated with type I collagen (25 μg/ml), fibronectin (25 μg/ml), or bovine serum albumin (0.5%). The percentage of cells that incorporated BrdU during the final 12 hours of incubation was determined. Results shown represent the mean ± SEM for at least three separate experiments.
To determine whether α2β1 integrin-mediated adhesion to type I collagen by normal human primary mammary epithelial cells would support cell-cycle progression, the ability of human primary mammary epithelial cells to enter S phase when adherent to type I collagen was determined by bromodeoxyuridine (BrdU) incorporation. The cells were serum-starved for 72 hours and plated onto dishes coated with type I collagen (25 μg/ml), fibronectin (25 μg/ml), or bovine serum albumin in serum-free, growth factor-enriched media. After a 36-hour incubation, BrdU was added to the culture media and the incorporation of BrdU into the cellular DNA was analyzed. As shown in Figure 1C ▶ , human primary mammary epithelial cells entered S phase to similar extents when adherent to either type I collagen or fibronectin. In contrast, few primary mammary epithelial cells entered S phase when plated on bovine serum albumin-coated dishes. Therefore, the α2β1 integrin can provide the necessary signals to support cell-cycle progression of normal human primary mammary epithelial cells adherent to type I collagen. These results obtained with primary mammary epithelial cells are consistent with our previous studies of murine mammary epithelial cell lines in which the α2β1 integrin supported cellular proliferation. 17
α2 Subunit Cytoplasmic Domain Requirement for Cell-Cycle Progression
To identify the functional and structural elements required for α2β1-mediated cell-cycle progression, we performed a series of experiments using stably transfected cell lines expressing the full-length human α2-integrin subunit cDNA (X2C2), a chimeric cDNA consisting of the α2 extracellular and transmembrane domains fused to the α1 cytoplasmic domain (X2C1) or the cDNA including the α2 subunit with a cytoplasmic domain truncated after the conserved GFFKR motif (X2C0). Murine mammary epithelial cells stably expressing the X2C2 or X2C1 integrin α-subunit cDNA constructs with an intact α1 or α2 cytoplasmic domain, but not transfectants expressing either the X2C0 or control cDNA constructs, proliferated on type I collagen matrices in the presence of growth factors. 17 To determine the importance of the α-integrin cytoplasmic domain in cell-cycle progression, the ability of the X2C2, X2C1, X2C0, and control transfectants, as well as transfectants expressing the full-length α1-integrin subunit (X1C1), to enter S phase was measured by BrdU incorporation. Synchronized cells were plated on dishes coated with either type I collagen or fibronectin (at 25 μg/ml) in serum-free growth factor-enriched media. After a 22-hour incubation, BrdU was added to the culture media and the incorporation of BrdU into the cellular DNA was analyzed 2 hours later. These assay conditions were used because preliminary experiments determined that the maximum number of cells entered S phase during this time frame. As shown in Figure 2A ▶ , the X2C2, X2C1, and X1C1 transfectants entered S phase as determined by BrdU incorporation when plated on type I collagen in growth factor-enriched media, as expected. Also as expected from our earlier studies, the X2C0 and control transfectants failed to enter S phase, even in the presence of growth factors. Proliferation on fibronectin was also assayed as a control. All of the transfectants expressed high and comparable levels of endogenous α5β1 integrin and entered S phase to a similar extent when plated on fibronectin substrates in serum-free, growth factor-enriched media (data not shown). These results indicate that the presence of either the α1 or α2 cytoplasmic domain is sufficient to support G1 progression in mammary epithelial cells when adherent to type I collagen in a media rich in growth factors.
Figure 2.
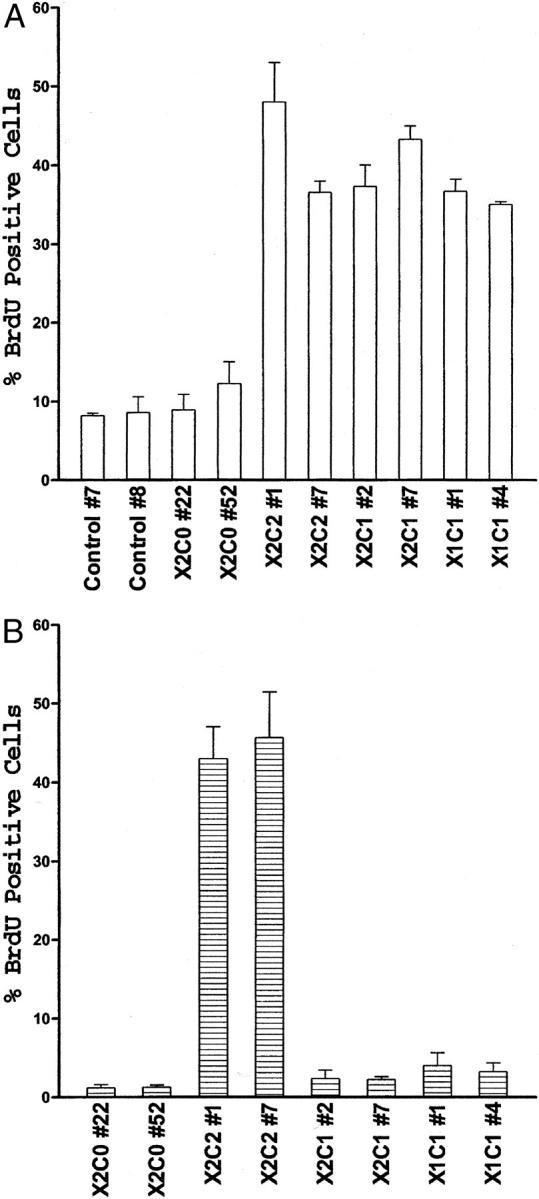
Entry into S phase supported by adhesion to type I collagen. A: Entry into S phase by clones of the X2C2, X2C1, X2C0, X1C1, and control transfectants was measured by BrdU incorporation 24 hours after plating the cells on type I collagen in serum-free MEBM containing recombinant human epidermal growth factor (10 ng/ml), bovine pituitary extract (13 mg/ml), insulin (5 μg/ml), and hydrocortisone (0.5 μg/ml). The percentage of cells that incorporated BrdU during the final 2 hours of incubation was determined. Results shown represent the mean ± SEM for at least three separate experiments. B: Entry into S phase by clones of the X2C0, X2C2, X2C1, and X1C1 transfectants was determined 24 hours after the serum-starved cells were plated on type I collagen in DMEM plus insulin (5 μg/ml). The percentage of cells that incorporated BrdU during the final 2 hours of incubation was determined. Results shown represent the mean ± SEM from at least three separate experiments.
