

Abstract
Chemokine production by tumors is a well-known phenomenon, but its role in tumor biology remains debatable. Although intratumoral injection of granulocyte chemotactic protein-2 (GCP-2) had no effect on tumor parameters, needle-free stable expression of the chemokine resulted in enhanced tumor growth. It is shown here that tumors that express a potent form of GCP-2 induce a strong influx and activation of tumor-associated neutrophils. The production of GCP-2 leads to intratumoral expression of gelatinase B and advantage for tumor growth by increased angiogenesis. These results are in line with the countercurrent principle of chemokine action and support the notion that paraneoplastic expression of ELR-positive CXC chemokines has to be blocked rather than stimulated in cancer therapy.
Chemokines have been associated with tumor biology for a long time. In fact, many chemokines have first been purified from tumor cells. 1-5 The production of chemokines by tumor cells may be a paraneoplastic coincidence and either help the host to eliminate the tumor by immunosurveillance mechanisms 6 or give an advantage to cancer progression. The latter is called the countercurrent principle and includes various biological effects by which the chemoattracted cells may help the tumor. 7 The advantages of chemokine production for the tumor are multiple. 8 First, tumor cell invasion and metastasis may be enhanced because the chemoattracted cells may degrade basement membranes and connective tissues when they arrive in the tumor from the nearest vessels. Thus chemokines may pave the way for invasion and metastasis. Second, the chemoattracted cells may provide trophic factors to the environment to enhance the tumor growth. Third, specific chemokines are angiogenic and not only may help to establish a better oxygen and nutrient supply for tumor growth, but they may also bring the blood or lymph vessels closer to the tumor. 9
Recently, the chemokines have been classified on the basis of structural elements in two large cytokine families, the CC or CXC ligands (CCLs or CXCLs), and two smaller groups (XCLs and CX3CL1). Accordingly, the receptors for these molecules are abbreviated 10 as CCRs, CXCRs, XCRs, and CX3CR1. Further to the gross dichotomy, in which CXCLs mainly attract polymorphonuclear cells, whereas CCLs chemoattract predominantly mononuclear cells, additional branching in the CXCL family of molecules is based on the presence or absence of the ELR motif. This three amino-acid motif, which is in front of the CXC, is critical for the granulocyte chemotactic activity 11-13 and is also a crucial feature for the CXCR2 recognition and the angiogenic activity of the chemokines. 9,14,15 In general, ELR+ CXC chemokines 16-18 are angiogenic, whereas ELR− CXC ligands are angiostatic. 14,15 Prototypic members of the ELR+ CXC chemokines in the human species are interleukin-8 (IL-8), 4, 13 and granulocyte chemotactic protein-2 (GCP-2). 16 Whereas IL-8 does not exist in the mouse, a homologue of GCP-2 has been identified as one of the most potent murine neutrophil attracting chemokines. 17,18 To evaluate the countercurrent principle in vivo, we studied the effects of intratumoral injection of GCP-2 and, using gene transfer technology, the needle-free GCP-2 supplied by the tumor itself in a xenotransplant model.
Materials and Methods
Preparation and Purification of Recombinant Murine GCP-2
Mouse GCP-2 cDNA was cloned from MO murine fibroblasts. 17 Confluent cell monolayers were obtained in Eagle’s Minimal Essential Medium (MEM) (Life Technologies, Gibco BRL, Paisley, Scotland) supplemented with 10% fetal calf serum (Life Technologies, Gibco BRL) and induced for 72 hours in medium containing 2% fetal calf serum supplemented with lipopolysaccharide (10 μg/ml) plus double-stranded RNA poly(rI:rC) (50 μg/ml). Total RNA was extracted with the RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) and quantified spectrophotometrically. One μg total RNA was reverse transcribed in 1× reverse transcriptase buffer (0.14 mol/L KCl, 8 mmol/L MgCl2, 50 mmol/L Tris-HCl, pH 8.1), 25 mmol/L dithiothreitol, 0.15 mmol/L dNTPs (Amersham Pharmacia Biotech, Rainham, UK), 1 μg random hexamer primers (Life Technologies, Gibco BRL), 50 U human placental ribonuclease inhibitor and 4 U RAV-2 reverse transcriptase (both from Amersham Pharmacia Biotech) up to a final volume of 50 μl. The reverse transcriptase mixture was incubated for 80 minutes at 42°C, followed by denaturation of the enzyme for 5 minutes at 95°C. The subsequent polymerase chain reaction (PCR) was done on 2.5 μl of the reverse transcriptase mixture in a total volume of 50 μl, containing 1× ULTMA buffer (Applied Biosystems, Foster City, CA), 100 μmol/L dNTPs, 2 mmol/L MgCl2, and 10 pmol of each primer. The two specific primers used were 5′-TACCATGGCCACGGAGCTGCGTTGTGTTTGC-3′ (forward) and 5′-TACTCGAGTTATCAAGCTTTCTTTTTGTCACTGCC-3′ (backward) (respective NcoI and XhoI sites are in italics), amplifying a 210-bp cDNA fragment, 18 encoding the murine GCP-2(9-78) protein. After 5 minutes of denaturation, 0.5 U ULTMA DNA polymerase (Perkin Elmer) was added and 35 cycles of PCR were performed (95°C for 35 seconds, 60°C for 50 seconds, 72°C for 35 seconds) followed by a final extension of 10 minutes at 72°C. The PCR reaction product was gel-purified, restriction-digested with NcoI and XhoI, and ligated into the corresponding sites of the pHEN1 expression vector. 19 Verification of the sequence of pHEN1 mGCP-2(9-78) was done on both strands by the dideoxynucleotide chain termination method with the use of thermosequenase (Amersham Pharmacia Biotech) and fluorescent-labeled forward and reverse primers on an automated laser fluorescence sequencer (Amersham Pharmacia Biotech). Recombinant protein was expressed in JM83 Escherichia coli cells in the same way 20 as described previously for recombinant human MCP-2.
The recombinant murine GCP-2(9-78) was, in a first step, purified by heparin-Sepharose affinity chromatography and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining as described before. 20 The relative molecular mass (Mr) markers used were: ovalbumin (Mr 43,000), carbonic anhydrase (Mr 29,000), β-lactoglobulin (Mr 18,400), lysozyme (Mr 14,300), and bovine trypsin inhibitor (Mr 6200). The fractions that contained the highest amount of ±6.5-kd protein on SDS-PAGE were dialyzed against 50 mmol/L formiate, 0.01% Tween 20, pH 4.0, and further purified by cation-exchange fast protein liquid chromatography. The purity and relative molecular masses of the proteins in the eluted fractions were determined by SDS-PAGE and silver-staining analysis. As a final purification step, fractions that contained the recombinant protein were purified by reverse phase-high pressure-liquid chromotography (HPLC) on a C-8 Aquapore RP-300 column. Proteins derived from reverse phase-HPLC were analyzed by mass spectrometry on an Esquire Electrospray ion trap mass spectrometer (Esquire, Bruker, Bremen, Germany) and analyzed for endotoxin contamination by the Limulus amebocyte lysate test (BioWhittaker, Walkersville, MD).
