

Abstract
Myofibroblast activation is a key event playing a critical role in the progression of chronic renal disease. Emerging evidence suggests that myofibroblasts can derive from tubular epithelial cells by an epithelial to mesenchymal transition (EMT); however, the details regarding the conversion between these two cell types are poorly understood. Here we dissect the key events during the process of EMT induced by transforming growth factor-β1. Incubation of human tubular epithelial cells with transforming growth factor-β1 induced de novo expression of α-smooth muscle actin, loss of epithelial marker E-cadherin, transformation of myofibroblastic morphology, and production of interstitial matrix. Time-course studies revealed that loss of E-cadherin was an early event that preceded other alterations during EMT. The transformed cells secreted a large amount of matrix metalloproteinase-2 that specifically degraded tubular basement membrane. They also exhibited an enhanced motility and invasive capacity. These alterations in epithelial phenotypes in vitro were essentially recapitulated in a mouse model of renal fibrosis induced by unilateral ureteral obstruction. Hence, these results indicate that tubular epithelial to myofibroblast transition is an orchestrated, highly regulated process involving four key steps including: 1) loss of epithelial cell adhesion, 2) de novo α-smooth muscle actin expression and actin reorganization, 3) disruption of tubular basement membrane, and 4) enhanced cell migration and invasion.
Progression of chronic renal disease is considered to be an irreversible process that eventually leads to end-stage renal failure characterized by wide-spread tissue fibrosis. 1-3 Regardless of the initial causes, renal fibrosis is characterized by massive interstitial myofibroblast activation that is believed to play a central role in the pathogenesis of renal interstitial fibrosis. 4-6 Although the exact origins of these α-smooth muscle actin (α-SMA)-positive myofibroblasts remain uncertain, 7-9 emerging evidence suggests that they may derive from tubular epithelial cells
by an epithelial to mesenchymal transition (EMT) process under pathological conditions. 10-12 However, the details regarding the conversion between these two distinct types of cells are poorly defined.
Because tubular epithelial cells and interstitial myofibroblasts dramatically differ in their morphology and phenotypes, and are located in separated tissue compartments within the kidneys, one can envision that there have to be remarkable alterations in the expression of many sets of genes to make this phenotypic conversion possible. Indeed, previous studies have identified altered expression patterns of several genes such as α-SMA and fibroblast-specific protein-1 (Fsp1) during tubular epithelial to myofibroblast transition (EMT). 13,14 However, the cause-effect relationship of these changes in EMT as well as the key events during the entire EMT course at cellular level remain to be fascinating unanswered questions.
Given the fact that several key obstacles have to be overcome to make epithelial to myofibroblast transition possible, we propose that tubular epithelial to myofibroblast transition at the cellular level is likely a step-wise process involving several crucial events that eventually lead to the completion of the entire course. It is conceivable that these tubular epithelial cells lose the key epithelial cell markers that make them epithelium in the first place, while acquiring de novo expression of myofibroblastic markers that define their newly adapted morphology and phenotypes. Likewise, these cells have to find a way to pass across the tubular basement membrane (TBM) that surrounds the renal tubule and finally enter their newly found home, the interstitial compartments of the kidneys.
In this study, we attempt to decipher the key events controlling the tubular epithelial to myofibroblast transition both in vitro and in vivo. Our data suggest that the entire EMT process consists of several key steps that depend on hyperactive transforming growth factor (TGF)-β1 signaling.
Materials and Methods
Cell Culture and Treatment
Human proximal tubular epithelial cells (HKC-8) were kindly provided by Dr. L. Racusen of Johns Hopkins University. 15 Cells were cultured in Dulbecco’s modified Eagle’s medium-F12 medium supplemented with 5% fetal bovine serum (Life Technologies, Inc., Grand Island, NY). 16 For cytokine treatment, HKC cells were seeded at ∼70% confluence in complete medium containing 5% fetal bovine serum. Twenty-four hours later, the cells were changed to serum-free medium, and recombinant TGF-β1 (R & D Systems, Minneapolis, MN) was added at a final concentration of 4 ng/ml except where otherwise indicated. The cells and conditioned media were collected at different time points for further characterization.
Animal Model
Male CD-1 mice weighing 20 to 22 g were obtained from Harlan Sprague-Dawley (Indianapolis, IN). Unilateral ureteral obstruction (UUO) was performed using an established procedure. 17 Briefly, under general anesthesia, complete ureteral obstruction was performed by double-ligating the left ureter using 4-0 silk after a midline abdominal incision. Sham-operated mice had their ureters exposed, manipulated but not ligated. Mice were sacrificed at different time points as indicated after surgery, and the kidneys were removed. One part of the kidneys was fixed in 10% phosphate-buffered formalin followed by paraffin embedding for histological and immunohistochemical studies. The remaining kidneys were snap-frozen in liquid nitrogen and stored at −80°C for protein extractions.