Signals from growth factor receptors and integrins act synergistically to promote progression through G1 and into the S phase of the cell cycle. 2,25,26 We were interested in examining the role of the α integrin cytoplasmic domain in regulating G1 progression. Therefore, we removed the growth factors, with the exception of insulin, from the media and evaluated the ability of the transfectants to enter S phase. In contrast to the results obtained in the presence of growth factors shown above, only cells expressing the full-length α2-integrin subunit entered S phase in the absence of growth factors other than insulin (Figure 2B) ▶ . Cells expressing the full-length α1-integrin subunit (X1C1) or the chimeric integrin α chain (X2C1) were no longer capable of entering S phase. In the absence of insulin only 4.2 ± 1.4% and 0.7 ± 0.2% of the X2C2 and X2C1 transfectants, respectively, entered S phase. On fibronectin in the presence of insulin, none of the transfectants entered S phase to an appreciable extent (data not shown). These results demonstrated that the α2, but not the α1, integrin cytoplasmic domain could supply the necessary signals, in conjunction with those from the insulin receptor, to support G1 progression in mammary epithelial cells adherent to type I collagen in the presence of insulin.
Activation of the cyclin D/cdk4/6 and cyclin E/cdk2 kinase complexes is the crucial step in controlling progression through the G1 phase of the cell cycle. 27 As shown in Figure 3A ▶ , the activity of the cdk6/cyclin D kinase complex was similar among all of the transfected cell lines. However, the kinase activity of the cyclin E/cdk2 complex was more than threefold greater in cells expressing the X2C2 construct than in cells expressing either the X2C1 or X2C0 constructs (Figure 3B) ▶ . Thus, our data suggest that binding of the full-length α2β1 integrin to type I collagen regulates cell-cycle progression in mammary epithelial cells by controlling the activity of the cyclin E/cdk2 complex.
Figure 3.
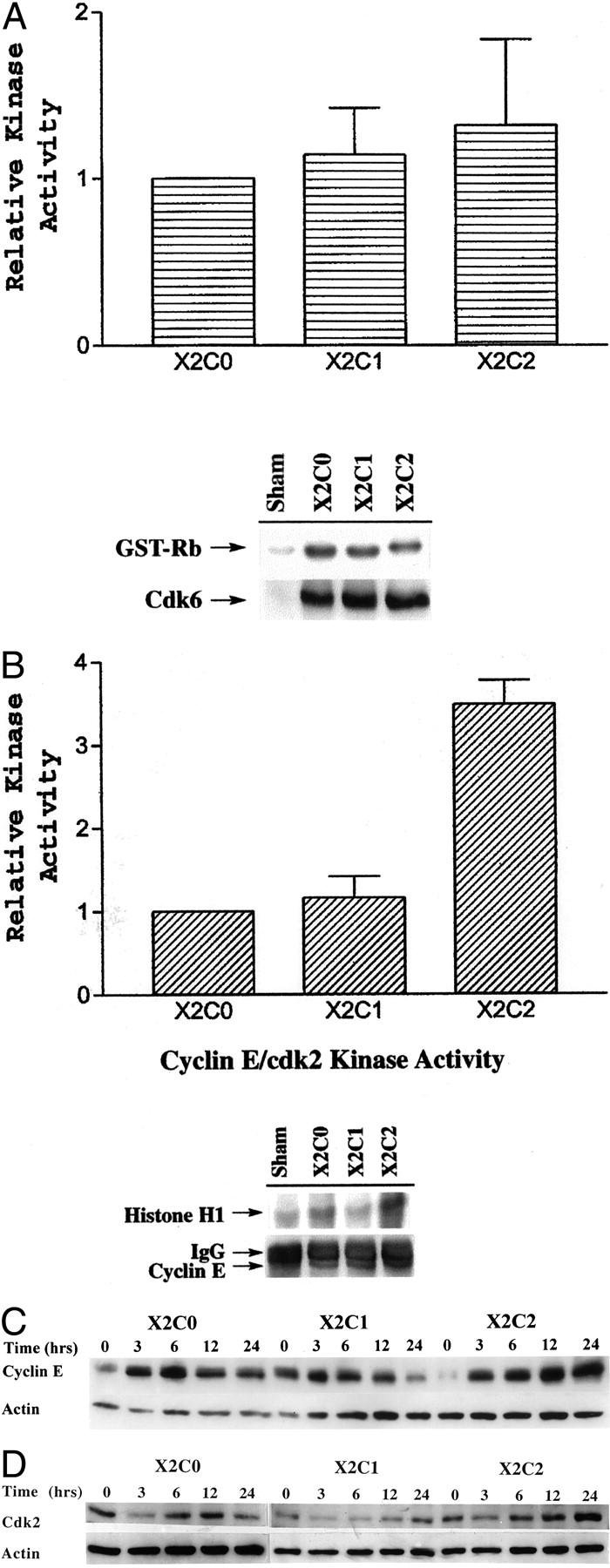
Regulation of cyclin E/cdk2 activity and cyclin E and cdk2 expression is dependent on the α2 cytoplasmic domain. A: Kinase assays were conducted on cyclin D/cdk4/6 complexes from the X2C2, X2C1, and X2C0 transfectants after serum starvation and adhesion to type I collagen for 20 hours. The cyclin D/cdk6 kinase activity of the X2C2 and X2C1 transfectants was normalized to the kinase activity of the X2C0 transfectants. The data represent the mean ± SEM of three separate experiments (top). A representative autoradiograph of the phosphorylated GST-Rb fusion protein, the substrate in the kinase reaction, is shown along with immunoblot showing levels of cdk6 immunoprecipitated for the kinase reaction (bottom). B: Kinase assays were performed on cyclin E/cdk2 kinase complexes from the X2C2, X2C1, and X2C0 transfectants after serum starvation and adhesion to type I collagen for 20 hours, as described above. The cyclin E/cdk2 kinase activity of the X2C2 and X2C1 transfectants was normalized to the kinase activity of the X2C0 transfectants. The data represent the mean ± SEM of three separate experiments (top). A representative autoradiograph of the phosphorylated histone-H1 protein, the