Intratumoral Injection of Bowes Melanoma Tumors with Recombinant Murine GCP-2(9-78)
Human Bowes melanoma cells (ATCC CRL 9607) were cultured in Eagle’s MEM supplemented with 10% fetal calf serum. Confluent monolayers were trypsinized, cells were washed twice with MEM and resuspended at a concentration of 2.5 × 10 7 cells/ml. Eight-week-old female athymic nu/nu mice (NMRI background) were injected intradermally with 5 × 10 6 melanoma cells resuspended in 200 μl of MEM. On day 3 and every Monday, Wednesday, and Friday onwards, animals were injected at the tumor site with 20 μl of sample, containing 500 ng of recombinant murine GCP-2(9-78) (prepared and purified as described above) diluted in 0.25% human serum albumin (endotoxin ≤ 0.1 ng/ml) (Sigma, St. Louis, MO) in saline (endotoxin-free 0.9% NaCl) (Baxter, Lessines, Belgium). The dosage of 500 ng per injection site was derived from published data with other chemokines, 4 preliminary experiments with various dosages of GCP-2 and calculation of in vivo obtainable levels in small-sized tumors. The latter were calculated from data on in vitro cellular GCP-2 production yields. In the control mice, the tumors were injected with diluent. All animals were observed twice a week and tumor size was estimated (in square millimeters) using the formula 3.14 × (a × b)/4, with a as the largest diameter and b as the smaller diameter.
Mammalian Expression Vector Construction
A 399-bp cDNA fragment, encoding the full-size murine GCP-2 protein [signal peptide (40 AA) + mature protein (92 AA) + stop codon], was amplified by PCR from cDNA of lipopolysaccharide-stimulated MO murine fibroblasts, prepared as described above. The murine GCP-2-specific primers used were 5′-TACTCGAGATGAGCCTCCAGCTCCGCAGCTCC-3′ (forward) and 5′-TACTCGAGCTATTGAACACTGGCCGTTCTTTCC-3′ (backward) (XhoI site is in italics). The specific PCR product was gel-purified, digested with XhoI, and cloned into the corresponding site of the pLXIN vector (Clontech Laboratories, Palo Alto, CA). Verification of the sequences of the sense (pLXIN-mGCP-2) and antisense (pLXIN-mGCP-2-REV) was done by the dideoxynucleotide chain termination method as described above.
Transfection of Bowes Melanoma Cells and Selection of Transfected Cell Lines
Aliquots of 20 μg of the constructs pLXIN-mGCP-2 and pLXIN-mGCP-2-REV were linearized with AccI (Roche Diagnostics GmbH, Mannheim, Germany) and gel-purified. One day before transfection, confluent monolayers of human Bowes melanoma cells, grown in Eagle’s MEM supplemented with 10% fetal calf serum, were plated in 24-well dishes at a density of 5 × 10 4 cells/well. Cells were transfected with (1, 2, or 3 μl) Fugene6 (Roche Diagnostics, GmbH) in combination with the linearized plasmid DNA (300, 600, or 900 ng). The procedure was performed as described by the manufacturer. Three days after transfection, cells were grown in the presence of 1 mg/ml G418 (Life Technologies, Gibco BRL). G418-resistant colonies were isolated and expanded. To verify whether the cell lines expressed human chemokines, subclones were tested 17 by specific enzyme-linked immunosorbent assay (ELISA) for the expression of human IL-8 and human GCP-2. For the human GCP-2 sandwich ELISA, microtiter plates were coated with 0.2 μg/ml of polyclonal anti-human GCP-2 antibody (PeproTech, Rocky Hill, NJ) and the antigen was captured with 0.25 μg/ml monoclonal anti-human GCP-2 antibody (R&D Systems, Abingdon, UK).
Analysis of Murine GCP-2 Expression by Northern and Western Blot
To analyze the murine GCP-2 mRNA (sense or antisense) levels of the individual transfected cell lines, monolayers of parental Bowes cells and transfectants were grown to confluency and total RNA was prepared with the use of the RNeasy Mini Kit (Qiagen). Samples of 10 μg of RNA were separated on a formaldehyde/agarose gel and blotted onto a nylon membrane. The membrane was hybridized with a murine GCP-2 probe (399-bp cDNA fragment, excised with XhoI from pLXIN-mGCP-2) using the ExpressHyb hybridization solution (Clontech Laboratories) at the highest stringency. Probe labeling was by random priming in the presence of [α-32P]dCTP (Amersham Pharmacia Biotech). Control of sample preparation and handling was done by ethidium bromide staining of the RNA after electrophoresis in the agarose gel. To test the GCP-2 protein expression by the individual Bowes melanoma cell subclones, cells were grown to confluency and the growth medium was replaced by medium without serum. The cell culture supernatants were collected 24 hours later and subjected to Western blot analysis. Supernatants (7.5 μl) were loaded on a 16.5% Tris-tricine Ready Gel (Bio-Rad, Hercules, CA) and subjected to SDS-PAGE. As a positive control, purified natural murine GCP-2 was loaded. 17 Proteins were transferred to a polyvinylidene difluoride membrane by electroblotting and the blot was blocked in 2% casein in NT-buffer (10 mmol/L Tris-HCl, pH 7.4, 100 mmol/L NaCl). After three wash steps in wash buffer (NT-buffer and 0.1% Tween-20), the blot was incubated for 2 hours with 1 μg of biotinylated anti-mouse LIX/GCP-2 antibody (R&D Systems) diluted in 5 ml of wash buffer and 0.1% casein. The blot was washed three times for 5 minutes and subsequently incubated for 1 hour with streptavidin-horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) at a 1:10,000 dilution in wash buffer and 0.1% casein. The blot was again washed (five times for 5 minutes) and protein was visualized after incubation of the blot in enhanced chemiluminescence solution according to the protocol of the manufacturer (Amersham Pharmacia Biotech), and exposure to a BioMax MR-2 film (Kodak, Rochester, NY).
