

Abstract
We investigated the effects of 3-nitropropionic acid (3NPA), a previously characterized neurotoxin, in four strains of mice to better understand the molecular basis of variable host responses to this agent. Unexpectedly, we found significant cardiac toxicity that always accompanied the neurotoxicity in all strains of mice in acute and subacute/chronic toxicity testing. Caudate putamen infarction never occurred without cardiac toxicity. All mouse strains tested are sensitive to 3NPA although the C57BL/6 and BALB/c mice require more exposure than 129SVEMS and FVB/n mice. Cardiac toxicity alone was found in 50% of symptomatic mice tested and morphologically, the cardiac toxicity is characterized by diffuse swelling of cardiomyocytes and multifocal coagulative contraction band necrosis. In subacute to chronic exposure, atrial thrombosis, cardiac mineralization, cell loss, and fibrosis are combined with cardiomyocyte swelling and necrosis. Ultrastructurally, mitochondrial swelling occurs initially, followed by disruption of myofilaments. Biochemically, isolated heart mitochondria from the highly sensitive 129SVEMS mice have a significant reduction of succinate dehydrogenase activity, succinate oxygen consumption rates, and heart adenosine triphosphate after 3NPA treatment. The severity of morphological changes parallels the biochemical alterations caused by 3NPA, consistent with cardiac toxicity being a consequence of the effects of 3NPA on succinate dehydrogenase. These experiments show, for the first time, that 3NPA has important cardiotoxic effects as well as neurotoxic effects, and that cardiac toxicity possibly resulting from inhibition of the succinate dehydrogenase in heart mitochondria, contributes to the cause of death in 3NPA poisoning in acute and subacute/chronic studies in mice.
3-Nitropropionic acid (3NPA) is a natural environmental toxin made by various plants and fungi. Human 3NPA intoxication has occurred in China via ingestion of fungal contaminated sugarcane, 1,2 yet 3NPA contamination of various foodstuffs (corn, peanuts, sugarcane) is not monitored by regulatory agencies. 3NPA induces neurodegeneration in the caudate putamen in humans and experimental animals, resembling Huntington’s disease. 3-8 Consequently, 3NPA poisoning is used primarily as an animal model of selective neurodegeneration. The mechanism of toxicity is thought to be because of the irreversible, covalent binding of 3NPA with subsequent inhibition of succinate dehydrogenase (SDH), an enzyme of the citric acid cycle that transfers electrons to the electron transport chain via its complex II function. 9,10 Thus, a major factor in 3NPA toxicity is because of cellular and mitochondrial stress seen with metabolic impairment.
Differential neurotoxic effects of 3NPA have been identified between rats and mice, 11 as mice seem resistant and require more 3NPA exposure, and between the mice carrying the Huntington’s disease mutation and their wild-type littermates. 12,13 Additionally, various strains of rats exhibit differential sensitivity to 3NPA, 14 however previous investigations of 3NPA toxicity have not considered strain differences in mouse models. Significant variation has been found between mouse strains in response to neurological injury resulting from other neurotoxins such as kainic acid and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, 15,16 as well as hypoxia and ischemia reperfusion mouse models. 17,18 We investigated the effects of 3NPA on the same strains of inbred mice, C57BL/6, BALB/c, FVB/n, and 129SVEMS strains, which showed differential sensitivity to the neurotoxin kainic acid. We were interested if certain strains of mice were less sensitive to neurological injury. The significance of this would be of great importance in strain background selection in genetic engineering of mice and the interpretation of these studies.
Furthermore, because the heart, similar to the brain, has a tremendous dependence on mitochondrial function and oxidative metabolism for the production of ATP, we investigated the effects of 3NPA on the heart in this experimental protocol. Our finding of 3NPA-induced cardiac pathology, which varied between mouse strains, was then followed up by investigation of biochemical mechanisms of cardiac toxicity in the most sensitive and least sensitive mouse strains.
Materials and Methods
Animals and Materials
BALB/c and C57BL/6 mice (8- to 10-week-old males ) were purchased from Hilltop Labs (Philadelphia, PA). FVB/n and 129SVEMS mice (8- to 10-week-old males) were purchased from the Jackson Laboratory (Bar Harbor, ME). All of the mice were housed in groups of five on a 12-hour light/dark schedule. Mice were allowed free access to mouse lab chow and water. All experiments were performed in accordance to the Guidelines for the Use and Care of Laboratory Animal (NIH Publication 85-23).
Drug Administration and Experimental Groups
3NPA (Aldrich Chemical Co., Milwaukee, WI) was made fresh daily and dissolved in isotonic saline (20 mg/ml) without neutralization, passed through a 0.2-μm filter to remove any bacterial contamination, and administered by intraperitoneal injection. Mice were numbered, weighed, and injected daily between 11:00 a.m. and 1:00 p.m. Two dosing protocols were used.
Protocol 1 Acute Toxicity
This 3NPA-dosing protocol was adapted from the original mouse protocol for Webster Swiss mice used by Gould and Gustine. 7 Saline or 3NPA was administered to 4 to 6 mice per strain/treatment (C57BL/6, BALB/c, 129SvEMS, and FVB/n) for two injections, 100 mg/kg 3NPA, 24 hours apart, and survival was assessed at 48 hours after the first injection. In this survival study, mice were monitored every 15 minutes throughout a 12-hour period and were euthanized using criteria described below based on clinical signs. In another series of structural and biochemical studies, this same protocol was used, except mice were euthanized at 24 hours after the first dose or 1 hour after the second dose to harvest tissues for electron microscopy (n = 10/strain/treatment), ATP analysis (n = 3 to 6/strain/treatment), or isolate heart mitochondria for metabolic studies (n = 3 to 4/strain/treatment).
Protocol 2 Subacute/Chronic Toxicity
3NPA (75 mg/kg/day) was given to the above strains of mice to model a subacute/chronic exposure. Mice were monitored every 15 minutes throughout a 12-hour period and euthanized based on clinical signs as described below.
3NPA-Induced Clinical Signs in Mice
With protocol 2, exposing mice to a more chronic, lower dose exposure of 3NPA, more consistent neurological signs were observed, accompanied by neuropathology. The majority of mice dosed with 3NPA developed a characteristic, progressive neurological disorder, clinically recognized in three stages referred to as stage I, II, and III, similar to the disorder seen in the 3NPA-treated rats. 8
In stage I, symptomatic mice were hypoactive but retained a normal posture and gait. These mice had minimal grooming activity and interaction with other mice. In stage II, there was an increase in spontaneous motor activity, which was characterized by a wobbly gait (ataxia), tremors, and a frequency to fall to one side with short episodes of paddling.
Finally, stage III was characterized by a reduction in motor hyperactivity with ventral or lateral recumbency (moribund state) and frequently bilateral hindlimb extension (more pronounced in protocol 2). Occasionally mice would have a seizure (<10% of FVB/n mice) when aroused.
Other clinical signs included weight loss (20%) that usually began 2 to 3 days before stage III signs were observed. Stage III mice routinely were hypothermic, with body temperatures as low as 29°C compared to 38°C in control mice. Body temperatures were monitored using a Mallinckrodt Mon-a-therm model 6510 system (Mallinckrodt Medical Inc., St Louis, MO) with rectal probe attachment. Additionally, hypothermic moribund mice, which never progressed through neurological stages, seemed to be in cardiopulmonary failure, characterized by respiratory signs (labored breathing) and possibly impaired perfusion (bluish distal extremities).
Criteria for euthanasia and evaluation: the moribund mice described above were immediately euthanized and histologically examined. Additionally, all mice with stage III neurological signs were euthanized and histologically examined. Mice that died acutely without significant clinical signs during the day of observation (15%) were also examined and each of the above groups were included in Figure 1 ▶ , Table 1 ▶ , or Table 2 ▶ .
Figure 1.

129SVEMS and FVB/n mice show increased sensitivity to 3NPA compared to C57BL/6 and BALB/c mice in protocol 2 (75 mg/kg/day 3NPA). Modified Kaplan-Meier survival curve. Mice included in this study were observed every 15 minutes throughout a 12-hour period for consecutive days of 3NPA dosing as described in Materials and Methods. These mice were euthanized because of stage III neurological signs (mice with heart and brain lesions from Table 2 ▶ ) or if clinically moribund (mice with only heart lesions from Table 2 ▶ ) as described in Materials and Methods. Strain differences in lesions are presented in Table 2 ▶ . Fifteen percent of BALB/c mice and 7.4% of C57BL/6 mice did not present clinical signs throughout the 22-day experiment demonstrating an intrastrain and interstrain variability in resistance to 3NPA.
Table 1.
Survival Rate (in %) of Four Mouse Strains after 3NPA Treatment (Protocol 1)
| Strain | Number of mice | Survival rate (48 hours), % |
|---|---|---|
| C57BL/6 | 5 | 100 |
| BALB/c | 6 | 84 |
| FVB/n | 4 | 25 |
| 129SVEMS | 5 | 20 |
Eight-week-old male mice (C57BL/6, BALB/c, FVB/n, and 129SVEMS strains) were intraperitoneally treated with 2 doses, 100 mg/kg 3NPA at 0 and 24 hours and survival rate was assessed at 48 hours after the first injection. This treatment protocol was based on the original mouse protocol developed by Gould and Gustine. 7 Mice included in this study were observed every 15 minutes over a 12-hour period for 48 hours after the first 3NPA dose and were euthanized using criteria described in the Materials and Methods section based on clinical signs. Chi-square Fisher’s exact analysis on these mice showed a significant difference in survival between C57BL/6 mice compared to FVB/n (P = 0.048) and C57BL/6 compared to 129SVEMS mice (P = 0.048). Comparison of Balb/c mice with other strains did not reach the 0.05 level of significance. Significant cardiac injury was found in all mice that were euthanized or died after exposure to 3NPA in this study.
Table 2.