substrate in the kinase reaction, is shown along with an immunoblot showing the levels of cyclin E immunoprecipitated for the kinase reaction (bottom). C: The levels of cyclin E and actin expressed by the X2C2, X2C1, and X2C0 transfectants after adhesion to type I collagen for 0, 3, 6, 12, and 24 hours in DMEM plus insulin were determined by immunoblot analysis. The results shown are representative of at least three separate experiments. D: The levels of cdk2 and actin expressed by the X2C2, X2C1, and X2C0 transfectants after adhesion to type I collagen for 0, 3, 6, 12, and 24 hours in DMEM plus insulin were determined by immunoblot analysis. The results shown are representative of at least three separate experiments.
One possible mechanism to explain the increase in cyclin E/cdk2 activity is an increase in the level of cyclin E and/or cdk2. To investigate this possibility the levels of cyclin E and cdk2 in the X2C0, X2C1, and X2C2 transfectants after adhesion to type I collagen were assessed by immunoblot analysis. The X2C0 and X2C1 transfectants transiently increased cyclin E expression 3 to 6 hours after adhesion to type I collagen in the presence of insulin (Figure 3C) ▶ . However, cyclin E protein levels increased 3 to 6 hours after adhesion to type I collagen and remained elevated between 12 and 24 hours only in the X2C2-expressing cells that had been plated on type I collagen. The increase in cyclin E by the X2C2 transfectants correlated with the ability of the cells to enter S phase. As shown in Figure 3D ▶ , cdk2 protein levels increased in a time-dependent manner and peaked 24 hours after plating the X2C2-transfected cells on type I collagen. The time course of cdk2 induction was similar to that of cyclin E. Cdk2 levels did not increase in the X2C1- or X2C0-transfected cells plated on type I collagen. Thus, these results suggest that the α2, but not the α1, integrin cytoplasmic domain provides the signals necessary to both increase the cyclin E and cdk2 protein levels and activate the cyclin E/cdk2 complex in mammary epithelial cells adherent to type I collagen in the presence of insulin.
Primary Mammary Epithelial Cell Expression of Cyclin E and Cdk2 during Cell-Cycle Progression
To validate the results obtained with the transfected cell lines, we assessed the levels of cell-cycle regulatory proteins expressed by the primary human mammary epithelial cells when entering the cell cycle. Primary mammary epithelial cells were serum-starved for 72 hours in DMEM and then plated onto dishes coated with type I collagen in growth factor-enriched media and lysed at various times after plating. As shown in Figure 4A ▶ , the expression of cyclin E increased within 6 hours after adhesion to type I collagen and exposure of the primary mammary epithelial cells to growth factors. Robust expression of cyclin E was observed for 48 hours (Figure 4A) ▶ . Expression of cdk2 increased gradually after adhesion to type I collagen and peaked at 48 hours (Figure 4B) ▶ . These findings are consistent with the results of studies performed with the X2C2-transfected cell line. Primary mammary epithelial cells up-regulated the expression of cyclin E and cdk2 during progression through G1 when adherent to type I collagen.