Injection of Stably Transfected Melanoma Cell Lines in nu/nu Mice and Histology
The in vivo study protocols were approved by the local ethical committee (National certification LA1210243, Belgium). Eight-week-old female athymic nu/nu mice (NMRI background) were injected intradermally at the left abdominal side with 2 × 10 6 or 5 × 10 6 exponentially growing parental or transfected human Bowes melanoma cells resuspended in 200 μl of MEM. Tumor diameters were measured once a week and the size was calculated as described above. For histological analysis, from each group of animals (parental, antisense, and sense transfectants) and from both developmental stages of the tumor (early tumors, late tumors) mice were killed and analyzed individually in two independent experiments. Biopsies from the primary tumor, brain, lung, and intercostal tissue were excised and fixed in Bouin’s fixative. Standard paraffin embedding, sectioning, and staining with hematoxylin and eosin (H&E) were performed for microscopic examination. S-100 staining for melanoma tumors was performed to analyze the presence of tumor metastases in different organs. To distinguish infiltrated neutrophils from tumor cells, paraffin-embedded sections of the tumors were subjected to myeloperoxidase (MPO) staining. Neutrophils were counted in 20 fields (magnification of ×400) of nonnecrotic tumor tissue in end-stage (9 weeks) or early (9 days) tumors. Results, scored by two independent researchers for whom the tumor sample labels were covered, were expressed as the mean ± SEM. Tumor tissue was also frozen at −70°C and frozen sections were immunostained for CD31 using the MEC 13.3 anti-CD31 monoclonal antibody (BD Pharmingen, San Diego, CA).
Preparation of Tumor Extracts and Analysis
Tumor tissue was homogenized in 9 volumes of phosphate-buffered saline and Complete (1 tablet/50 ml buffer) (Roche Diagnostics, GmbH) and cell debris was removed by centrifugation. Total protein content was determined by Bradford analysis using bovine serum albumin as a standard protein. 21 Murine GCP-2 was detected with a sandwich ELISA, using recombinant murine GCP-2(9-78) as a standard. In this test, coating of the plate was done with 1 μg/ml of anti-mouse LIX/GCP-2 antibody (R&D Systems) and capturing of the antigen was with 0.1 μg/ml of biotinylated anti-mouse LIX/GCP-2 antibody (R&D Systems). The detection limit for the muGCP-2 ELISA was 0.1 ng/ml. Gelatinase B/MMP9 activity in the tumor extracts was determined by SDS-PAGE gelatin zymography as described previously. 22
Results
Expression and Purification of Recombinant Murine GCP-2(9-78)
Because it had been shown previously 23 that the amino- and carboxy-terminally truncated natural isoform GCP-2(9-78) is more potent than intact murine GCP-2(1-92), the recombinant GCP-2(9-78) form was expressed in E. coli and purified to homogeneity. Recombinant mouse GCP-2(9-78) possessed the expected size (±6.2 kd on SDS-PAGE; 7626.8 d by electrospray mass spectrometry) and was resuspended in saline at a concentration of 100 μg/ml (Figure 1) ▶ . This preparation possessed in vitro neutrophil chemotactic activity and its endotoxin level was <25 pg/ml.
Figure 1.

Biochemical characterization of recombinant muGCP-2(9-78). A: The silver-stained SDS-PAGE gel of reverse phase-HPLC-purified recombinant muGCP-2(9-78). The sample (S) is compared with a mixture of protein standards (M) of which the molecular sizes are indicated on the left in kd. Different fractions of reverse phase-HPLC purified recombinant muGCP-2(9-78) were subjected to electrospray mass spectrometry analysis. The unprocessed (B) and charge-deconvoluted (C) spectra are shown. Fractions containing pure 7626.8 d muGCP-2(9-78) were pooled and used for further studies.
Local Injection of Recombinant Murine GCP-2(9-78) Has No Effect on Tumor Growth
The influence of murine GCP-2(9-78) on the growth of human Bowes melanoma cells in vivo was evaluated by intratumoral injection. Nude mice were subcutaneously xenografted with 5 × 10 6 Bowes melanoma cells at the abdominal side. From day 3 onwards (when small tumors appeared) and for a period of 3 weeks, 500 ng of muGCP-2(9-78) or diluent were injected into the tumors three times per week. In two independent experiments (each with two groups of 10 animals) no differences were observed in tumor growth, nor were effects detectable in the host. From the pooled data (Figure 2) ▶ , it was concluded that intratumoral administration of muGCP-2(9-78) did not alter the growth of melanoma tumors significantly. It is difficult to obtain a homogeneous distribution of the injected GCP-2 to target the complete tumor tissue. Because we anticipated that it would be better to have a more continuous, needle-free, and homogeneous supply of GCP-2 to evaluate the effect on tumor growth, stably muGCP-2-transfected Bowes melanoma cell lines were generated and used.
Figure 2.

Intratumoral administration of murine GCP-2 in melanoma tumors. Parental human Bowes melanoma cells (5 × 106) were inoculated subcutaneously into the left abdominal side in nude mice. From 2 days after inoculation onwards, 500 ng of recombinant murine GCP-2(9-78) or diluent alone (control) was administered three times a week into the tumor. Tumor diameters were monitored and the tumor size was calculated. Pooled data (means and standard errors of the mean) from two independent experiments, in which each group consisted of 10 animals, are shown.
Mouse GCP-2 Gene Transfer Enhances the Development of Bowes Melanoma Tumors
Sense and antisense transfectants of the murine GCP-2 cDNA in Bowes melanoma cells were generated. To test the mRNA production levels in the individual transfected subclones, Northern blot analysis was performed (Figure 3A) ▶ . Total RNA of the parental melanoma cells was used as a negative control. Several clones, which produced relatively high levels of sense or antisense muGCP-2 mRNA were selected and further tested for the production of muGCP-2 protein by Western blot analysis (Figure 3B) ▶ . The Western blot analysis showed the presence of multiple bands that co-migrated with those present in the natural mouse GCP-2. These bands correspond to the various truncation variants by amino- and carboxyterminal processing, as previously described. 23 The subclones GB+3 (sense, based on protein expression) and GB-8 (antisense) were selected and expanded for further in vivo studies. At several time intervals, throughout a period of more than 12 months, the supernatants were tested for murine GCP-2 production to provide evidence that the expression was constitutive and stable. The subclones (with a human cell line origin) were also tested for human IL-8 and human GCP-2 expression by specific ELISAs and scored negative in both tests (data not shown).
Figure 3.