Western Immunoblot Analysis
HKC cells and cytokine-treated cells were lysed with sodium dodecyl sulfate (SDS) sample buffer (62.5 mmol/L Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mmol/L dithiothreitol, and 0.1% bromophenol blue). Kidney tissue was homogenized by a polytron homogenizer (Brinkmann Instruments, Westbury, NY) and the supernatant was collected after centrifugation at 13,000 × g at 4°C for 20 minutes, as described previously. 18 After protein concentration was determined using a bicinchoninic acid protein assay kit (Sigma Chemical Co., St. Louis, MO), the tissue lysate was mixed with an equal amount 2× SDS sample buffer. Samples were heated at 100°C for ∼5 to 10 minutes before loading and separated on precasted 10% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA). The proteins were electrotransferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL) in transfer buffer containing 48 mmol/L Tris-HCl, 39 mmol/L glycine, 0.037% SDS, and 20% methanol at 4°C for 1 hour. Nonspecific binding to the membrane was blocked for 1 hour at room temperature with 5% Carnation nonfat milk in TBS buffer (20 mmol/L Tris-HCl, 150 mmol/L NaCl, and 0.1% Tween 20). The membranes were incubated for 16 hours at 4°C with various primary antibodies in TBS buffer containing 5% milk at the dilutions specified by the manufacturers. The monoclonal α-SMA antibody was purchased from Sigma Chemical Co. The antibody for E-cadherin was obtained from Transduction Laboratories (Lexington, KY). The antibody against β-actin was purchased from Santa Cruz Biochemicals (Santa Cruz, CA). The anti-human matrix metalloproteinase (MMP)-2 antibody was purchased from Oncogene Research Products (Cambridge, MA). Binding of primary antibodies was followed by incubation for 1 hour at room temperature with the secondary horseradish peroxidase-conjugated IgG in 1% nonfat milk. The signals were visualized by the enhanced chemiluminescence system (ECL, Amersham), as described previously. 18
Immunostaining
Indirect immunofluorescence staining was performed using an established procedure on HKC cells cultured on coverslips. Briefly, control or cytokine-treated HKC cells were washed with cold phosphate-buffered saline (PBS) twice, and fixed with cold methanol:acetone (1:1) for 10 minutes on ice. After extensive washing with PBS containing 0.5% bovine serum albumin, the cells were blocked with 20% normal donkey serum in PBS buffer for 30 minutes at room temperature, and then incubated with specific primary antibodies described above, except the rat monoclonal anti-E-cadherin (clone DECMA-1) that was obtained from Sigma. The cells were then routinely stained with fluorescein isothiocyanate-conjugated secondary antibodies (Sigma). Cells were also stained with 4′,6-diamidino-2-phenylindole, HCl to visualize the nuclei. For visualizing F-actin, cells were stained with tetramethylrhodamine isothiocyanate-conjugated phalloidin (Sigma). Stained cells were mounted with anti-fade mounting medium (Vector Laboratories, Burlingame, CA) and viewed on a Nikon Eclipse E600 Epi-fluorescence microscope (Nikon, Melville, NY). For immunostaining renal tissue, kidney sections from paraffin-embedded tissues were prepared at 4-μm thickness using a routine procedure. 10 Immunohistochemical localization was performed using the Vector M.O.M. immunodetection kit (Vector Laboratories). The primary antibodies used were anti-E-cadherin and anti-α-SMA (Sigma), anti-TGF-β1 and anti-TGF-β type I receptor (Santa Cruz Biochemicals). As a negative control, the primary antibody was replaced with either nonimmune mouse or rabbit IgG, corresponding to species of the primary antibodies.
Gelatin Zymographic Analysis
Zymographic analysis of the MMP proteolytic activity in the supernatant of cultured cells or kidney tissue homogenates was performed according to the method described previously. 10,19 Briefly, an equal number of the HKC cells were seeded on 6-well plates at a density of 4 × 105/well in Dulbecco’s modified Eagle’s medium-F12 medium containing 5% fetal bovine serum. Twenty-four hours later, culture medium was changed to 0.7 ml of serum-free medium, and TGF-β1 was added to the cultures. At different time points as indicated, conditioned media were collected and centrifuged at 13,000 × g for 5 minutes to remove any cell debris. The protein concentration was determined using a protein assay kit with bovine serum albumin as a standard (Sigma). Kidney tissue homogenates were prepared essentially according to the methods described by Kim and colleagues. 19 A constant amount of protein from the conditioned media (15 μg) or kidney tissue homogenates (30 μg) was loaded into 10% SDS-polyacrylamide gel containing 1 mg/ml gelatin (Bio-Rad). After electrophoresis, SDS was removed from the gel by incubation in 2.5% Triton X-100 at room temperature for 30 minutes with gentle shaking. The gel was washed well with distilled water to remove detergent and incubated at 37°C for ∼16 to 36 hours in a developing buffer containing 50 mmol/L Tris-HCl, pH 7.6, 0.2 mol/L NaCl, 5 mmol/L CaCl2, and 0.02% Brij 35. The gel was then stained with a solution of 30% methanol, 10% glacial acetic acid, and 0.5% Coomassie Blue G250, followed by destaining in the same solution without dye. Proteinase activity was detected as unstained bands on a blue background representing areas of gelatin digestion.