Figure 4.

Cyclin E and cdk2 levels in primary mammary epithelial cells adherent to type I collagen. Primary mammary epithelial cells were serum-starved for 72 hours, plated on type I collagen (25 μg/ml) in serum-free, growth factor-enriched media and lysed at the indicated time points. The levels of cyclin E, cdk2, and actin expression were evaluated by immunoblot analysis. The results shown are representative of at least three separate experiments.
Requirement for Co-Expression of Cyclin E and Cdk2 for X2C1 Cell-Cycle Progression
If increased expression of cyclin E and cdk2 by the X2C2-transfected cells is important for G1 progression, we reasoned that overexpression of cyclin E or cdk2, but not cyclins D1, D2, or D3 would be sufficient to overcome the cell-cycle block in the X2C1-expressing cells. However, the results shown in Figure 5 ▶ demonstrated that constitutive overexpression in stable cell lines of either cyclin D1, D2, D3, or E or cdk2 alone was unable to stimulate robust G1 progression in the X2C1-transfected cells. Overexpression of cyclin D1 in the X2C1 transfectants had a modest effect on the ability of the cells to enter S phase.
Figure 5.

Overexpression of G1 cyclins or cdk2 failed to rescue S-phase entry by the X2C1 transfectants. a: The ability of cyclin D1, D2, D3, E, or cdk2 overexpression to stimulate entry into S phase was evaluated by determining the percentage of X2C2 and X2C1 transfectants and the percentage of X2C1-cyclin D1, X2C1-cyclin D2, X2C1-cyclin D3, X2C1-cyclin E, and X2C1-cdk2 co-transfectants that incorporated BrdU during the final 2 hours of a 24-hour incubation on type I collagen in DMEM plus insulin. b: The level of cyclin D1, D2, D3, E, or cdk2 protein expressed by the X2C1-cyclin D1, D2, D3, E, or cdk2 co-transfectants was compared to the level of endogenous protein expressed by the X2C2 transfectants plated on type I collagen for 24 hours.
To co-express both cyclin E and cdk2 in the X2C1 transfectants we transduced cdk2 into the X2C1-cyclin E co-transfectants using a TAT-cdk2 fusion protein. The TAT-fusion protein technology is a rapid means to efficiently introduce a protein into the majority of targeted cells. 28 Using a fusion protein consisting of a portion of the HIV-TAT protein spliced to the amino terminus of the human cdk2 protein we were able to transduce the human cdk2 protein into the transfectants and to reconstitute an active kinase complex. Figure 6A ▶ demonstrates that an active cyclin E/cdk2 kinase complex was immunoprecipitated from cells overexpressing cyclin E transduced with the TAT-cdk2 fusion protein. Formation of an active kinase complex required the presence of both cyclin E and the TAT-cdk2 fusion protein because the X2C1-cyclin E-expressing cells exhibited low levels of cyclin E-associated kinase activity in the absence of the TAT-cdk2 fusion protein and the TAT-cdk2 fusion protein exhibited substantially less kinase activity in the absence of cyclin E (Figure 6A ▶ , and data not shown). In addition, formation of the active kinase complex was sufficient to support G1 progression in the X2C1-cyclin E-expressing cells, as shown in Figure 6B ▶ . The TAT-cdk2 fusion protein had no effect on the ability of the X2C1 transfectants lacking cyclin E to enter S phase (X2C1 plus cdk2) (Figure 6B) ▶ . Expression of both proteins in these cells did not result in integrin-independent kinase activation or G1 progression because similar experiments on polylysine-coated dishes did not result in formation of an active kinase complex or entry into S phase (data not shown). These data demonstrate that the α2 cytoplasmic domain controls entry into S phase by regulating the activity of the cyclin E/cdk2 kinase complex by inducing the expression of both proteins. The G1 regulatory point can be overcome in the X2C1 transfectants by expressing both cyclin E and cdk2.
Figure 6.

Simultaneous expression of cyclin E and cdk2 by the X2C1 transfectants rescued entry into S phase. A: Cyclin E/cdk2 kinase activity of the X2C2 and cyclin E and X2C1 and cyclin E transfectants, in the presence and absence of the TAT-cdk2 fusion protein was determined after adhesion to type I collagen. Cyclin E/cdk2 kinase assays were performed on cyclin E immunoprecipitates from serum-starved cells after adhesion to type I collagen for 20 hours. The X2C2 and cyclin E transfectants with (+) or without (−) the TAT-cdk2 fusion protein and the X2C1 and cyclin E transfectants transduced with the TAT-cdk2 fusion protein formed active kinase complexes. B: The TAT-cdk2 fusion protein stimulated entry into S phase when expressed in X2C1 and cyclin E co-transfectants (X2C1 and E and cdk2) but not in the X2C1 transfectants (X2C1 and cdk2) adherent to type I collagen. The percentage of cells incorporating BrdU was measured during the final 2 hours of a 24-hour incubation. Neither cyclin E overexpression alone nor the TAT-cdk2 fusion protein alone was sufficient to promote entry into S phase in the X2C1-expressing cells (*, P > 0.05). However, expression of both cyclin E and TAT-cdk2 in the X2C1 transfectants was sufficient to mediate progression through G1 when the cells were adherent to type I collagen (++, P < 0.0001).