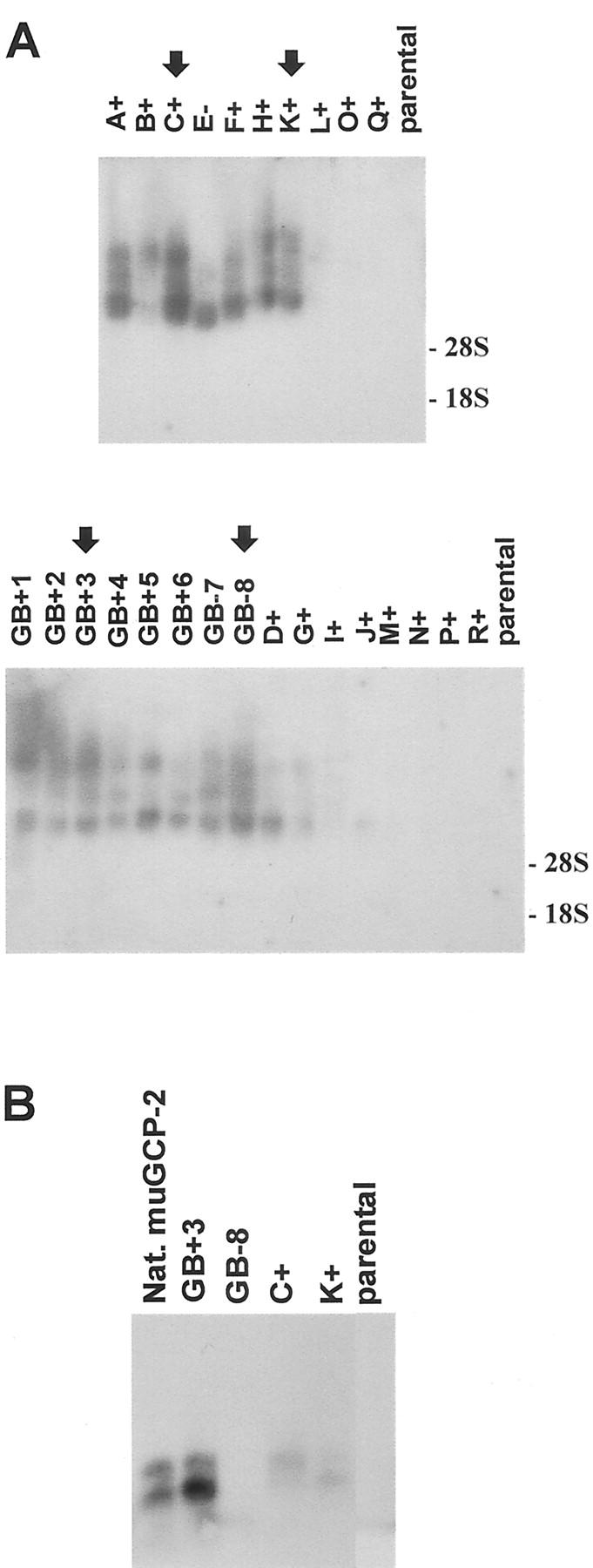
mRNA and protein levels of murine GCP-2 in Bowes melanoma transfectant cell lines. A: Northern blot analysis. Bowes melanoma cell clones with sense (A+, B+, C+, F+, H+, K+, L+, O+, Q+, GB+1, GB+2, GB+3, GB+4, GB+5, GB+6, D+, G+, I+, J+, M+, N+, P+, R+) or antisense (E−, GB-7, GB-8) muGCP-2 constructs and parental Bowes melanoma cells were tested for specific mRNA levels. Total RNA was extracted, purified, and equivalent amounts of 10 μg were subjected to Northern blot analysis. The membrane was hybridized with a muGCP-2-specific probe and the film was exposed overnight. Size-standardization and analysis of the loading of equal amounts of sample were by ethidium bromide-staining analysis after the electrophoresis. The 28S and 18S ribosomal RNAs are indicated on the figure. The arrows indicate the cell lines from several clones that were tested for protein expression. B: Western blot analysis. Confluent monolayers of subclones GB+3, C+, K+ (sense), and GB-8 (antisense) and of the parental Bowes melanoma cells were grown to confluency. Consecutively, the growth medium was replaced with serum-free medium. After 24 hours, cell culture supernatants were collected and 7.5-μl fractions were subjected to Western blot analysis to visualize the muGCP-2 protein. Purified natural murine GCP-2 (Nat. muGCP-2) was used as a positive control.
The effects of muGCP-2 sense and antisense gene transfection in the subclones were assessed and compared with those of parental (untransfected) Bowes melanoma cells. In several independent experiments, nude mice were subcutaneously injected with the various tumor cell lines and tumor growth was monitored. From week 8 onwards, the tumor sizes of the GB+3 tumors were clearly larger than those of GB-8 (antisense control) and parental Bowes tumors (Figure 4, A and B) ▶ . To assess whether the increase in tumor size was caused by a direct effect of muGCP-2 on tumor cell growth, the in vitro proliferation of Bowes melanoma cells was determined in the presence of recombinant murine GCP-2(9-78). Murine GCP-2(9-78) (50 ng/ml) was added to Bowes melanoma cells and cell numbers were determined after 3 days. Murine GCP-2 did not have an influence on the in vitro proliferation rate of the tumor cell lines GB+3, GB-8, and parental Bowes cells (data not shown).
Figure 4.
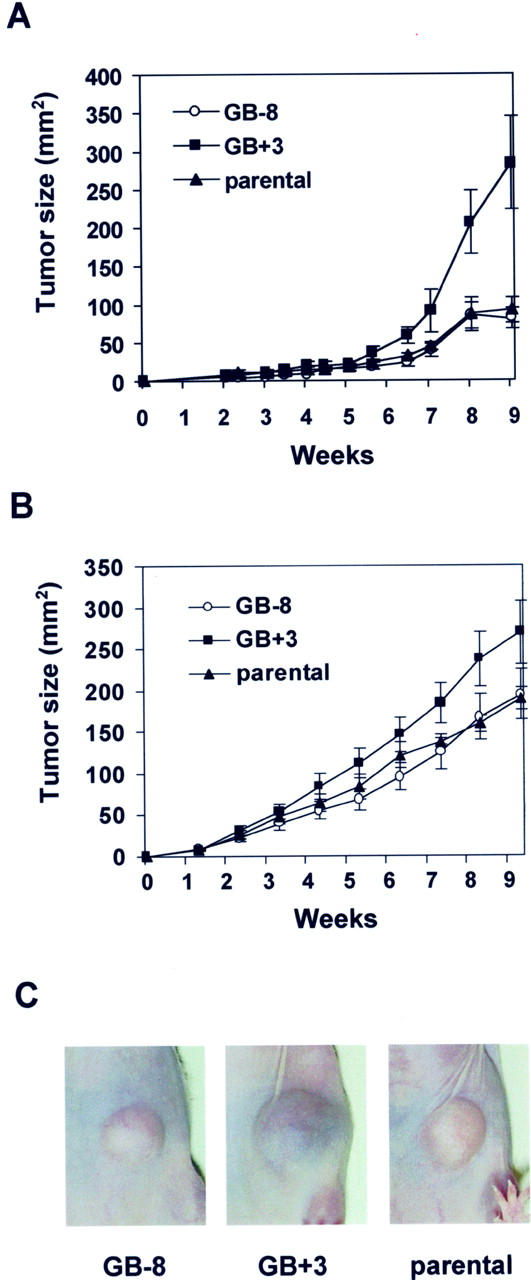
Growth of murine GCP-2 Bowes melanoma transfectants in nude mice. Nude mice were injected subcutaneously into the left abdominal side with 2 × 10 6 (A) and 5 × 10 6 (B) parental or muGCP-2-transfected sense (GB+3) or antisense (GB-8) Bowes melanoma cells. Tumor diameters were monitored in groups of 10 animals and the tumor sizes were calculated (means and standard errors). C: Photographs of a representative tumor of each group from the first experiment, taken at week 8.