Incidence of Lesions and Tissues Injured (Caudate Putamen, Heart, or Both) in Mice Treated with 75 mg/kg/day of 3NPA (Protocol 2)
| Mouse strain | Number of mice | Type of lesion | ||
|---|---|---|---|---|
| Heart only | Brain only | Heart and brain | ||
| C57BL/6 | 17 | 10 | 0 | 7 |
| BALB/c | 16 | 3 | 0 | 13 |
| FVB/n | 8 | 6 | 0 | 2 |
| 129SVEMS | 8 | 4 | 0 | 4 |
Mice included in this study were observed every 15 minutes over a 12-hour period for consecutive days of 3NPA dosing as described in the Materials and Methods section. Mice included in this table were euthanized due to stage III neurological signs (mice with heart and brain lesions above) or if clinically moribund (mice with only heart lesions above) as described in the Materials and Methods section and in Figure 1 ▶ .
Light Microscopy
For necropsy, mice exhibiting the above clinical signs were anesthetized with metaphane, perfused via left ventricle with 10% buffered formalin or tissues were fixed in formalin by immersion-fixation. Tissues were processed for standard hematoxylin and eosin staining (H&E). Because of the acute deaths in mice using protocol 1, with inconsistent neurological signs, multiple organs were examined in protocols 1 and 2 (heart, lungs, liver, kidneys, pancreas, and intestines) to identify other organ pathological injury and the likely cause of sudden death. After it was determined that only the heart and brain (specifically caudate putamen) were affected and no other organs showed significant pathology, only the brain and heart were examined in subsequent studies.
Characterization and Grading of 3NPA-Induced Cardiac Toxicity
C57BL/6 mice (n = 7) were compared to 129SVEMS mice (n = 7) treated with two doses of 3NPA (100 mg/kg; 0 and 24 hours) and perfused with a 1% paraformaldehyde and 1.25% glutaraldehyde solution 1 hour after the second injection. Saline-treated controls (n = 6 per strain) were compared to the 3NPA treatment groups. This time point was used because 129SVEMS mice would commence to die shortly (1 to 2 hours) after the second injection. The degree of cardiac cellular swelling, necrosis, mineralization, and hemorrhage were assessed on toluidine blue-stained histological sections. Twenty representative heart sections per mouse were graded semiquantitatively on a scale of 0 to 4 (0, absent; 1, minimal; 2, mild; 3, moderate; 4, severe) and averaged according to previously published criteria. 19 For cellular swelling the following scores were used: 1, microscopic foci of cellular swelling that involve a few cardiomyocytes in one location in the atria, ventricles, and septum; 2, cellular swelling foci consisting of a few cardiomyocytes involving more than one of the above locations; 3, small localized, multiple foci of cellular swelling involving more that one area; and 4, large diffuse cellular swelling involving the ventricular walls and septum. Necrosis was scored as follows: 1, microscopic foci of necrosis that involve a few cardiomyocytes in one location in the atria, ventricles, and septum; 2, necrotic foci consisting of a few cardiomyocytes involving more than one of the above locations; 3, small localized, multiple foci of necrosis involving more that one area; and 4, large diffuse necrotic foci involving the ventricular walls and septum. Mineralization was scored as 1, microscopic foci of mineralization that involve a few cardiomyocytes in one location in the atria, ventricles, and septum: 2, mineralization foci consisting of a few cardiomyocytes involving more than one of the above locations; 3, small localized, multiple foci of mineralization involving more that one area; and 4, large diffuse mineralization foci involving the ventricular walls and septum. Hemorrhage was scored as follows: 1, occasional erythrocytes in the interstitium; 2, small groups of erythrocytes in the interstitium; 3, large groups of erythrocytes in the interstitium; and 4, diffuse large accumulation of erythrocytes in the interstitium.
Electron Microscopy
129SVEMS and C57BL/6 mice were compared by ultrastructural analysis of caudate putamen and heart at the time point that was used for mitochondrial biochemical analysis and whole heart adenosine triphosphate (ATP) evaluation. Ten mice per group were treated intraperitoneally with saline or 100 mg/kg of 3NPA given at 0 and 24 hours and sacrificed at 1 hour after the second injection. Mice were anesthetized with metaphane and perfused by intracardiac perfusion via the left ventricle. One percent paraformaldehyde in phosphate buffer was used to clear the blood from vessels followed by a 1% paraformaldehyde and 1.25% glutaraldehyde in phosphate buffer using a 20 ml/minute flow rate. The brain and hearts were removed and placed in the glutaraldehyde fixative overnight. Tissues were trimmed to a 1 to 2 mm3, postfixed in osmium, processed, and embedded in epoxy resin. Blocks were cut in 1-μm sections, stained with toluidine blue, and screened by light microscopy. Thin sections were cut, stained with lead acetate and uranyl acetate, and representative samples were viewed with a Jeol electron microscope.
Heart Mitochondrial Isolation
Various biochemical parameters were compared in isolated mitochondria from saline- or 3NPA-treated (protocol 1) 129SVEMS and C57BL/6 mice. Mitochondria were isolated from mice (single hearts) using Nargarse digestion as described by Hansford and colleagues. 20 Mice were euthanized by cervical dislocation and decapitation. Hearts were quickly removed and washed free of blood in 0.25 mol/L sucrose, 10 mmol/L hepes and 1 mmol/L EGTA isolation buffer. Hearts were then cut into 1-mm3 pieces and homogenized for 8 minutes (3 strokes/minute) in a glass-Teflon homogenizer with 10 ml of sucrose buffer and 0.7 mg Nargarse enzyme per heart. The homogenate was spun at 8500 × g for 8 minutes, the pellet was resuspended in 5 ml of isolation buffer, and rehomogenized (10 strokes total). The homogenate was spun at 500 × g for 12 minutes. The supernatant containing mitochondria was centrifuged at 9500 × g for 9 minutes to pellet mitochondria. The pellet was gently resuspended and spun at 8500 × g to pellet only unbroken mitochondria. The last pellet was resuspended in the above described sucrose/hepes buffer without the addition of EGTA. Mitochondrial proteins were measured using the method of Lowery and colleagues. 21
SDH Activity in Heart Mitochondria
SDH activity was measured in isolated heart mitochondria from three to four mice per strain in the saline controls and the 3NPA-treated mice (protocol 1). Heart mitochondrial protein (100 μg) was solubilized in a 0.01% Triton X solution in an incubation buffer of 0.05 mol/L potassium phosphate, 0.02 mol/L succinate, 50 μmol/L 2,6-dichlorophenolindophenol, 2 μg/ml antimycin A, rotenone 2 μg/ml, 2 mmol/L KCN, and 50 μmol/L decylubiquinone. 22 Briefly, SDH activity was measured by the rate of reduction of decylubiquinone using the substrate succinate by following the secondary reduction of the dye 2,6-dichlorophenolindophenol. The reaction was followed spectrophotometrically by a decrease in absorbance at 600 nm for 3 minutes at 30°C.
Oxygen Consumption
Oxygen consumption rates in heart mitochondria (protocol 1) were measured at 28°C with a Clark-type O 2 electrode. Mitochondria (0.25 mg) were added to a 1-ml aliquot of respiration buffer containing 0.12 mol/L KCl, 20 mmol/L K Hepes, pH 7.4, 5 mmol/L K phosphate, 5 mmol/L succinate, and 1 μmol/L rotenone. Respiration was measured without ADP (state IV) and after the addition of 0.5 mmol/L ADP (state III). 22
Cardiac ATP Analysis
Using protocol 1, C57BL/6 and 129SVEMS mice, 3NPA (n = 6 per strain) or saline (n = 3) was administered at 0 and 24 hours. One hour after the second injection, mice were anesthetized with 0.5 mg/g of chloral hydrate and endotracheally intubated with a 22-gauge catheter (2N1116; Baxter, Deerfield, IL). Mice were ventilated using a Harvard apparatus rodent ventilator (no. 680) on room air with a respiratory rate 150/minute and tidal volume set at 0.125 ml. 23 The tidal volume was adjusted to ensure an arterial carbon dioxide tension within the physiological range. The thorax was opened and while the lungs were being ventilated, the hearts were freeze-clamped by a precooled metal clamp that was immediately immersed into liquid nitrogen. The heart samples were ground to fine powder under liquid nitrogen and extracted and homogenized in ice cold 0.4 mol/L perchloric acid. The denatured protein was pelleted and reserved for protein analysis. 21 The acid extract was neutralized with equal volumes of 0.4 mol/L KHCO3. Each extract was subjected to nucleotide analysis using gradient ion-pair reversed-phase liquid chromatography. 24 HPLC separation was performed using an ESA (Chelmsford, MA) solvent delivery system with a 3-μm symmetry C18 column (3.9 × 150 mm inner diameter) from Waters Corporation (Milford, MA). Separation was performed by reverse-phase chromatography using an isocratic mobile phase consisting of buffer A (35 mmol/L KH2PO4, 6 mmol/L tetrabutylammonium hydrogensulfate, pH 6.0, 125 mmol/L ethylenediaminetetraacetic acid) and buffer B (a mixture of buffer A and HPLC-grade acetonitrile in a ratio of 1:1, v/v), filtered through a 0.2-μm membrane filter and helium degassed. The flow rate was set at 1.0 ml/minute and detection was performed at 260 nm using an ESA variable wavelength UV/V is absorbance detector.
Statistical Analysis
Survival data from protocol 1 was compared by chi-square Fisher’s exact analysis. SDH activity and ATP analysis were analyzed by analysis of variance followed by Bonferroni’s post hoc test. Heart lesion severity scores (cellular swelling, necrosis, hemorrhage, and mineralization) were analyzed by the Kruskal-Wallis rank test. A P value <0.05 was considered statistically significant. All data are presented as mean ± SEM.
Results
3NPA-Induced Mortality Is Mouse Strain- and Dose-Dependent
In 3NPA protocol 1 (100 mg/kg, two doses at 0 and 24 hours) C57BL/6 mice and BALB/c mice had significantly better survival at 48 hours compared to the FVB/n mice and 129SVEMS mice (Table 1) ▶ . Frequent monitoring of mice was performed (every 15 minutes) throughout a 12-hour period. Mice were euthanized when first observed in the recumbent hypothermic condition (more prevalent in FVB/n and 129SVEMS mice) because it was found that disease progression occurs rapidly in these strains and mice in this state die suddenly.