Integrity of Insulin Receptor Signaling in X2C1 Transfectants
One possible confounding explanation for the inability of the X2C1 transfectants to progress through G1 in media containing insulin, but not other growth factors, is that signaling through the insulin receptor is reduced in the X2C1 transfectants relative to the X2C2 transfectants. Previous studies have shown that EGF-mediated activation of the MAPK-ERK pathway is defective in the absence of adhesion. 29 Therefore, we considered the possibility that insulin receptor signaling was impaired in the X2C1 transfectants. The ability of the X2C2 and X2C1 transfectants to phosphorylate Akt/PKB in the presence and absence of insulin was determined. As shown in Figure 7 ▶ , adhesion to type I collagen by both the X2C1 and X2C2 transfectants resulted in low levels of Akt/PKB phosphorylation in the absence of insulin. Insulin markedly and rapidly stimulated phosphorylation of Akt/PKB in both the X2C1 and X2C2 transfectants, demonstrating that this signaling pathway downstream of the insulin receptor was intact in both cell lines. 30 Therefore, it seems that a combination of signals from both the α2 cytoplasmic domain and the insulin receptor are responsible for inducing the expression of both cyclin E and cdk2 and regulating progression through G1 in the X2C2 transfectants.
Figure 7.

Intact insulin receptor signaling. X2C2 and X2C1 transfectants were serum-starved and plated on collagen type I-coated dishes in DMEM with or without insulin. The cells were lysed at the indicated time points and the level of Akt/PKB phosphorylation and expression was determined by immunoblot analysis with antibodies against phosphorylated and total Akt/PKB.
Discussion
The findings reported herein establish novel and critical roles for integrin collagen receptor α-chain cytoplasmic domains in supporting mammary epithelial cell proliferation. These studies support a role for the α2β1 integrin in mediating cell-cycle progression by mammary epithelial cells adherent to type I collagen. When mammary epithelial cells are adherent to collagen via either the α1β1 or α2β1 integrin in mitogen-enriched media the presence of an α cytoplasmic domain is sufficient to support entry into S phase. Adhesion to type I collagen via an integrin lacking an α subunit cytoplasmic domain is insufficient to support entry into S phase even in the presence of growth factors. Furthermore, the full-length α2-integrin collagen receptor subunit with the α2 cytoplasmic domain, but neither the chimeric α2-integrin containing the α1 cytoplasmic domain nor the full-length α1-integrin subunit can support G1 progression in media limited in growth factors, ie, in the presence of insulin. Thus, our results also demonstrate for the first time that the α2 cytoplasmic domain plays a unique and critical role in supporting G1 progression in mammary epithelial cells in the absence of growth factors other than insulin.
Previous studies have demonstrated that signals from the α1β1 integrin regulate cell proliferation in a caveolin-dependent manner by signaling through Fyn and Shc whereas the α2β1 integrin is incapable of activating Shc. 20,31 In unpublished studies, we have confirmed the results of Wary and colleagues. 20 Transfectants expressing the α1β1 integrin (X1C1) activated Shc whereas those expressing the X2C2, X2C1, or X2C0 constructs were unable to do so. These findings are consistent with the ability of the α1 subunit to interact through its transmembrane, but not cytoplasmic, domain with the caveolin/Shc signaling complex. 20,31 In contrast, the results reported here show that regulation of G1 progression by the α2β1 integrin in mammary epithelial cells required the cytoplasmic domain of the α2 subunit. Additionally, studies in myoblasts have determined that the α5 cytoplasmic domain was permissive for proliferation whereas the α6A cytoplasmic domain inhibited proliferation. 32,33 Those findings in combination with our results provide strong evidence that certain integrin α subunit cytoplasmic domains can exert a positive influence on cellular proliferation whereas others cannot. In addition, our findings support the observation that the α1-integrin subunit requires the transmembrane domain for effector functions whereas the α2-integrin subunit requires the cytoplasmic domain. The results of cell proliferation using mammary epithelial cells adherent to type I collagen differ significantly from those of fibroblasts. α1 null fibroblasts, when adherent to type I collagen via the α2β1 integrin, fail to proliferate even in 2% serum. 34 The difference may be because of differences in regulation of the cell-cycle machinery in distinct cell types, as discussed below, or to differing levels of integrin expression. The mammary epithelial cells used in this study express high levels of the α2β1 integrin. In contrast, mouse embryo fibroblasts express only low levels of the α2β1 integrin (data not shown).