All tumors (Figure 4A) ▶ were photographed at week 8 and a representative tumor of each group is shown in Figure 4C ▶ . Obviously, not only the size of the sense (GB+3) tumors differed from that of the parental and antisense (GB-8) controls, but also the GB+3 tumors had a dark bluish color that was not observed in the others.
In Vivo Neutrophil Chemotaxis and Activation
At the end of the experiment (9 weeks), the animals were autopsied. Macroscopically, the structures of the GB+3 tumors were totally different from the controls. Tumors of GB-8 and parental Bowes melanoma cells were solid and easy to distinguish from the surrounding tissue, whereas the GB+3 tumors were soft, bloody, and connected with the underlying tissue. Different organs (lung, brain, intercostal tissue, and liver) were inspected but macroscopically no differences were observed between controls (GB-8 and parental) and GB+3 tumors. Biopsies of the primary tumors, the brains, lungs, and intercostal tissues, were histologically analyzed. Staining for S-100 protein showed microscopic lung metastases of the melanomas in all three animal groups, whereas the other studied tissues were not affected (data not shown). In the three tumor groups, necrotic regions were reproducibly found and distinguished from nonnecrotic tissue. Figure 5 ▶ (left) represents tumor tissue of parental, GB-8 and GB+3 tumors after H&E staining. By staining for MPO, we observed a neutrophil load in the nonnecrotic regions of the tumor that was 7 to 10 times higher in the GB+3 compared to the GB-8 or parental tumors (Figure 5 ▶ , right, and Figure 6A ▶ ). In necrotic regions, many neutrophils were present in all of the parental, GB-8, and GB+3 tumors.
Figure 5.
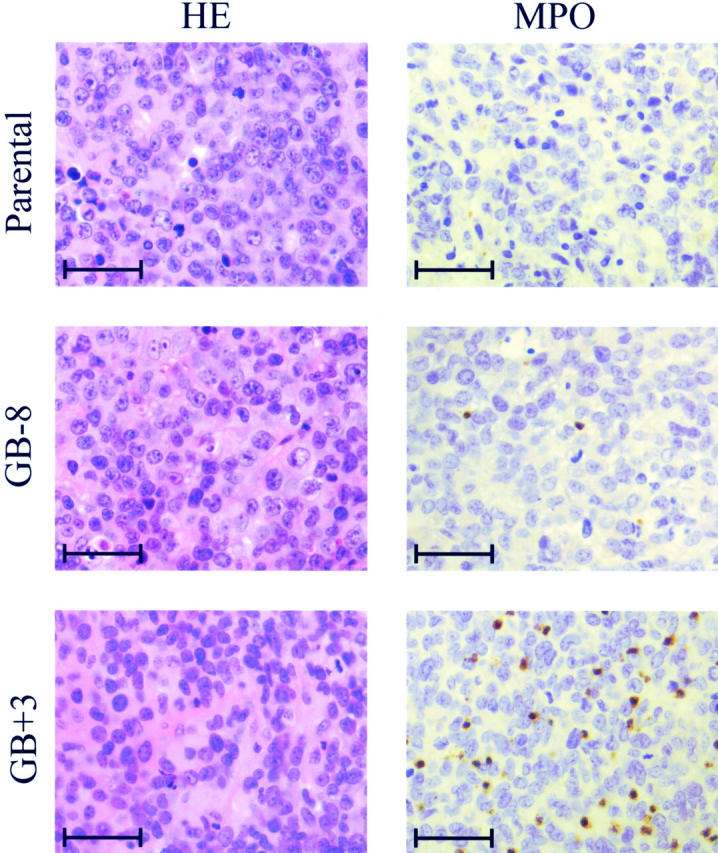
Histological analysis of murine GCP-2-transfected tumors. Left: H&E staining of parental, GB-8, and GB+3 tumors in vivo. Right: MPO staining of similar paraffin-embedded sections. Scale bars, ∼50 μm.
Figure 6.