When comparing mouse strains to each other, neurological signs were inconsistently seen between strains in mice treated with this 3NPA treatment paradigm adapted from the original 3NPA mouse study. 7 In the original pilot study (n = 4 to 6 mice) presented in Table 1 ▶ , none of the mice presented neurological signs up to 48 hours after the initial injection. The majority of FVB/n and 129SVEMS mice became acutely moribund after the second injection. These mice either died during the observation period or were euthanized based on clinical signs. In later studies using the protocol 1 dosing paradigm, 40% of the C57BL/6 mice progressed to stage II (ataxia and tremors) 1 hour after the second injection, while 129SVEMS mice would infrequently show neurological signs because of a moribund condition (recumbency, nonambulatory state).
Because our initial goal was to study 3NPA-induced neurodegeneration between strains of mice, we reasoned that the cause of death, which prevented the development of overt neurotoxicity, may be avoided if we lowered the 3NPA dose. In a second study (protocol 2), a lower daily dose of 3NPA was used (75 mg/kg) for consecutive days until specific neurological signs (ataxia and tremors initially with progression to hindlimb extension) were observed. More reproducible neurological signs were observed with this protocol and neurodegeneration was found to accompany and precede mortality in all mouse strains tested. When comparing the four strains of mice, signs of toxicity were evident in mice as early as day 5 to day 14 with daily exposure to 75 mg/kg. In contrast, in the 100 mg/kg (2 doses, 24 hours apart) study, signs of toxicity occur 1 hour after the second dose especially in the 129SVEMS and FVB/n mice.
Survival curves for protocol 2 (Figure 1) ▶ illustrate the time course variability between the mouse strains in survival after 3NPA treatment. By day 9, 100% of the 129SVEMS and FVB/n mice had died or were euthanized. In contrast, 30% of the C57BL/6 mice and 26% of the BALB/c mice remained without exhibiting any clinical signs in the 3NPA treatment group. Between days 15 to 22, no additional mice in the remaining strains (C57BL/6 and BALB/c) showed clinical signs. On day 22 of dosing, the saline-treated and 3NPA-treated mice that had not presented clinical signs were euthanized and histologically analyzed. Fifteen percent of BALB/c mice and 7.4% of C57BL/6 mice (presented in Figure 1 ▶ ) did not present clinical signs throughout the 22-day experiment demonstrating an intrastrain and interstrain variability in resistance to 3NPA. In contrast, all of the 129SVEMS and FVB/n mice treated in this protocol with 3NPA either showed clinical signs and were euthanized or died suddenly by day 9 of 3NPA treatment, demonstrating a lack of resistance in these strains to 3NPA.
3NPA Causes Strain- and Dose-Dependant Caudate Putamen and Cardiac Toxicity
A histological survey from protocol 1 revealed toxicity in the heart, a finding that had not been previously reported in any species. One hundred percent of the mice that died or were euthanized within the first 48 hours of the first dose (Table 1) ▶ had only acute cardiac injury (see Figure 3B ▶ ). There was no evidence of histological injury in saline-treated controls.
Figure 3.
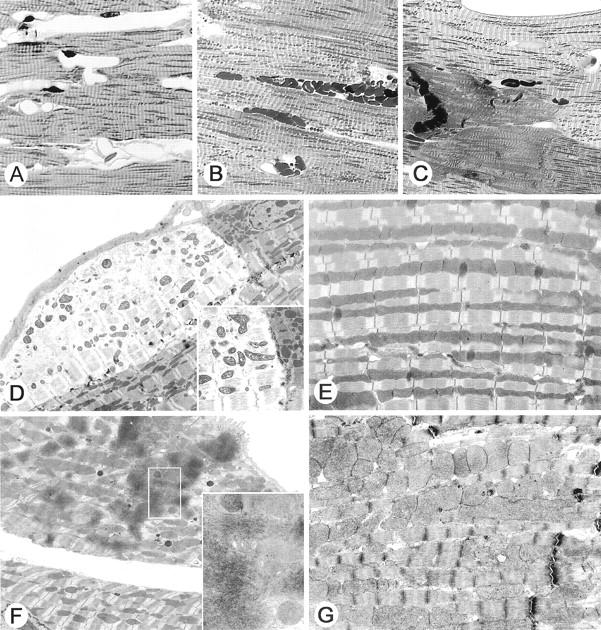
3NPA treatment induces cardiac injury in mice. A to C are representative light micrographs of toluidine blue-stained 1-μm-thick plastic-embedded sections of mouse hearts (original magnification, ×600). A: Normal heart saline-treated mouse. B: Myocardial cellular swelling, mitochondrial enlargement, and focal hemorrhage. C: Myocardial contraction band necrosis. D to G are representative examples of electron micrographs from mouse hearts. D: Epicardial myocyte showing myofilament disruption and swollen mitochondria adjacent to less affected cell (original magnification, ×3000). The inset shows a comparison of the pale myocyte to an adjacent myocyte separated by an intercalated disk (original magnification, ×5000). Note the dark electron-dense mitochondria with abnormal morphology within the affected myocyte. E: Normal control myocyte (original magnification, ×6000). F: Myocyte showing contraction band necrosis and swollen mitochondria (original magnification, ×4000). The inset shows the aggregated disrupted myofilament (original magnification, ×12,000). G: Swollen mitochondria are present in this cardiac myocyte. These mitochondria are larger than control, but they do not show the abnormal morphology and electron density noted in the myocytes with myofilament disruption shown in D. Note the good preservation of the sarcomeres.
In attempts to avoid the acute mortality and predominant cardiac toxicity seen in protocol 1, the 3NPA daily dose was decreased to 75 mg/kg for another series of studies (protocol 2). A histological survey of multiple organs was performed to correlate with clinical signs and the cause of death in 3NPA-treated mice. With this protocol, a much higher incidence in caudate putamen infarction was observed, although cardiac injury was still a confounding problem and was present in 100% of the mice tested. 3NPA treatment either induced heart lesions alone (myocardial cellular swelling and necrosis with frequent atrial thrombosis) (Figure 2) ▶ (23 of 49 mice) or in combination with caudate putamen infarction (26 of 49 mice). There were no examples of mice with only caudate putamen infarction. Table 2 ▶ summarizes the incidence and tissues injured in mice treated with 3NPA from the four mouse strains tested in the protocol 2.
Figure 2.
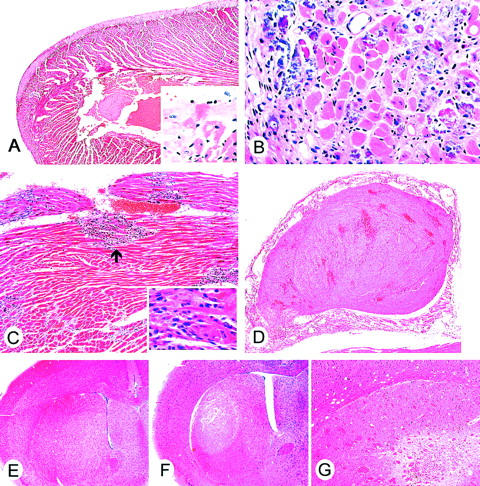
3NPA treatment induces cardiac and caudate putamen injury in mice. A to D are representative light micrographs of 3NPA-induced lesions in the heart (H&E stain). A: Left ventricle showing a band of marked epicardial pallor (original magnification, ×24). The inset represents an area of transition between the epicardial swollen (pale) myocytes and the deeper (darker) myocytes (original magnification, ×100). B: An area of cardiac myocyte loss, interstitial fibrosis, and mineralization present in a subacute-treated mouse is shown (original magnification, ×100). C: Several foci of inflammatory mononuclear cell infiltrates (arrow) are noted (original magnification, ×30). Myocyte necrosis and cellular infiltrate are shown (inset; original magnification, ×80). D: Organizing thrombus filling the atrial cavity (original magnification, ×24). E to G are representative light micrographs of the caudate putamen (H&E stain). E: Normal caudate putamen from saline-treated mouse (original magnification, ×10). F: Caudate putamen from 3NPA-treated mouse with infarct (note area of pallor) in the dorsal lateral aspect (original magnification, ×10). G: Caudate putamen from 3NPA-treated mouse with cellular loss, glial proliferation, and hemosiderin-laden mononuclear cells. Note the white matter tracts are positioned closer together (resolving infarct) (original magnification, ×60).
3NPA-Induced Caudate Putamen Pathology
One hundred percent of the mice exhibiting hindlimb extension (euthanized in stage III) had severe damage in the caudate putamen including widespread cellular swelling, necrosis, edema, and mild to moderate hemorrhage (infarction) (Figure 2F) ▶ . Occasional small vessel microthrombi were found in the caudate putamen in areas adjacent to infarcts. Saline-treated mice showed no morphological injury in the caudate putamen (Figure 2E) ▶ . Infarcts were bilateral in the majority of mice. The infarcts were centered on the dorsal-lateral caudate putamen. In some mice, that showed transient inconsistent neurological signs, removed from the study and examined 7 days later, the caudate putamen also showed evidence of chronic injury with white matter bundles positioned in close proximity to each other because of substantial neuronal loss (Figure 2G) ▶ . Ten percent of the treated mice (FVB/n and 129SVEMS) were observed to have seizures that were associated with unilateral infarcts that extended into the globus pallidus, thalamus, hippocampus, cingulate and motor cortex, as well as caudate putamen.
3NPA Acute Toxicity Induces Cardiomyocyte Cellular Swelling and Necrosis
In protocol 1, two morphological patterns were observed in cardiomyocytes, frequently seen in adjacent cells (cellular swelling and necrosis). Cardiomyocytes exhibited multifocal to diffuse cellular swelling, characterized by increased pallor (Figure 2A ▶ and Figure 3D ▶ ) and prominent eosinophilic granulation (swollen mitochondria) and occasional microvacuolation. Swollen mitochondria are easily seen on the toluidine blue sections (Figure 3B) ▶ . Multifocal coagulative necrosis of cardiomyocytes was characterized by cardiomyocytes with homogeneous intensively eosinophilic cytoplasm (hematoxylin and eosin), loss of striations, and irregular contraction bands (Figure 3C ▶ , toluidine blue).