Signals from both the extracellular matrix and growth factors are required for cells to progress through the G1 phase of the cell cycle. The pathways leading from integrins and growth factor receptors converge at several points to stimulate progression through G1. 26,35,36 For example, growth factors and adhesion have been shown to act cooperatively to stimulate the expression of cyclin D1 in fibroblasts. 3,4,37 However, this is not the case for primary mammary epithelial cells or mammary epithelial cell lines. Primary mammary epithelial cells and the transfected mammary epithelial cells used in our study constitutively express cyclin D1 in the presence or absence of growth factors and/or integrin-dependent adhesion (data not shown). Others have reported that forced expression of cyclin D1 is sufficient to induce anchorage-independent entry into S phase in 3T3 cells. 4,38 However, we find that in NMuMG cells exogenous overexpression of cyclin D1 (or the other D-type cyclins) is not sufficient to induce anchorage-independent progression through G1. This point is illustrated by the inability of the X2C1 transfectants overexpressing cyclin D1, D2, or D3 to enter S phase when plated on type I collagen (Figure 5) ▶ . Because cyclin D/cdk4/6 complex activity was not increased in an α cytoplasmic domain-dependent manner, it is possible that cyclin D/cdk4/6 activity could be dependent on signals from the β-integrin cytoplasmic domain, and therefore activated in all of the transfectants, whereas signals from the α cytoplasmic domain may regulate cyclin E and cdk2 expression in mammary epithelial cells.
Adhesion to type I collagen by the X2C2 transfectants led to increased expression of cyclin E and cdk2 in addition to activation of the cyclin E/cdk2 complex. The ability of the α2-integrin cytoplasmic domain to mediate increased expression of both cyclin E and cdk2 seems to be the mechanism whereby the α2 cytoplasmic domain controls G1 progression in these cells. We also found that the levels of cyclin E and cdk2 increased in a time-dependent manner when normal human mammary epithelial cells progressed through G1 and into the S phase of the cell cycle. Generally, as cells progress through the cell cycle the levels of the cdks remain constant and only the levels of the cyclins fluctuate. However, Gong and colleagues 39 also found that the levels of cyclin E and cdk2 increased as colonic epithelial cells progressed through the cell cycle. Increased cdk2 levels have also been observed in ovarian and epidermal cells, 40,41 suggesting that the up-regulation of cdk2 maybe an under-appreciated method of regulating G1 progression in epithelial cells. Romanou and colleagues 42 recently reported that normal mammary epithelial changes do not undergo the same process of senescence as primary human fibroblasts suggesting that the cell-cycle regulatory mechanisms are not equivalent in epithelial cells and fibroblasts. This may also contribute to the differences observed between our studies of mammary epithelial cells and to those of α1 null fibroblasts.
Regulation of the cyclin E/cdk2 complex is dependent not only on the expression levels of its constituents, but also on expression of members of the p21 family of cdk inhibitors whose expression is regulated by adhesion to the extracellular matrix. 4-6 Koyama and colleagues found that arterial smooth muscle cells proliferate on monomeric type I collagen while they growth arrest on polymerized type I collagen due, in part, to increased levels of p21 and p27. Similarly, we found that mammary epithelial cells proliferated on monomeric type I collagen in an α2 cytoplasmic domain-dependent manner. However, in preliminary studies we found that p27 levels are decreased after adhesion to type I collagen irrespective of which α subunit cytoplasmic domain is present and that p21 levels remained low in all three transfectants (data not shown). Thus, a decrease in p27 or p21 levels cannot explain the ability of the X2C2, but not the X2C1, transfectants to enter S phase on type I collagen in the presence of insulin. Rather, progression through G1 may require the combination of a decrease in p27 levels and an increase in cyclin E and cdk2 levels in mammary epithelial cells adherent to type I collagen, as evidenced by the ability of both the X2C2 transfectants and the X2C1-cyclin E co-transfectants transduced with cdk2 to enter S phase (Figure 6) ▶ .
Overexpression of each of the G1 phase cyclins or cdk2 alone was insufficient to overcome the G1 block exhibited by the X2C1 transfectants after adhesion to type I collagen. However, co-expression of cyclin E and a TAT-cdk2 fusion protein was able to form an active cyclin E/cdk2 complex and to promote G1 progression in the X2C1 transfectants adherent to type I collagen (Figure 6) ▶ . The ability of cyclin E and the TAT-cdk2 fusion protein to form an active kinase complex in both the X2C2 and X2C1 transfectants suggests that the activity of the cyclin-activating kinase that activates the complex is either constitutively active, activated by adhesion, activated by insulin, or activated by some other mechanism during G1. Our results strongly suggest that the α2 cytoplasmic domain regulates G1 progression by increasing the levels of both cyclin E and cdk2 that partner to form an active kinase complex and initiate entry into S phase in mammary epithelial cells adherent to type I collagen.
In summary, we have shown that the presence of an α integrin cytoplasmic tail is necessary to support entry into S phase in mammary epithelial cells adherent to type I collagen via the α2β1 integrin in mitogen-enriched media. In contrast, the α2, but not the α1, integrin cytoplasmic domain can support entry into S phase after adhesion to type I collagen in a mitogen-poor medium containing only insulin. Our results illustrate that as the mammary epithelial cells progress through G1 phase and into S phase signals from the α2-integrin cytoplasmic domain are required to induce sustained expression of cyclin E as well as increased expression of cdk2. Increased expression of cyclin E and cdk2 is sufficient to support entry into S phase in murine mammary epithelial cells adherent to type I collagen in the presence of insulin.