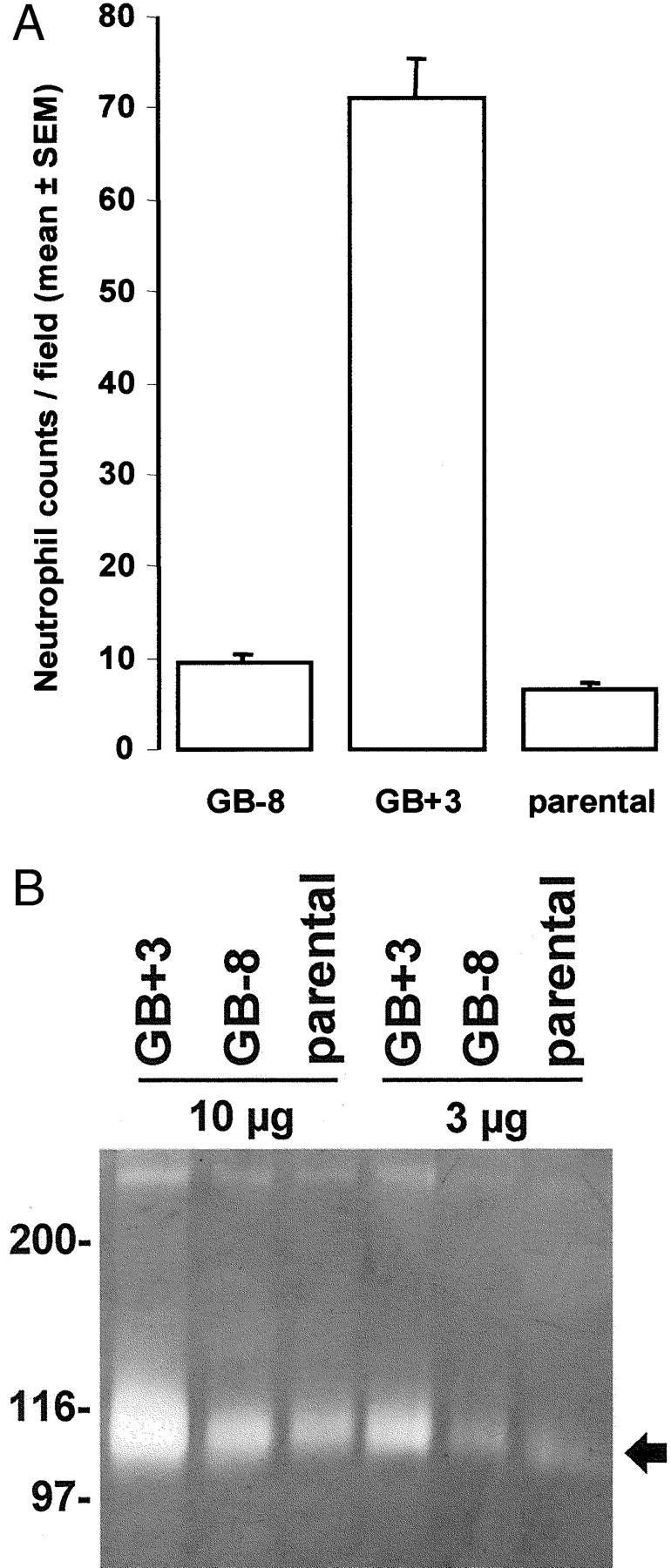
GCP-2 transfection induces neutrophil chemotaxis and activation in vivo. Expression of MPO was used as a marker for neutrophils. To evaluate the neutrophil chemotaxis in vivo, specific cell counts were done by histomorphometry. The MPO-positive cells were counted in 20 microscopic fields of in vivo grown tumors and compared for the parental Bowes and the antisense (GB-8) and sense (GB+3) transfectants (A). Data were expressed as means and standard errors of the mean. Degranulation of gelatinase B from neutrophils is induced by muGCP-2 in vivo. Tumor extracts of 9-day-old melanoma tumors were subjected to Bradford analysis and equivalent amounts of total protein (10 μg and 3 μg) were subsequently subjected to SDS-PAGE zymography (B). Left: A molecular weight standardization in kd is included, whereas the arrow at the right indicates the zymolysis zones obtained by the action of mouse gelatinase B (112 kd).
Individual tumors of each group, obtained from independent experiments, were dissected, and frozen at −70°C for further freeze-section analysis. Tumor extracts were tested for the presence of mouse GCP-2 with the use of a specific ELISA. In GB+3 tumor tissue, a level of ∼25 ng muGCP-2 per mg total protein was detected, which was two to three times higher than in the controls (GB-8 and parental) tumors. This result, together with the finding of 7 to 10 times more polymorphonuclear cells in the nonnecrotic tumor tissue shows that the GB+3 transfected Bowes melanoma tumor cells stably produce active muGCP-2 protein.
Tumor Angiogenesis in Vivo by Mouse GCP-2
To determine whether tumor angiogenesis is a mechanism by which muGCP-2 influenced Bowes melanoma growth, the vascularity was assessed by immunohistochemistry with CD31-specific antibodies on sections of frozen tissues from 9-week-old tumors. No significant differences in the numbers of cells staining positive for CD31 in the three types of late-stage tumors were detected. Similarly, no differences were observed when the numbers of vessel transections were counted per microscopic field (data not shown). To exclude that the angiogenic effects of GCP-2 gene transfer in late stage tumors were faded out by the necrosis and endogenous mouse chemokine production, we also assessed growing tumors at an early stage before necrosis was evident. Nude mice were subcutaneously injected with 5 × 10 6 parental or muGCP-2 transfected sense (GB+3) or antisense (GB-8) Bowes melanoma cells. After 9 days, tumor tissue was dissected and freeze-sections were prepared. After MPO staining, the neutrophil load in the GB+3 tumor was 7 to 10 times higher than in the control tumors, which is in agreement with the neutrophil load of nonnecrotic regions of tumors at 9 weeks. Gelatinase B activity in such tumor extracts was determined by SDS-PAGE zymography (Figure 6B) ▶ , and in the GB+3 (sense) tumors, found to be at least three times higher than in the GB-8 and parental tumors. This is in agreement with the finding that in GB+3 tumors many more neutrophils are present, and indicates that the in vivo chemotaxis was accompanied by neutrophil activation.
Histopathology of tumors at a late stage (9 weeks) indicated a considerable neutrophil influx in the GCP-2-producing tumors (see Figure 5 ▶ , MPO-staining) and, macroscopically, these tumors were larger, shining bluish through the skin and, when dissected, contained hemorrhagic pus. Lacunar vessel growth was evidenced by the softness and the fact that the red and white blood cells leaked out of the GCP-2 transfected tumors, whereas in the antisense-transfected and parental tumors, this was not observed. To show these elements by histopathology, we also dissected tumors at an early stage (day 9). Figure 7 ▶ illustrates the growth characteristics of these tumors as well as the microscopic peritumoral angiogenesis in the GCP-2 sense transfectant GB+3. In parental and GCP-2 antisense-transfected tumors, the solid tumors were confined and some CD31-positive vessels were observed, mainly in coexistence with normal tissue at the tumor front. As a contrast, in the GCP-2-producing tumors (GB+3) an extensive vascularization was observed at the tumor periphery. These vessels were not penetrating into the tumor. Furthermore, within the GB+3 neoplastic tissue, there was, already at this early stage, evidence of necrosis mainly at the center. When we counted the CD31-positive vessels (per microscopic field) within the tumors, no significant differences were found between the parental, sense, and antisense groups (data not shown). At the tumor perimeter the vessel growth was significantly stimulated by GCP-2 expression (Figure 7) ▶ . In conclusion, tumor-derived GCP-2 induced peritumoral angiogenesis in vivo and assisted in tumor progression.
Figure 7.