In the protocol 2, in addition to cellular swelling and necrosis, bilateral atrial thrombosis (Figure 2D) ▶ , cardiomyocyte mineralization, cellular loss, and fibrosis (Figure 2B) ▶ were present. Neutrophils were associated with the superficial atrial endocardium and endothelium attached to atrial thrombi. With the exception of thrombi, inflammatory cells were rarely seen in acute toxicity associated with cellular swelling and myocardial necrosis.
In the several mice that showed transient inconsistent neurological signs and were removed from the study, and examined 7 days after the last 3NPA injection, cardiac macrophage and fibroblastic remodeling after necrosis was evident (Figure 2C) ▶ . In two C57BL/6 mice that survived to day 22 in protocol 2, without neurological signs, mild to moderate multifocal cardiomyocyte cellular loss and fibrosis was observed but no caudate putamen pathology was found in these mice.
129SVEMS Mice Are More Vulnerable to 3NPA Cardiac Toxicity Compared to C57BL/6 Mice
Because C57BL/6 and 129SVEMS mice show the extremes of interstrain variability to 3NPA-induced mortality in various dosing protocols, we hypothesized that 129SVEMS mice would have more significant cardiac injury compared to the more resistant C57BL/6 mice when compared at the same time point using protocol 1. Four morphological components were recognized and separately scored (Tables 3 and 4) ▶ ▶ by a semiquantitative method described in Materials and Methods. The incidence and severity of lesions was greater in the 129SVEMS mice compared to the C57BL/6 mice. Only 3NPA-treated 129SVEMS mice lesions scores were significantly (P < 0.001) more severe than those of control mice compared to C57BL/6 mice.
Table 3.
Incidence and Severity of Cardiac Myocyte Cellular Swelling and Necrosis in the Most Sensitive (129SVEMS) and Least Sensitive (C57BL/6) Strain
| Strain/treatment group | Number of mice | Cellular swelling scores | Necrosis scores | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||
| 129 mice/3NPA | 7 | 0 | 0 | 0 | 0 | 7* | 0 | 0 | 0 | 1 | 6* |
| 129 mice/saline | 6 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 |
| C57 mice/3NPA | 7 | 0 | 3 | 2 | 2 | 0 | 2 | 2 | 2 | 1 | 0 |
| C57 mice/saline | 6 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 |
129SVEMS and C57BL/6 mice were treated with 3NPA (100 mg/kg, 2 doses 0 and 24 hours), sacrificed 1 hour after the 2nd dose, and lesions were scored and analyzed.
*Lesion scores significantly more severe than controls (P < 0.05 by Kruskal-Wallis rank test).
Table 4.
Incidence and Severity of Myocardial Hemorrhage and Mineralization in the Most Sensitive (129SVEMS) and Least Sensitive (C57BL/6) Strain
| Strain/treatment group | Number of mice | Hemorrhage scores | Mineralization scores | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 0 | 1 | 2 | 3 | 4 | ||
| 129 mice/3NPA | 7 | 3 | 1 | 2 | 1 | 0 | 5 | 2 | 0 | 0 | 0 |
| 129 mice/saline | 6 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 |
| C57 mice/3NPA | 7 | 6 | 0 | 1 | 0 | 0 | 7 | 0 | 0 | 0 | 0 |
| C57 mice/saline | 6 | 6 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 |
129SVEMS and C57BL/6 mice were treated with 3NPA (100 mg/kg, 2 doses 0 and 24 hours), sacrificed 1 hour after the 2nd dose, and lesions were scored and analyzed.
Electron Microscopy Shows that Mitochondria in Brain and Heart Are Major Targets for 3NPA Toxicity
Ultrastructural evaluation of the heart revealed 129SVEMS mice had more severe damage compared to the C57BL/6 mice. 129SVEMS mice showed extensive diffuse myocardial cellular swelling compared to mild, multifocal areas myocardial swelling in the hearts of C57BL/6 mice. Acute ultrastructural changes included cellular swelling, disrupted myofilaments and markedly swollen mitochondria. Mitochondrial swelling may have occurred early in the progression of pathology, as it was found alone in many cardiomyocytes without cardiomyocyte myofilament disruption (Figure 3, B and G) ▶ . Mitochondrial swelling always accompanied cellular swelling and disruption of myofilaments (Figure 3F) ▶ .
Ultrastructural assessment of heart and brain revealed a temporal relationship between the injury in the two organs. Heart ultrastructural lesions appeared before caudate putamen infarction. Some mice had severe heart damage and no or mild caudate putamen injury. In the 129SVEMS mice, in cases in which the heart damage was extensive, caudate putamen neurons were affected as described below (Figure 4, A and B) ▶ .
Figure 4.
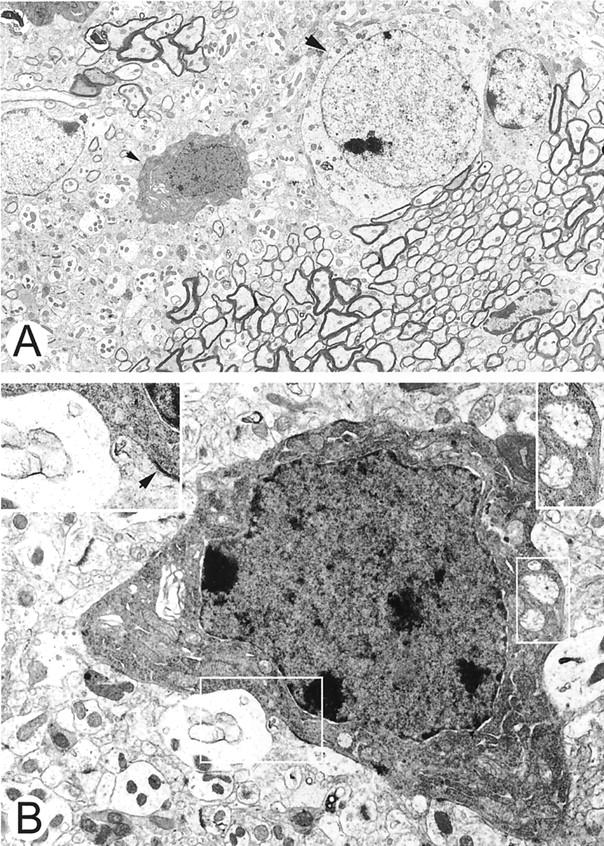
3NPA treatment induces neuronal degeneration in 129SVEMS mice. A and B are electron micrographs from the caudate putamen of a 129SVEMS mouse with severe myocardial injury. A: A dark shrunken degenerating caudate putamen neuron (small arrowhead) adjacent to a normal neuron (large arrowhead). Adjacent normal myelinated axons are at the bottom right of the micrograph (original magnification, ×4000). B: Dark shrunken neuron (original magnification, ×10,000) with swollen mitochondria (small box) (original magnification, ×11,000) and adjacent to a swollen astroglial process (large box) (original magnification, ×17,000). This cell can be identified as a neuron because of the axosomatic synaptic junction (inset, top left, arrowhead).
Caudate putamen from 3NPA-treated mice had a distinctive ultrastructural pathology at a timepoint that preceded infarction. Damage was more prominent in 129SVEMS mice compared to C57BL/6 mice. The major ultrastructural abnormalities occurred in neurons and could be classified as cell body or axonal degeneration. The pathology in neuronal cell bodies predominated in the dorsolateral caudate putamen. The neurons were angular rather than round, suggesting shrinkage (Figure 4, A and B) ▶ . These degenerating neurons had a very dark and granular cytoplasmic matrix because of fine particles, possibly free ribosomes, within the cytoplasm. Cytoplasmic vacuoles were present. Most of the vacuoles were derived from swollen mitochondria, because remnants of cristae were present within the vacuole. Other mitochondria in surrounding neuropil structures appeared normal. The Golgi apparatus was also swollen in these neurons. These abnormal neuronal cell bodies typically had a nucleus with a dark nucleoplasmic matrix with nascent chromatin condensation along the nuclear envelope. Many dark neurons were surrounded partially by swollen astroglial processes characterized by a pale cytoplasmic matrix, few organelles, and membranous cisterns. In the caudate putamen of C57BL/6 mice, axonal pathology was found in white matter bundles. Individual myelinated axons, found among normal axons, were prominently dark. These abnormal axons had a normal caliber and normal myelin sheath and were not dystrophic but contained neurofilament accumulations.
3NPA Induces a Reduction in Heart Mitochondrial SDH Activity and Oxygen Consumption Rates
Because extensive ultrastructural mitochondrial swelling was observed in the heart, a variety of mitochondrial metabolic endpoints were examined at the time point used in the ultrastructural studies to verify the metabolic significance of the mitochondrial swelling. SDH activity was measured in isolated heart mitochondria from saline- and 3NPA-treated 129SVEMS and C57BL/6 mice in protocol 1. Compared to the controls, 3NPA-treated 129SVEMS mice had a significant decrease in SDH activity compared to the 3NPA-treated C57BL/6 mice (Figure 5A) ▶ . Control 129SVEMS mice SDH activity was significantly higher compared to the control C57BL/6 mice. Oxygen consumption rates using succinate as a substrate (SDH substrate) were also higher in the 129SVEMS mouse controls compared to the C57BL/6 mice (Figure 5B) ▶ . This is consistent with the finding that the control 129SVEMS mice have a higher SDH activity. SDH enzyme activity and oxygen consumption rates were significantly reduced in both C57BL/6 and 129SVEMS mice after two doses of 100 mg/kg 3NPA compared to the controls. Oxygen consumption rates were significantly lower in the 129SVEMS mice compared to the C57BL/6 mice after only one dose of 3NPA suggesting an increased metabolic vulnerability in the 129SVEMS strain at this earlier time point.