It is likely that the findings of this study account for the association of high-level expression of the α2β1 integrin with the orderly, regulated proliferation of epithelial cells observed in our early studies. 43 As the α2β1 integrin recognizes similar, if not identified determinants, on multiple collagen types our findings should also be applicable to α2β1 integrin-mediated adhesive interactions with other collagens. In this regard, it is noteworthy that the regulatory mechanism we describe does not require the formation of collagen fibrils. Poorly differentiated adenocarcinomas of the breast that are typically characterized by very low or absent α2β1 integrin expression 44 have escaped and circumvented the α2β1 integrin-dependent regulation of cell-cycle progression. Future studies will evaluate the signaling pathway downstream of the α2-integrin cytoplasmic domain that is activated after adhesion to collagens that is necessary to up-regulate cyclin E and cdk2 levels.
Acknowledgments
We thank Bruce Linders, George Li, and Glenda White for expert technical assistance; Dr. Douglas Dean for providing constructs and advice; and Mary Beth Flynn for expert secretarial assistance.
Footnotes
Address reprint requests to Mary M. Zutter, Washington University School of Medicine, Department of Pathology & Immunology, 660 S. Euclid, Campus Box 8118, St. Louis, MO 63110.
Supported in part by National Institute of Health grants CA70275, CA83690, and HL63446.
References
- 1.Clark EA, Brugge JS: Integrins and signal transduction pathways: the road taken. Science 1995, 268:233-239 [DOI] [PubMed] [Google Scholar]
- 2.Giancotti FG, Ruoslahti E: Integrin signaling. Science 1999, 285:1028-1032 [DOI] [PubMed] [Google Scholar]
- 3.Böhmer RM, Scharf E, Assoian RK: Cytoskeletal integrity is required throughout the mitogen stimulation phase of the cell cycle and mediates the anchorage-dependent expression of cyclin D1. Mol Biol Cell 1996, 7:101-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X, Ohtsubo M, Böhmer RM, Roberts JM, Assoian RK: Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2, and phosphorylation of the retinoblastoma protein. J Cell Biol 1996, 133:391-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R: Fibrillar collagen inhibits arterial smooth muscle proliferation through regulation of Cdk2 inhibitors. Cell 1996, 87:1069-1078 [DOI] [PubMed] [Google Scholar]
- 6.Fang F, Orend G, Watanabe N, Hunter T, Ruoslahti E: Dependence of cyclin E-CDK2 kinase activity on cell anchorage. Science 1996, 271:499-502 [DOI] [PubMed] [Google Scholar]
- 7.Santoro SA: Identification of a 160,000 dalton platelet membrane protein that mediates the initial divalent cation-dependent adhesion of platelets to collagen. Cell 1986, 46:913-920 [DOI] [PubMed] [Google Scholar]
- 8.Staatz WD, Rajpara SM, Wayner EA, Carter WG, Santoro SA: The membrane glycoprotein Ia-IIa (VLA-2) complex mediates the Mg++-dependent adhesion of platelets to collagen. J Cell Biol 1989, 108:1917-1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staatz WD, Fok KF, Zutter MM, Adams SP, Rodriguez BA, Santoro SA: Identification of a tetrapeptide recognition sequence for the alpha 2 beta 1 integrin in collagen. J Biol Chem 1991, 266:7363-7367 [PubMed] [Google Scholar]
- 10.Staatz WD, Walsh JJ, Pexton T, Santoro SA: The alpha 2 beta 1 integrin cell surface collagen receptor binds to the alpha 1 (I)-CB3 peptide of collagen. J Biol Chem 1990, 265:4778-4781 [PubMed] [Google Scholar]
- 11.Hall DE, Reichardt LF, Crowley E, Holley B, Moezzi H, Sonnenberg A, Damsky CH: The alpha 1/beta 1 and alpha 6/beta 1 integrin heterodimers mediate cell attachment to distinct sites on laminin. J Cell Biol 1990, 110:2175-2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ignatius MJ, Large TH, Houde M, Tawil JW, Barton A, Esch F, Carbonetto S, Reichardt LF: Molecular cloning of the rat integrin alpha 1-subunit: a receptor for laminin and collagen. J Cell Biol 1990, 111:709-720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camper L, Hellman U, Lundgren-Akerlund E: Isolation, cloning, and sequence analysis of the integrin subunit alpha10, a beta1-associated collagen binding integrin expressed on chondrocytes. J Biol Chem 1998, 273:20383-20389 [DOI] [PubMed] [Google Scholar]
- 14.Velling T, Kusche-Gullberg M, Sejersen T, Gullberg D: cDNA cloning and chromosomal localization of human alpha(11) integrin. A collagen-binding, I domain-containing, beta(1)-associated integrin alpha-chain present in muscle tissues. J Biol Chem 1999, 274:25735-25742 [DOI] [PubMed] [Google Scholar]
- 15.Langholz O, Rockel D, Mauch C, Kozlowska E, Bank I, Krieg T, Eckes B: Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by alpha 1 beta 1 and alpha 2 beta 1 integrins. J Cell Biol 1995, 131:1903-1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zutter MM, Santoro SA, Staatz WD, Tsung YL: Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci USA 1995, 92:7411-7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zutter MM, Santoro SA, Wu JE, Wakatsuki T, Dickeson SK, Elson EL: Collagen receptor control of epithelial morphogenesis and cell cycle progression. Am J Pathol 1999, 155:927-940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riikonen T, Westermarck J, Koivisto L, Broberg A, Kahari VM, Heino J: Integrin alpha 2 beta 1 is a positive regulator of collagenase (MMP-1) and collagen alpha 1(I) gene expression. J Biol Chem 1995, 270:13548-13552 [DOI] [PubMed] [Google Scholar]