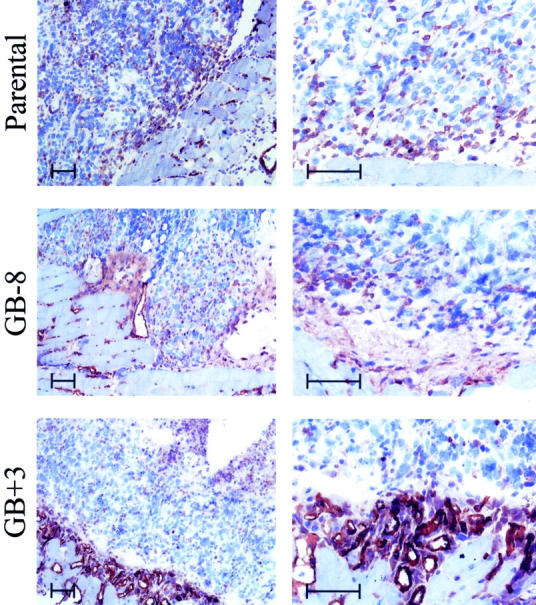
GCP-2 gene transfection induces peritumoral angiogenesis in vivo. Tumor samples were collected at 9 days after inoculation of 5 × 10 6 cells and processed as frozen sections. These were stained for mouse CD31 as a marker for endothelial cells. The left column shows low-power magnification of parental, antisense GB-8, and sense GB+3 transfectants at the front of the tumors with the normal tissue. CD31-positive cells are stained in red. The right column shows similar pictures of other sections at a higher magnification to visualize in more detail the peritumoral network of vessels formed in the GCP-2-expressing tumor variant GB+3. Scale bars, ∼50 μm. The peritumoral vessel transection counts per microscopic field (original magnification, ×400) in this specific experiment were 16.6 ± 1.7 for GB+3, 1.8 ± 0.2 for GB-8, and 2.8 ± 0.6 for the parental Bowes cells (means ± standard errors, n = 5).
Discussion
Many tumors produce chemokines, which may explain the presence of tumor-associated leukocytes. However, the role of these chemokines in tumor biology is not clear. They may assist in immunosurveillance and help to eliminate the tumor. 6 Alternatively, they may help the tumor for its growth and invasion. We coined the latter “the countercurrent principle” 7 and recently reviewed three key elements in this model: invasion promotion by enhancing the protease load of the tumor, tumor growth promotion by provision of trophic factors, and angiogenesis. 8 The control of angiogenesis has been defined as an important function of chemokines. 14,15,24 Recently, it has also been shown that the expression of IL-8 regulates angiogenesis and induces metastasis of specific tumors. 25,26
By using gene transfer technology, we here provide in vivo proof that a solid tumor indeed may have an advantage by producing a chemokine. For this, a tumor model of human melanoma cells that were xenotransplanted in nude mice was used. Evaluating the role of GCP-2 by intratumoral injection of the recombinant protein, we did not observe any significant effect on tumor parameters including tumor growth and survival of the host (data not shown). To evaluate the long-term effects in a needle-free setting and to study whether the tumor itself may acquire an advantage by the production of a chemokine, we stably transfected the murine GCP-2 cDNA into a melanoma cell line and compared various parameters in vivo. These included GCP-2 expression, tumor growth, neutrophil chemotaxis, gelatinase B degranulation from neutrophils, in vivo metastasis to the lungs, and tumor angiogenesis. Evidence of the countercurrent principle was found by the observation that GCP-2 expressing tumors, already at an early stage, had a higher neutrophil influx and showed a significantly increased angiogenesis. This angiogenesis was peritumoral allowing for a larger tumor size and a hemangioma-like macroscopic appearance to develop.
To investigate if this resulted in an elevated protease load, tumor extracts were subjected to zymography. No significant differences in gelatinase B levels were detected in extracts of large end-stage tumors with large necrotic areas, when compared with parental Bowes melanoma tumors and antisense transfected tumors (data not shown). This may be the result of the strong inflammatory component. In line with this, we observed areas of necrosis in the late-stage tumors with parental, sense- and antisense-transfected Bowes cells. Microscopically, these necrotic tumor areas contained in all instances pus. In the early developmental stages of the tumor, however, significantly higher levels of gelatinase B were observed in GCP-2-expressing tumors.
Obviously, in this model the vasculature remained peritumoral, which may imply that the high GCP-2 expression levels (chemokines follow a bell-shaped dose-response effect) prohibited vessel growth or that other factors play a role at the tumor front to allow or prohibit the blood vessels to grow into the tumor. Therefore, the model with GB+3 melanoma cells may also become useful to define and study molecules that allow the blood vessels to grow into the tumor or substances that inhibit the GCP-2-induced angiogenesis. The angiogenic effect of ELR-positive CXC chemokines has been established in inflammation and obviously also plays a role in tumor biology. This example of neutrophil chemotaxis and activation to release gelatinase B into the tumor site places the recent studies of potentiation of CXC chemokines by gelatinase B also central in tumor biology. 27 The role played by neutrophils may be studied by neutrophil depletion, the latter of which also occurs in patients under chemotherapy. However, we tried to gain insights in the natural tumor evolution without leukocyte depletion. Further studies with syngeneic tumor models may be better to evaluate the roles played by various leukocyte types.
Studies on skin carcinogenesis also show evidence for the countercurrent model and the contribution of leukocytes, as well as gelatinase B, in tumor progression. 28 In addition, MGSA/GRO proteins (CXCL1 and CXCL3) with melanocyte growth-promoting activity also lead to tumor angiogenesis. 29 Chemokines, chemoattracted leukocytes, as well as induced metalloproteinases, are to be considered as important players in the control of tumor vessel growth, 24,30 aside of other well-known (lymph)angiogenic and angiostatic factors in tumor biology. 31-34
Acknowledgments
We thank P. Fiten for DNA sequence analysis and image analysis, Brigitte Maes for light microscopy and photography, and P. Proost for mass spectrometry analysis.
Footnotes
Address reprint requests to Ghislain Opdenakker, Rega Institute, Minderbroedersstraat 10, B-3000 Leuven, Belgium. E-mail: ghislain.opdenakker@rega.kuleuven.ac.be.
Supported by the National Fund for Scientific Research (FWO-Vlaanderen), the Cancer Research Foundation of Fortis AB, the Belgian Federation against Cancer, and the Geconcerteerde Onderzoeksacties (GOA), Belgium.