Figure 5.
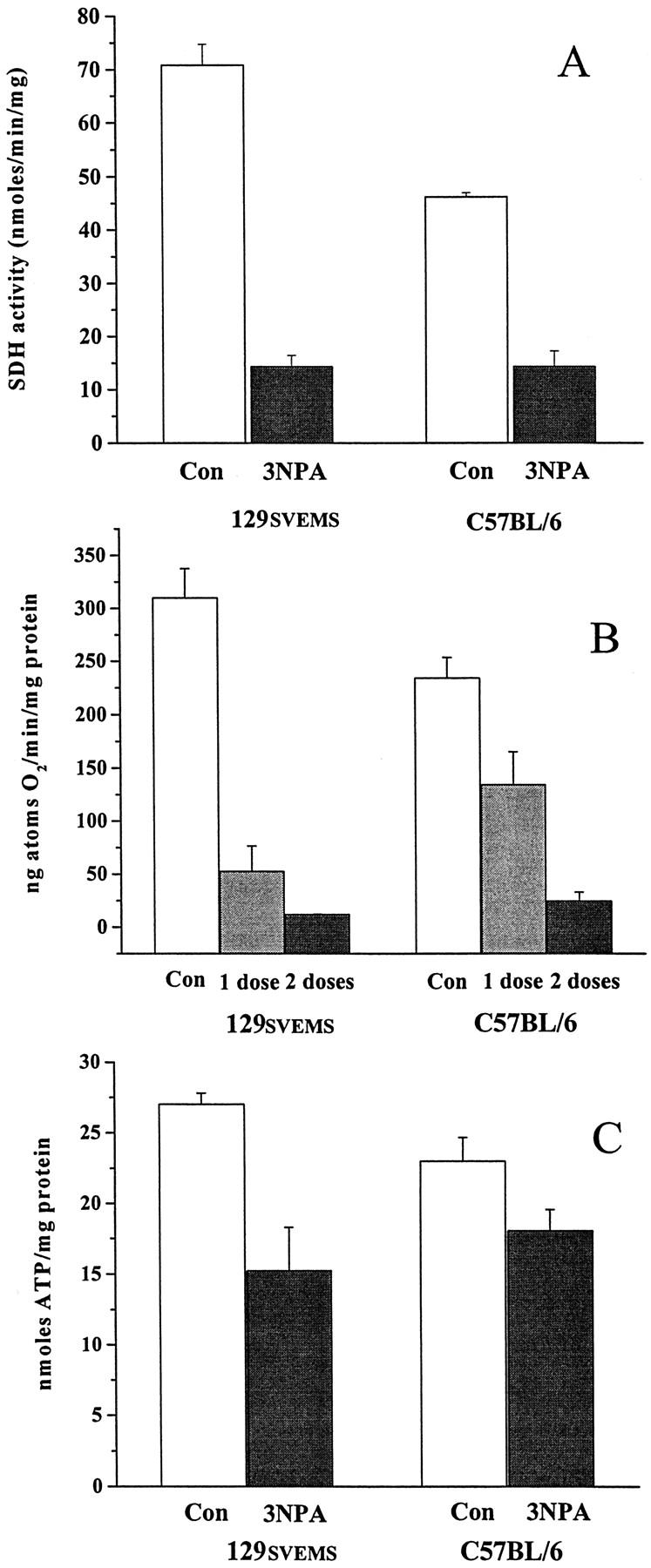
3NPA induces a reduction in heart mitochondrial SDH activity, oxygen consumption, and heart ATP in mice. 129SVEMS and C57BL/6 mice were treated with 3NPA (protocol 1) and heart mitochondria were isolated and analyzed for SDH activity and succinate oxygen consumption rates. 129SVEMS and C57BL/6 mice were treated with 3NPA (protocol 1) and heart ATP was extracted and analyzed by HPLC. A: Significant differences were found in SDH activity, control 129SVEMS versus 129SVEMS 3NPA-treated (P < 0.001), control C57BL/6 versus C57BL/6 3NPA-treated (P < 0.001), control 129SVEMS versus control C57BL/6 (P < 0.001). B: Significant differences were found in oxygen consumption, control 129SVEMS versus 129SVEMS 3NPA one dose, and two doses (P < 0.001), control C57BL/6 versus C57BL/6 3NPA two doses (P < 0.001). C: Significant difference was found in heart ATP, control 129SVEMS versus 129SVEMS 3NPA two doses (P < 0.05).
3NPA Induces a Significant Reduction of Cardiac ATP in 129SVEMS Mice
Other metabolic indices were analyzed to better understand mechanisms underlying interstrain mortality and morphological differences. We hypothesized that heart ATP values could predict the more susceptible strain. Because there was no current example in the literature for freeze-clamping the mouse heart from an in vivo preparation to analyze ATP, we used a method developed for rats. Intubated and room air-ventilated rats have been used to assess whole heart ATP levels. 25 We used this method, because mice that were not intubated in our pilot studies had low ATP values in all treatment groups. Figure 5C ▶ summarizes the mean ATP values from 3NPA-treated mice in protocol 1. Compared to 129SVEMS controls, only 3NPA-treated 129SVEMS mice had significantly lower ATP values. 3NPA-treated C57BL/6 mice did show the trend for ATP reduction, although it did not reach a 0.05 level of significance.
Discussion
Genetic differences between mouse strains can modulate responses to various perfusion insults or toxins. 15,26-29 We have determined that mouse strain differences can also modulate the response to 3NPA toxicity based on mortality, pathology, and biochemical analysis. 129SVEMS and FVB/n mice are more susceptible to 3NPA compared to the C57BL/6 and BALB/c mice in acute and subacute/chronic exposures. This pattern of vulnerability among strains is the same pattern found in kainic acid toxicity. 15 However, unlike the kainic acid study, the finding of cardiac toxicity with 3NPA most likely influences the pattern of mortality seen between strains.
We used a mouse 3NPA-dosing protocol adapted from that of Gould and Gustine 7 from the original 3NPA Swiss Webster mouse study. We modified their protocol by lowering the 3NPA dose from 120 mg/kg to 100 mg/kg and used only two doses given 24 hours apart. For our protocol, as in the Gould and Gustine protocol, we also did not neutralize 3NPA solutions before injection. The acute 3NPA dose for the mouse is higher than the acute 3NPA dose for the rat (30 mg/kg for two or three injections on consecutive days until neurological signs appear). The mechanisms for the marked differences in species (rat versus mouse) in 3NPA dose exposure required for toxicity are not known. The existence of a 3NPA transporter protein with differential expression between species, between mouse strains and tissues could explain the differences observed in 3NPA toxicokinetics. Additionally, differences in mitochondrial respiration/metabolism and in the requirement for ATP varies between species and possibly between strains, as it does between tissues and this may have an effect on the species differences seen after 3NPA exposure. In terms of potential cardiac toxicity, the heart rate is 600 to 700 beats/minute in the mouse and 300 beats/minute in the rat, thus the mouse may be more susceptible to reductions in cardiac high energy phosphates induced by 3NPA. In humans poisoned by 3NPA, there is a marked interindividual variability in the final neuropathological outcome ranging from diffuse cerebral edema to focal striatal damage. 2,30-33 This marked interindividual variability in humans reflects that seen in other species and is likely related to differences in 3NPA exposure, toxicokinetics, genetic variability, and pre-existing disease.
We found that the mouse strain susceptibility, based on mortality and pathology, is likely driven by 3NPA cardiotoxicity in these four commonly used mouse strains. All mouse strains are sensitive to 3NPA, although the cumulative dose needed to cause cardiac toxicity varies between strains. The ultrastructural changes, including mitochondrial swelling, cellular edema, and myofilament disruption, are more severe in the hearts of the highly sensitive 129SVEMS mice compared to C57BL/6 mice, suggesting the increased cardiac damage may be associated with the increased mortality of this strain. The light microscopic findings of cardiomyocyte cellular swelling, contraction band necrosis, mineralization, and atrial thrombosis are consistent with significant pathological abnormalities seen in other examples of cardiac toxicities. 19,34-41 The cardiac toxin doxorubicin also induces atrial thrombosis in mice. 42 The formation of atrial thrombi in doxorubicin-induced cardiomyopathy is thought to be associated with decreased cardiac contractility, effecting blood flow dynamics and toxic injury to the atrial cardiomyocytes and endothelial surfaces. These mechanisms of injury are likely involved in 3NPA cardiac toxicity.
We found that the mouse strain susceptibility, based on mitochondrial biochemical studies, is likely driven by 3NPA cardiotoxicity when comparing two mouse strains with differential sensitivity. 129SVEMS mice are metabolically more impaired at an earlier time point compared to C57BL/6 mice. The analysis of multiple mitochondrial biochemical endpoints of toxicity at the time point used in the ultrastructural studies revealed significant differences between the strains supporting vulnerability. SDH enzyme activity is significantly reduced in both strains after 3NPA treatment, although 129SVEMS mice have a greater percent reduction compared to controls. Notably, the SDH activity is constitutively higher in this strain that correlates with the higher oxygen consumption rates observed in 129SVEMS mice oxidizing succinate (the substrate for SDH). These data suggest that the 129SVEMS mouse strain may rely more on cardiac SDH activity. 3NPA-treated 129SVEMS mice oxidizing succinate have a significant reduction in oxygen consumption after one dose of 3NPA compared to C57BL/6 mice. In addition, heart ATP levels are significantly reduced in 129SVEMS mice treated with 3NPA compared to controls. These findings correlate well with the 129SVEMS mice strain being more susceptible to 3NPA, although ATP reduction is probably not the only factor involved in this strain’s increased sensitivity to 3NPA compared to C57BL/6 mice.