- 19.Ivaska J, Reunanen H, Westermarck J, Koivisto L, Kahari VM, Heino J: Integrin alpha2beta1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the alpha2 cytoplasmic tail. J Cell Biol 1999, 147:401-416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG: The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 1996, 87:733-743 [DOI] [PubMed] [Google Scholar]
- 21.Santoro SA, Zutter MM, Staatz WD, Saelman EUM, Keely PJ: Methods Enzymol 1995, 245:147–183 [DOI] [PubMed]
- 22.Tlsty T, Briot A, Poulose B: Analysis of cell cycle checkpoint status in mammalian cells. Methods Enzymol 1995, 254:125-133 [DOI] [PubMed] [Google Scholar]
- 23.Keely PJ, Fong AM, Zutter MM, Santoro SA: Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense alpha 2 integrin mRNA in mammary cells. J Cell Sci 1995, 108:595-607 [DOI] [PubMed] [Google Scholar]
- 24.Zutter MM, Santoro SA: Widespread histologic distribution of the alpha 2 beta 1 integrin cell-surface collagen receptor. Am J Pathol 1990, 137:113-120 [PMC free article] [PubMed] [Google Scholar]
- 25.Bottazzi ME, Assoian RK: The extracellular matrix and mitogenic growth factors control G1 phase cyclin and cyclin-dependent kinase inhibitors. Trends Cell Biol 1997, 7:348-352 [DOI] [PubMed] [Google Scholar]
- 26.Howe A, Aplin AE, Alahari SK, Juliano RL: Integrin signaling and cell growth control. Curr Opin Cell Biol 1998, 10:220-231 [DOI] [PubMed] [Google Scholar]
- 27.Roberts JM: Evolving ideas about cyclins. Cell 1999, 98:129-132 [DOI] [PubMed] [Google Scholar]
- 28.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF: Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med 1998, 12:1449-1452 [DOI] [PubMed] [Google Scholar]
- 29.Aplin AE, Short SM, Juliano RL: Anchorage-dependent regulation of the mitogen-activated protein kinase cascade by growth factors is supported by a variety of integrin alpha chains. J Biol Chem 1999, 274:31223-31228 [DOI] [PubMed] [Google Scholar]
- 30.Burgering BM, Coffer PJ: Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376:599-602 [DOI] [PubMed] [Google Scholar]
- 31.Wary KK, Mariotti A, Zurzolo C, Giancotti FG: A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 1998, 94:625-634 [DOI] [PubMed] [Google Scholar]
- 32.Sastry SK, Lakonishok M, Thomas DA, Muschler J, Horwitz AF: Integrin alpha subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J Cell Biol 1996, 133:169-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sastry SK, Lakonishok M, Wu S, Truong TQ, Huttenlocher A, Turner CE, Horwitz AF: Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J Cell Biol 1998, 142:587-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pozzi A, Wary KK, Giancotti F, Gardner H: Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol 1998, :587-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Assoian RK: Anchorage-dependent cell cycle progression. J Cell Biol 1997, 136:1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz MA: Integrins, oncogenes, and anchorage independence. J Cell Biol 1997, 139:575-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roovers K, Davey G, Zhu X, Bottazzi ME, Assoian RK: Alpha5beta1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol Biol Cell 1999, 10:3197-3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shulze A, Zerfass-Thome K, Berges J, Middendorp S, Jansen-Durr P, Henglein B: Anchorage-dependent transcription of the cyclin A gene. Mol Cell Biol 1996, 16:4632-4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong J, Ko TC, Brattain MG: Disruption of fibronectin binding to the alpha 5 beta 1 integrin stimulates the expression of cyclin-dependent kinases and DNA synthesis through activation of extracellular signal-regulated kinase. J Biol Chem 1998, 273:1662-1669 [DOI] [PubMed] [Google Scholar]
- 40.Marone M, Scambia G, Giannitelli C, Ferrandina G, Masciullo V, Bellacosa A, Benedetti-Panici P, Mancuso S: Analysis of cyclin E and CDK2 in ovarian cancer: gene amplification and RNA overexpression. Int J Cancer 1998, 75:34-39 [DOI] [PubMed] [Google Scholar]
- 41.Balasubramanian S, Ahmad N, Jeedigunta S, Mukhtar H: Alterations in cell cycle regulation in mouse skin tumors. Biochem Biophys Res Commun 1998, 243:744-748 [DOI] [PubMed] [Google Scholar]
- 42.Romanou SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD: Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001, 409:633-637 [DOI] [PubMed] [Google Scholar]
- 43.Zutter MM, Santoro SA: Widespread histologic distribution of the α2β1 integrin cell-surface collagen receptor. Am J Pathol 1990, 137:113-120 [PMC free article] [PubMed] [Google Scholar]
- 44.Zutter MM, Mazoujian G, Santoro SA: Decreased expression of integrin adhesive protein receptors in adenocarcinoma of the breast. Am J Pathol 1990, 137:863-870 [PMC free article] [PubMed] [Google Scholar]