A. W. is senior research assistant of the FWO-Vlaanderen.
References
- 1.Bottazzi B, Polentarutti N, Acera N, Balsari A, Boraschi D, Ghezzi P, Salmona M, Mantovani A: Regulation of the macrophage content of neoplasms by chemoattractants. Science 1983, 220:210-212 [DOI] [PubMed] [Google Scholar]
- 2.Graves DT, Jiang YL, Williamson MJ, Valente AJ: Identification of monocyte chemotactic activity produced by malignant cells. Science 1989, 245:1490-1493 [DOI] [PubMed] [Google Scholar]
- 3.Bottazzi B, Colotta F, Sica A, Nobili N, Mantovani A: A chemoattractant expressed in human sarcoma cells (tumor-derived chemotactic factor, TDCF) is identical to monocyte chemoattractant protein-1/monocyte chemotactic and activating factor (MCP-1/MCAF). Int J Cancer 1990, 45:795-797 [DOI] [PubMed] [Google Scholar]
- 4.Van Damme J, Van Beeumen J, Opdenakker G, Billiau A: A novel, NH2-terminal sequence-characterized human monokine possessing neutrophil chemotactic, skin-reactive, and granulocytosis-promoting activity. J Exp Med 1988, 167:1364-1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Damme J, Proost P, Lenaerts JP, Opdenakker G: Structural and functional identification of two human, tumor-derived monocyte chemotactic proteins (MCP-2 and MCP-3) belonging to the chemokine family. J Exp Med 1992, 176:59-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L: The origin and function of tumor-associated macrophages. Immunol Today 1992, 13:265-270 [DOI] [PubMed] [Google Scholar]
- 7.Opdenakker G, Van Damme J: Chemotactic factors, passive invasion and metastasis of cancer cells. Immunol Today 1992, 13:463-464 [DOI] [PubMed] [Google Scholar]
- 8.Opdenakker G, Van Damme J: Novel monocyte chemoattractants in cancer. Rollins BJ eds. Chemokines and Cancer. 1999, :pp 51-69 Humana Press Inc., Totawa [Google Scholar]
- 9.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM: Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258:1798-1801 [DOI] [PubMed] [Google Scholar]
- 10.Zlotnik A, Yoshie O: Chemokines: a new classification system and their role in immunity. Immunity 2000, 12:121-127 [DOI] [PubMed] [Google Scholar]
- 11.Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M: Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci USA 1993, 90:3574-3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hébert CA, Vitangcol RV, Baker JB: Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. J Biol Chem 1991, 266:18989-18994 [PubMed] [Google Scholar]
- 13.Baggiolini M, Dewald B, Moser B: Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv Immunol 1994, 55:97-179 [PubMed] [Google Scholar]
- 14.Strieter RM, Polverini PJ, Arenberg DA, Kunkel SL: The role of CXC chemokines as regulators of angiogenesis. Shock 1995, 4:155-160 [DOI] [PubMed] [Google Scholar]
- 15.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D: The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 1995, 270:27348-27357 [DOI] [PubMed] [Google Scholar]
- 16.Proost P, De Wolf-Peeters C, Conings R, Opdenakker G, Billiau A, Van Damme J: Identification of a novel granulocyte chemotactic protein (GCP-2) from human tumor cells. In vitro and in vivo comparison with natural forms of GRO, IP-10, and IL-8. J Immunol 1993, 150:1000-1010 [PubMed] [Google Scholar]
- 17.Wuyts A, Haelens A, Proost P, Lenaerts JP, Conings R, Opdenakker G, Van Damme J: Identification of mouse granulocyte chemotactic protein-2 from fibroblasts and epithelial cells. Functional comparison with natural KC and macrophage inflammatory protein-2. J Immunol 1996, 157:1736-1743 [PubMed] [Google Scholar]
- 18.Smith JB, Herschman HR: Glucocorticoid-attenuated response genes encode intercellular mediators, including a new C-X-C chemokine. J Biol Chem 1995, 270:16756-16765 [DOI] [PubMed] [Google Scholar]
- 19.Hoogenboom HR, Griffiths AD, Johnson KS, Chiswell DJ, Hudson P, Winter G: Multi-subunit proteins on the surface of filamentous phage: methodologies for displaying antibody (Fab) heavy and light chains. Nucleic Acids Res 1991, 19:4133-4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Coillie E, Proost P, Van Aelst I, Struyf S, Polfliet M, De Meester I, Harvey DJ, Van Damme J, Opdenakker G: Functional comparison of two human monocyte chemotactic protein-2 isoforms, role of the aminoterminal pyroglutamic acid and processing by CD26/dipeptidyl peptidase IV. Biochemistry 1998, 37:12672-12680 [DOI] [PubMed] [Google Scholar]
- 21.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 22.Masure S, Billiau A, Van Damme J, Opdenakker G: Human hepatoma cells produce an 85 kDa gelatinase regulated by phorbol 12-myristate 13-acetate. Biochim Biophys Acta 1990, 1054:317-325 [DOI] [PubMed] [Google Scholar]
- 23.Wuyts A, D’Haese A, Cremers V, Menten P, Lenaerts JP, De Loof A, Heremans H, Proost P, Van Damme J: NH2- and COOH-terminal truncations of murine granulocyte chemotactic protein-2 augment the in vitro and in vivo neutrophil chemotactic potency. J Immunol 1999, 163:6155-6163 [PubMed] [Google Scholar]
- 24.Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, Strieter RM: CXC chemokines in angiogenesis. J Leukoc Biol 2000, 68:1-8 [PubMed] [Google Scholar]
- 25.Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD, Yano S, Bar-Eli M, Radinsky R, Pettaway CA, Dinney CP: Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res 2000, 6:2104-2119 [PubMed] [Google Scholar]
- 26.Inoue K, Slaton JW, Kim SJ, Perrotte P, Eve BY, Bar-Eli M, Radinsky R, Dinney CP: Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res 2000, 60:2290-2299 [PubMed] [Google Scholar]
- 27.Van Den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G: Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96:2673-2681 [PubMed] [Google Scholar]
- 28.Coussens LM, Tinkle CL, Hanahan D, Werb Z: MMP-9 supplied by bone-marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103:481-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haghnegahdar H, Du J, Wang DZ, Strieter RM, Burdick MD, Nannay LB, Cardwell N, Luan J, Shattuck-Brandt R, Richmond A: The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukoc Biol 2000, 67:53-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balkwill F, Mantovani A: Inflammation and cancer: back to Virchow? Lancet 2001, 357:539-545 [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 32.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J: Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88:277-285 [DOI] [PubMed] [Google Scholar]
- 33.Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo M, Jackson DG, Nishikawa S-I, Kubo H, Achen MG: VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001, 7:186-191 [DOI] [PubMed] [Google Scholar]
- 34.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M: Induction of tumor angiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med 2001, 7:192-198 [DOI] [PubMed] [Google Scholar]