Mitochondrial toxicity is key to 3NPA neurotoxicity 8,43-54 and seems critical in 3NPA cardiac toxicity. 3NPA induces only mitochondrial swelling in some cardiomyocytes. In more severely damaged cells, in addition to mitochondrial swelling, there is also severe disruption in myofilaments. This suggests that 3NPA-induced toxicity in the cardiomyocyte first involves damage to mitochondria, as an early event in toxicity. Further damage to other organelles in this model, may be a sequelae of mitochondrial dysfunction.
There have been no previously published studies evaluating 3NPA in vivo cardiac toxicity; yet, 3NPA has been studied in various in vitro cardiac systems. 3NPA is a competitive inhibitor of succinoferricyanide oxidoreductase (another name for SDH) activity of Keilin-Hartree mitochondrial particles from rat heart. 55 3NPA is slowly oxidized by SDH and its oxidation product instantaneously and irreversibly inhibits cardiac SDH. 7,9 Isolated rat atria incubated with 3NPA (10−4 mol/L) in an organ bath for 15 minutes produced bradycardia and significant ATP depletion compared to controls. 56 This finding is consistent with our in vivo experiments showing that mice treated with 3NPA have a significant cardiac ATP depletion (129SVEMS mice). Isolated control heart mitochondria exposed to 3NPA 10−2 mol/L for a 30-minute incubation produced significant reduction of O2 consumption in the presence of the substrates malate/glutamate or succinate. 56 This in vitro finding also parallels our experiments showing in vivo 3NPA treatment reduces oxygen consumption rates in heart mitochondria isolated from treated mice.
Indirect physiological evidence of 3NPA cardiac toxicity has been reported such as models showing bradycardia and a reduction in contractile force in the guinea pig atria, 57 vasodilation in rabbit aortic rings, and hypotension and bradycardia in dogs. 58 Consequently, in light of our more direct evidence of 3NPA inducing cardiac damage, there is substantial evidence that 3NPA cardiac toxicity has the potential to be responsible for cardiac dysfunction and death in animals exposed to this toxin. It is not known if sudden death in mice and rats treated with 3NPA in acute and chronic models (with and without neurological signs) reported by others investigators were because of 3NPA-induced cardiac toxicity 7,11,13,14,51,59,60 . In our protocol, comparable to Gould and Gustine’s 7 acute protocol, ultrastructural evidence of cardiac injury is observed at the time point when 129SVEMS mice begin to die acutely, before any significant caudate putamen damage is observed. Caudate putamen infarction never occurred in our studies (acute and subacute/chronic) in the absence of cardiac toxicity. Any potential relationship between the two lesions needs to be addressed in future studies. The data from our studies show that cardiac toxicity is found in all cases of acute and subacute/chronic 3NPA toxicity in the mouse. The influence of cardiac toxicity (inducing decreased cardiac output) on the development of neurological lesions in the mouse is not known. The incidence of cardiac toxicity in acute and chronic models of 3NPA toxicity in the rat is unknown. Gould and Gustine 7 suggested in the original mouse study, that because 3NPA induced a dramatic decrease in cardiac SDH activity, 3NPA-induced cardiac injury, if severe enough, may decrease cardiac output and cause subsequent caudate putamen ischemic hypoxia-induced striatal damage. Gould and Gustine reported that SDH activity in the mouse heart was reduced to 13% of the controls whereas brain SDH activity was reduced to 20% of the controls. Cardiac pathological lesions were not reported in this study, although it was not clear at what time point heart samples were collected and examined, possibly only after one dose. 7 It should also be noted that the sites of lesions found in the Gould and Gustine 7 3NPA mouse study, as they point out, are similar to those found as a result of hypoxia. 61 In one of the first studies using 3NPA in rats, Hamilton and Gould 50 observed that the neuronal damage produced by systemic 3NPA histologically resembled that produced either by kainic acid, hypoglycemia, or ischemia. Interestingly, a recent model of global brain ischemia in rats produces cell loss in the striatum that resembles that seen in Huntington’s disease. 62
In general, the striatum is exquisitely sensitive to ischemia and hypoxia. 62-69 The proposed reasons for this increased sensitivity of the striatum to hypoxia and ischemia are: 1) glutamatergic excitotoxicity via NMDA receptor has been attributed to the neuronal cell death and oxidative stress in this area 70,71 ; 2) the architecture of the blood supply to the striatum is predominantly end-arterial with few collateral vessels, so this region is susceptible to changes in nutrient perfusion and oxygen delivery 72,73 ; 3) the unique connectivity and the level of oxidative metabolism of the striatum 74 ; and 4) the striatum is a region in the brain where massive glutamateric inputs and dopaminergic inputs converge. Dopamine and glutamate are important neurotransmitters in the brain, but when concomitantly over released, each acts as a potent neurotoxin. 75 In fact, the mechanisms attributed to the striatum’s generalized vulnerability to ischemia and hypoxia are consistent with those mechanisms found responsible in the pathogenesis of 3NPA striatal neurotoxicity.
Global brain ischemia/hypoxia with striatal damage 76 and increased blood brain barrier permeability 77 can be induced by heart failure. Oxidative stress is a potential outcome of brain hypoperfusion. 78-80 Interestingly, increased protein carbonyls groups indicative of oxidative stress are demonstrated in the cortex and the striatum of 3NPA-treated rats 81 suggesting a more global brain 3NPA effect. Thus, 3NPA intrinsic striatal neurotoxicity needs to be addressed in future studies in the absence of 3NPA cardiac toxicity to rule out the potential influence of cardiac dysfunction and any subsequent possible striatal hypoperfusion (ischemia/hypoxia) that may occur in the 3NPA neurotoxicity mouse model. It is not known if 3NPA cardiac toxicity-induced deaths observed in mice in a chronic model of 3NPA toxicity developed by Ouary and colleagues, 14 although we observed chronic cardiac injury in mice exposed to 3NPA daily for 21 days.
3NPA may have an intrinsic toxic effect (direct or indirect) on the striatum through its effects on the function of the lateral striatal artery. The potential for a focal disruption of blood flow dynamics involving the lateral striatal artery should be considered. The blood brain barrier of the lateral striatal artery and its tributaries is disrupted in 3NPA toxicity models 44,73 as well as in cardiac arrest/resuscitation models 77 demonstrating the increased sensitivity of this artery to conditions of cardiac dysfunction induced hypoxia/ischemia and 3NPA metabolic inhibition. The mechanisms involved in this blood brain barrier disruption are unknown. Additionally, intermittent cortical vasospasms have been observed after cardiac arrest and global brain ischemia 82,83 and the potential of striatal vasospasms was not evaluated in these studies. 3NPA produces bradycardia and systemic hypotension and vasodilation, 57,84 although regional striatal blood flow dynamics have not been studied. It would be important to evaluate striatal blood flow in 3NPA toxicity, especially in light of cardiac dysfunction, to possibly shed light on the heightened vulnerability of the lateral striatal artery and the striatum.
The finding of strain dependency in 3NPA-induced toxicity is significant because the mouse strains tested in this study are commonly used as background strains for transgenic and knockout mice. For the construction of knockout mice, 129SVEMS mice are most commonly used as the source of the embryonic stem cells. After successful gene targeting, embryonic stem cells are routinely implanted in the blastocysts of C57BL/6 mice. 85 FVB/n mice are also used for the construction of transgenic mice because they have large ova that are easily microinjected with DNA. BALB/c mice are also used as a background strain for nude mice. Differences in the mouse strain genetic background, especially if more than a single parent strain is included, can greatly influence the expression of transgenes and phenotype of knockout mice. Thus, a better understanding of mouse genetics and phenotypes will improve our design and interpretation of studies using genetically engineered mice.
This 3NPA model of cardiac toxicity could be a valuable tool for understanding many issues in cardiac pathophysiology including the mitochondria’s role in cardiac cell death (apoptosis and necrosis), oxidative stress, and chemical preconditioning. 3NPA is known for its profound preconditioning effects at low doses (1/25 of the LD-50 dose, ie, gerbils) to protect against subsequent brain ischemia in various rodent models. 86-90 Recently it was found that low doses of 3NPA (1 mg/kg) induces preconditioning and protection in the heart when given before ischemia/reperfusion in rabbits. 91 The stress signals (ie, ATP depletion and reactive oxygen species production) that are induced in 3NPA toxicity may likely be the same signals that induce the preconditioning effect. Thus at high doses, 3NPA is toxic to the brain and heart and at low doses, 3NPA may be beneficial to the brain and heart. The plant Astragalus called “Huang-Qi” in Chinese herbal medicine, known for containing 3NPA, has been used for centuries to precondition the heart. It is not known if the 3NPA in Astragalus is inducing this cardiac protective effect.
Remarkedly, there are few models of chemical cardiac toxicity. Of those that are well studied, mitochondrial toxicity seems to be of great importance in the heart. Fluoroacetate (1080), a rodenticide and an inhibitor of mitochondrial citric acid cycle enzymes aconitase and SDH, similar to 3NPA, induces damage in the heart and the brain depending on the species studied. 37,92,93 A second cardiac toxin, Doxorubicin (adriamycin) has been shown to redox cycle at complex I producing mitochondrial superoxide, which subsequently damages various complexes of the electron transport chain. 94,95 Thus for its effects on the heart, 3NPA may be another useful compound to study cardiac mitochondrial toxicity and preconditioning.
Acknowledgments
We thank Brian Schofield of the Johns Hopkins University School of Public Health for his assistance in the preparation of the light microscopy figures, and Frank Barksdale of the Johns Hopkins University School of Medicine for his help with the electron microscopy processing and sectioning.
Footnotes
Address reprint requests to Kathleen L. Gabrielson, D.V.M., Ph.D., 720 Rutland Ave., Ross 459, Comparative Medicine, School of Medicine, Baltimore, Maryland 21205. E-mail: kgabriel@jhmi.edu.
Supported by T32 ES07141 (to K. L. G.), the National Institute of Environmental Health Sciences (grant ES03819, NIEHS), the National Institutes of Health (grant NIA AG16282 to L. J. M.), the Department of Defense, United States Army Medical Research and Material Command (DAMD 17-99-1-9553 to L. J. M.), and RO1 ESO8785 NIEHS (to J. B.).
References
- 1.He FZS, Zhang C: Mycotoxin-Induced Encehalopathy and Dystonia in Children. 1990. Taylor and Francis, London
- 2.Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M: 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci 1991, 18:492-498 [DOI] [PubMed] [Google Scholar]
- 3.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT: Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 1993, 13:4181-4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexi T, Hughes PE, Faull RL, Williams CE: 3-Nitropropionic acid’s lethal triplet: cooperative pathways of neurodegeneration. Neuroreport 1998, 9:R57-R64 [DOI] [PubMed] [Google Scholar]
- 5.Binienda Z, Johnson JR, Tyler-Hashemi AA, Rountree RL, Sapienza PP, Ali SF, Kim CS: Protective effect of L-carnitine in the neurotoxicity induced by the mitochondrial inhibitor 3-nitropropionic acid (3-NPA). Ann NY Acad Sci 1999, 890:173-178 [DOI] [PubMed] [Google Scholar]
- 6.Borlongan CV, Koutouzis TK, Sanberg PR: 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev 1997, 21:289-293 [DOI] [PubMed] [Google Scholar]
- 7.Gould DH, Gustine DL: Basal ganglia degeneration, myelin alterations, and enzyme inhibition induced in mice by the plant toxin 3-nitropropanoic acid. Neuropathol Appl Neurobiol 1982, 8:377-393 [DOI] [PubMed] [Google Scholar]
- 8.Hamilton BF, Gould DH: Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: a type of hypoxic (energy deficient) brain damage. Acta Neuropathol 1987, 72:286-297 [DOI] [PubMed] [Google Scholar]
- 9.Coles CJ, Edmondson DE, Singer TP: Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem 1979, 254:5161-5167 [PubMed] [Google Scholar]
- 10.Alston TA, Mela L, Bright HJ: 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci USA 1977, 74:3767-3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexi T, Hughes PE, Knusel B, Tobin AJ: Metabolic compromise with systemic 3-nitropropionic acid produces striatal apoptosis in Sprague-Dawley rats but not in BALB/c ByJ mice. Exp Neurol 1998, 153:74-93 [DOI] [PubMed] [Google Scholar]
- 12.Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF: Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington’s disease. J Neurochem 1998, 71:2642-2644 [DOI] [PubMed] [Google Scholar]
- 13.Hickey MA, Morton AJ: Mice transgenic for the Huntington’s disease mutation are resistant to chronic 3-nitropropionic acid-induced striatal toxicity. J Neurochem 2000, 75:2163-2171 [DOI] [PubMed] [Google Scholar]
- 14.Ouary S, Bizat N, Altairac S, Menetrat H, Mittoux V, Conde F, Hantraye P, Brouillet E: Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: implications for neuroprotection studies. Neuroscience 2000, 97:521-530 [DOI] [PubMed] [Google Scholar]
- 15.Schauwecker PE, Steward O: Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 1997, 94:4103-4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonsalla PK, Heikkila RE: The influence of dose and dosing interval on MPTP-induced dopaminergic neurotoxicity in mice. Eur J Pharmacol 1986, 129:339-345 [DOI] [PubMed] [Google Scholar]
- 17.Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN: Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab 1993, 13:683-692 [DOI] [PubMed] [Google Scholar]
- 18.Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA: Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke 1997, 28:1805-1811 [DOI] [PubMed] [Google Scholar]
- 19.Herman EH, Zhang J, Chadwick DP, Ferrans VJ: Age dependence of the cardiac lesions induced by minoxidil in the rat. Toxicology 1996, 110:71-83 [DOI] [PubMed] [Google Scholar]
- 20.Hansford RG, Hogue BA, Mildaziene V: Dependence of H202 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr 1997, 29:89-95 [DOI] [PubMed] [Google Scholar]
- 21.Lowery O, Rosebrough N, Farr A, Randall R: Protein measurement with the folin phenol reagent. J Biol Chem 1951, 184:265-275 [PubMed] [Google Scholar]
- 22.Trounce IA, Kim YL, Jun AS, Wallace DC: Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 1996, 264:484-509 [DOI] [PubMed] [Google Scholar]
- 23.Harkness JE, Wagner JE: The Biology and Medicine of Rabbits and Rodents. 1995. Lea and Febiger, Philadelphia
- 24.Ally A, Park G: Rapid determination of creatine, phosphocreatine, purine bases and nucleotides (ATP, ADP, AMP, GTP, GDP) in heart biopsies by gradient ion-pair reversed-phase liquid chromatography. J Chromatogr 1992, 575:19-27 [DOI] [PubMed] [Google Scholar]
- 25.Williams JP, Headrick JP: Differences in nucleotide compartmentation and energy state in isolated and in situ rat heart: assessment by 31P-NMR spectroscopy. Biochim Biophys Acta 1996, 1276:71-79 [DOI] [PubMed] [Google Scholar]
- 26.Deschner EE, Hakissian M, Long FC: Genetic factors controlling inheritance of susceptibility to 1,2-dimethylhydrazine. J Cancer Res Clin Oncol 1989, 115:335-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng YG, Clayton EC, Means LW, Ramsdell JS: Repeated independent exposures to domoic acid do not enhance symptomatic toxicity in outbred or seizure-sensitive inbred mice. Fundam Appl Toxicol 1997, 40:63-67 [DOI] [PubMed] [Google Scholar]
- 28.Hamre K, Tharp R, Poon K, Xiong X, Smeyne RJ: Differential strain susceptibility following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration acts in an autosomal dominant fashion: quantitative analysis in seven strains of Mus musculus. Brain Res 1999, 828:91-103 [DOI] [PubMed] [Google Scholar]
- 29.Inoue H, Castagnoli K, Van Der Schyf C, Mabic S, Igarashi K, Castagnoli N, Jr: Species-dependent differences in monoamine oxidase A and B-catalyzed oxidation of various C4 substituted 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinyl derivatives. J Pharmacol Exp Ther 1999, 291:856-864 [PubMed] [Google Scholar]
- 30.He F, Zhang S, Lui L: Extrapyramidal lesions caused by mildewed sugarcane poisoning (with 3 case reports). Chin J Med 1987, 67:395-396 [PubMed] [Google Scholar]
- 31.He F, Zhang S, Qian F, Zhang C: Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid). Neurology 1995, 45:2178-2183 [DOI] [PubMed] [Google Scholar]
- 32.Lui X, Luo X, Hu W: Studies on the epidemiology and etiology of moldy sugarcane poisoning in China. Biomed Environ Sci 1992, 5:161-177 [PubMed] [Google Scholar]
- 33.Ming L: Moldy sugarcane poisoning—a case report with a brief review. Clin Toxicol 1995, 33:363-367 [DOI] [PubMed] [Google Scholar]
- 34.de la Asuncion JG, del Olmo ML, Sastre J, Millan A, Pellin A, Pallardo FV, Vina J: AZT treatment induces molecular and ultrastructural oxidative damage to muscle mitochondria. Prevention by antioxidant vitamins. J Clin Invest 1998, 102:4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies KJ, Doroshow JH: Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 1986, 261:3060-3067 [PubMed] [Google Scholar]
- 36.Doroshow JH, Davies KJ: Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 1986, 261:3068-3074 [PubMed] [Google Scholar]
- 37.Keller DA, Roe DC, Lieder PH: Fluoroacetate-mediated toxicity of fluorinated ethanes. Fundam Appl Toxicol 1996, 30:213-219 [DOI] [PubMed] [Google Scholar]
- 38.Lewis W, Dalakas MC: Mitochondrial toxicity of antiviral drugs. Nat Med 1995, 1:417-422 [DOI] [PubMed] [Google Scholar]
- 39.Pai VB, Nahata MC: Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention. Drug Saf 2000, 22:263-302 [DOI] [PubMed] [Google Scholar]
- 40.Tisdale MJ, Brennan RA: Role of fluroacetate in the toxicity of 2-fluroethylnitrosoureas. Biochem Pharmacol 1985, 34:3323-3327 [DOI] [PubMed] [Google Scholar]
- 41.Yuan C, Acosta D, Jr: Cocaine-induced mitochondrial dysfunction in primary cultures of rat cardiomyocytes. Toxicology 1996, 112:1-10 [DOI] [PubMed] [Google Scholar]
- 42.Fujihira S, Yamamoto T, Matsumoto M, Yoshizawa K, Oishi Y, Fujii T, Noguchi H, Mori H: The high incidence of atrial thrombosis in mice given doxorubicin. Toxicol Pathol 1993, 21:362-368 [DOI] [PubMed] [Google Scholar]
- 43.Schulz JB, Henshaw DR, MacGarvey U, Beal MF: Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem Int 1996, 29:167-171 [DOI] [PubMed] [Google Scholar]
- 44.Sato S, Gobbel GT, Li Y, Kondo T, Murakami K, Sato M, Hasegawa K, Copin JC, Honkaniemi J, Sharp FR, Chan PH: Blood-brain barrier disruption, HSP70 expression and apoptosis due to 3-nitropropionic acid, a mitochondrial toxin. Acta Neurochir 1997, 70(Suppl):S237-S279 [DOI] [PubMed] [Google Scholar]
- 45.Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF: Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J Neurosci 1998, 18:156-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthews RT, Yang L, Browne S, Baik M, Beal MF: Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA 1998, 95:8892-8897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ludolph AC, Seelig M, Ludolph AG, Sabri MI, Spencer PS: ATP deficits and neuronal degeneration induced by 3-nitropropionic acid. Ann NY Acad Sci 1992, 648:300-302 [DOI] [PubMed] [Google Scholar]
- 48.Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF: Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci 2000, 20:1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH: Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab 2000, 20:119-129 [DOI] [PubMed] [Google Scholar]
- 50.Hamilton BF, Gould DH: Correlation of morphologic brain lesions with physiologic alterations and blood-brain barrier impairment in 3-nitropropionic acid toxicity in rats. Acta Neuropathol 1987, 74:67-74 [DOI] [PubMed] [Google Scholar]
- 51.Gould DH, Wilson MP, Hamar DW: Brain enzyme and clinical alterations induced in rats and mice by nitroaliphatic toxicants. Toxicol Lett 1985, 27:83-89 [DOI] [PubMed] [Google Scholar]
- 52.Bogdanov MB, Ferrante RJ, Mueller G, Ramos LE, Martinou JC, Beal MF: Oxidative stress is attenuated in mice overexpressing BCL-2. Neurosci Lett 1999, 262:33-36 [DOI] [PubMed] [Google Scholar]
- 53.Beal MF, Henshaw DR, Jenkins BG, Rosen BR, Schulz JB: Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol 1994, 36:882-888 [DOI] [PubMed] [Google Scholar]
- 54.Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP: Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci 1998, 18:4439-4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hylin J, Matsumoto H: Inhibition of succinate dehydrogenase by 3-nitropropionate. Toxicol Appl Pharmacol 1964, 6:168-171 [Google Scholar]
- 56.Lopez PS, Castillo CH, Pastelin GH, Hernandez MR, Suarez MJ, Sanchez ML, Escalante BA: Characterization of 3-nitropropionic acid-induced bradycardia in isolated atria. Toxicol Appl Pharmacol 1998, 148:1-6 [DOI] [PubMed] [Google Scholar]
- 57.Castillo C, Reyes G, Rosas-Lezama MA, Valencia I, Hong E: Analysis of the cardiodepressor action of 3-nitropropionic acid. Proc West Pharmacol Soc 1994, 37:41-42 [PubMed] [Google Scholar]
- 58.Hong E, Castillo C, Rivero I, Somanathan R: Vasodilator and antihypertensive actions of 3-nitropropionic acid. Proc West Pharmacol Soc 1990, 33:209-211 [PubMed] [Google Scholar]
- 59.Guyot MC, Hantraye P, Dolan R, Palfi S, Maziere M, Brouillet E: Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 1997, 79:45-56 [DOI] [PubMed] [Google Scholar]
- 60.Miller PJ, Zaborszky L: 3-Nitropropionic acid neurotoxicity: visualization by silver staining and implications for use as an animal model of Huntington’s disease. Exp Neurol 1997, 146:212-229 [DOI] [PubMed] [Google Scholar]
- 61.Brierley JB: Cerebral Hypoxia. 1976. Edward Arnold, London
- 62.Meade CA, Figueredo-Cardenas G, Fusco F, Nowak TS, Jr, Pulsinelli WA, Reiner A: Transient global ischemia in rats yields striatal projection neuron and interneuron loss resembling that in Huntington’s disease. Exp Neurol 2000, 166:307-323 [DOI] [PubMed] [Google Scholar]
- 63.Burke RE, Karanas AL: Quantitative morphological analysis of striatal cholinergic neurons in perinatal asphyxia. Ann Neurol 1990, 27:81-88 [DOI] [PubMed] [Google Scholar]
- 64.Burke RE, Kent J, Kenyon N, Karanas A: Unilateral hypoxic-ischemic injury in neonatal rat results in a persistent increase in the density of striatal tyrosine hydroxylase immunoperoxidase staining. Brain Res Dev Brain Res 1991, 58:171-179 [DOI] [PubMed] [Google Scholar]
- 65.Chesselet MF, Gonzales C, Lin CS, Polsky K, Jin BK: Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol 1990, 110:209-218 [DOI] [PubMed] [Google Scholar]
- 66.Ferriero DM, Arcavi LJ, Sagar SM, McIntosh TK, Simon RP: Selective sparing of NADPH-diaphorase neurons in neonatal hypoxia-ischemia. Ann Neurol 1988, 24:670-676 [DOI] [PubMed] [Google Scholar]
- 67.Johnston MV, Hudson C: Effects of postnatal hypoxia-ischemia on cholinergic neurons in the developing rat forebrain: choline acetyltransferase immunocytochemistry. Brain Res 1987, 431:41-50 [DOI] [PubMed] [Google Scholar]
- 68.Mallard EC, Waldvogel HJ, Williams CE, Faull RL, Gluckman PD: Repeated asphyxia causes loss of striatal projection neurons in the fetal sheep brain. Neuroscience 1995, 65:827-836 [DOI] [PubMed] [Google Scholar]
- 69.Uemura Y, Kowall NW, Beal MF: Selective sparing of NADPH-diaphorase-somatostatin-neuropeptide Y neurons in ischemic gerbil striatum. Ann Neurol 1990, 27:620-625 [DOI] [PubMed] [Google Scholar]
- 70.Choi DW: Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1:623-634 [DOI] [PubMed] [Google Scholar]
- 71.Simpson JR, Isacson O: Mitochondrial impairment reduces the threshold for in vivo NMDA-mediated neuronal death in the striatum. Exp Neurol 1993, 121:57-64 [DOI] [PubMed] [Google Scholar]
- 72.Hier DB, Davis KR, Richardson EP, Jr, Mohr JP: Hypertensive putaminal hemorrhage. Ann Neurol 1977, 1:152-159 [DOI] [PubMed] [Google Scholar]
- 73.Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, Ooiwa T, Baba H: The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma 2000, 17:251-260 [DOI] [PubMed] [Google Scholar]
- 74.Portera-Cailliau C, Price DL, Martin LJ: Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol 1997, 378:70-87 [PubMed] [Google Scholar]
- 75.Onley JW: Neurotoxicity of Excitotoxicity Amino Acids. 1978. Raven Press, New York
- 76.Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ: Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol 1997, 42:335-348 [DOI] [PubMed] [Google Scholar]
- 77.Pluta R, Lossinsky AS, Wisniewski HM, Mossakowski MJ: Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res 1994, 633:41-52 [DOI] [PubMed] [Google Scholar]
- 78.Karibe H, Chen SF, Zarow GJ, Gafni J, Graham SH, Chan PH, Weinstein PR: Mild intraischemic hypothermia suppresses consumption of endogenous antioxidants after temporary focal ischemia in rats. Brain Res 1994, 649:12-18 [DOI] [PubMed] [Google Scholar]
- 79.Juurlink BH: Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neurosci Biobehav Rev 1997, 21:151-166 [DOI] [PubMed] [Google Scholar]
- 80.de la Torre J: Cerebral hypoperfusion, capillary degeneration, and development of Alzheimer disease. Alzheimer Dis Assoc Disord 2000, 14 (Suppl 1):S72-S81 [DOI] [PubMed] [Google Scholar]
- 81.La Fontaine MA, Geddes JW, Banks A, Butterfield DA: 3-nitropropionic acid induced in vivo protein oxidation in striatal and cortical synaptosomes: insights into Huntington’s disease. Brain Res 2000, 858:356-362 [DOI] [PubMed] [Google Scholar]
- 82.Wisniewski HM, Pluta R, Lossinsky AS, Mossakowski MJ: Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol 1995, 90:432-440 [DOI] [PubMed] [Google Scholar]
- 83.Arai T, Tsukahara I, Nitta K, Kojo H, Amakawa K: Responsiveness of cerebral vessels to changes of blood pressure and partial pressure of carbon dioxide after a transient period of cardiac arrest in dogs. Resuscitation 1985, 12:237-245 [DOI] [PubMed] [Google Scholar]
- 84.Castillo C, Valencia I, Reyes G, Hong E: An analysis of the antihypertensive properties of 3-nitropropionic acid, a compound from plants in the genus Astragalus. Arch Inst Cardiol Mex 1993, 63:11-16 [PubMed] [Google Scholar]
- 85.Gerlai R: Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci 1996, 19:177-181 [DOI] [PubMed] [Google Scholar]
- 86.Wiegand F, Liao W, Busch C, Castell S, Knapp F, Lindauer U, Megow D, Meisel A, Redetzky A, Ruscher K, Trendelenburg G, Victorov I, Riepe M, Diener HC, Dirnagl U: Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab 1999, 19:1229-1237 [DOI] [PubMed] [Google Scholar]
- 87.Weih M, Bergk A, Isaev NK, Ruscher K, Megow D, Riepe M, Meisel A, Victorov IV, Dirnagl U, Dirnagi U: Induction of ischemic tolerance in rat cortical neurons by 3-nitropropionic acid: chemical preconditioning. Neurosci Lett 1999, 272:207-210 [DOI] [PubMed] [Google Scholar]
- 88.Sugino T, Nozaki K, Takagi Y, Hashimoto N: 3-Nitropropionic acid induces ischemic tolerance in gerbil hippocampus in vivo. Neurosci Lett 1999, 259:9-12 [DOI] [PubMed] [Google Scholar]
- 89.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, Hugon J: Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning.” J Cereb Blood Flow Metab 1997, 17:257-264 [DOI] [PubMed] [Google Scholar]
- 90.Riepe MW, Ludolph AC: Chemical preconditioning: a cytoprotective strategy. Mol Cell Biochem 1997, 174:249-254 [PubMed] [Google Scholar]
- 91.Ockaili RA, Bhargava P, Kukreja RC: Chemical preconditioning with 3-nitropropionic acid in hearts: role of mitochondrial K(ATP) channel. Am J Physiol 2001, 280:H2406-H2411 [DOI] [PubMed] [Google Scholar]
- 92.Arellano M, Malet-Martino M, Martino R, Gires P: The anti-cancer drug 5-fluorouracil is metabolized by the isolated perfused rat liver and in rats into highly toxic fluoroacetate. Br J Cancer 1998, 77:79-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jones T, Hunt R: Veterinary Pathology, ed 5. 1983. Lea and Febiger, Philadelphia
- 94.Papadopoulou LC, Theophilidis G, Thomopoulos GN, Tsiftsoglou AS: Structural and functional impairment of mitochondria in adriamycin-induced cardiomyopathy in mice: suppression of cytochrome c oxidase II gene expression. Biochem Pharmacol 1999, 57:481-489 [DOI] [PubMed] [Google Scholar]
- 95.Yen HC, Oberley TD, Gairola CG, Szweda LI, St Clair DK: Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch Biochem Biophys 1999, 362:59-66 [DOI] [PubMed] [Google Scholar]