Determination of Basement Membrane Integrity by Bacterial Translocation
Bacterial translocation was performed for evaluating TBM integrity using a two-compartment Boyden chamber with transwell filters containing 3-μm diameter pores (Corning Co., Corning, NY). Matrigel (Becton Dickinson Labware, Bedford, MA), a solubilized basement membrane matrix consisting of laminin (56%), collagen IV (31%), entactin (8%), and heparan sulfate proteoglycan (perlecan), was added onto the transwell filters to form matrix gels at 37°C that essentially reconstitute the TBM in vivo. 20 Matrigel at a concentration of 22 μg/cm 2 produced a matrix gel layer at 15-μm depth, which represents 100-fold thickness of native TBM (150 nm). 21 Preliminary studies showed that the matrix gel on the transwell filters maintained its structural integrity for >7 days at 37°C that completely blocked bacterial translocation through the gel (data not shown). The transwell filters with the Matrigel were then incubated with the conditioned media (rich in MMP-2) from HKC cells treated with or without TGF-β1 at 37°C for 4 days. Escherichia coli DH5α (Life Technologies) was grown in Luria Broth (LB) medium at 37°C overnight and bacterial concentration was estimated by reading at an optical density of 600 nm with 1 optical density equivalent to 10 9 bacteria/ml. 22 Approximately 10 8 bacteria in 100 μl were added to the upper compartment of the Boyden chamber in a final volume of 400 μl. Aliquots (20 μl) were removed at 0.5 and 2 hours, respectively, from the bottom compartment of the chamber containing 1 ml of media. Dilutions from each aliquot were plated on LB agar plates and incubated at 37°C for 16 hours and colonies were counted. The entire experiments were performed in triplicate for each time points per treatment.
Boyden Chamber Motility Assay
Cell motility and migration were evaluated using Boyden chamber motogenicity assay with tissue culture-treated transwell filters (Costar). 23 HKC cells (1 × 104) were seeded onto the filters (8-μm pore size, 0.33-cm 2 growth area) in the top compartment of the chamber. After 2 or 5 days of incubation with or without TGF-β1 at 37°C, filters were fixed with 3% paraformaldehyde in PBS, and stained with 0.1% Coomassie Blue in 10% methanol and 10% actic acid, and the upper surface of the filters was carefully wiped with a cotton-tipped applicator. Cells that passed through the pores were counted in five nonoverlapping ×20 fields and photographed with a Nikon microscope.
Matrigel Invasion Assay
Matrigel (1.43 mg/cm2) was added onto the transwell filters (8-μm pore size, 0.33-cm 2 growth area) of the Boyden chamber to form matrix gels at 1.0-mm depth. HKC cells (1 × 104) in a volume of 100 μl were added onto the top of the gels. After 2 and 5 days of incubation with or without TGF-β1 at 37°C, filters were fixed with 3% paraformaldehyde in PBS, and stained with 0.1% Coomassie Blue in 10% methanol and 10% actic acid, and the upper surface of the filters was carefully wiped with a cotton-tipped applicator. Cells that invaded and migrated across the Matrigel and passed the transwell filter pores toward the lower surface of the filters were counted in five nonoverlapping ×10 fields. The experiments were performed in triplicate cultures.
Determination of Tissue TGF-β1 Levels by Enzyme-Linked Immunosorbent Assay
For measurement of tissue TGF-β1 level, kidneys were homogenized in the extraction buffer containing 20 mmol/L Tris-HCl, pH 7.5, 2 mol/L NaCl, 0.1% Tween-80, 1 mmol/L ethylenediaminetetraacetic acid, and 1 mmol/L phenylmethyl sulfonyl fluoride, and the supernatant was recovered after centrifugation at 19,000 × g for 20 minutes at 4°C. Kidney tissue TGF-β1 level was determined by using a commercial Quantikine TGF-β1 enzyme-linked immunosorbent assay kit in accordance with the protocol specified by the manufacturer (R & D Systems). This kit measures the abundance of active TGF-β1 protein that binds to its soluble type II receptor precoated onto a microplate. The concentration of tissue TGF-β1 in kidneys was expressed as pg/mg total protein.
Statistical Analysis
All data examined were expressed as mean ± SE. For Western blot analysis, quantitation was performed by scanning and analyzing the intensity of the hybridization signals using NIH Imagine software. Statistical analysis of the data were performed by the Student-Newman-Keuls test using SigmaStat software (Jandel Scientific, San Rafael CA). A P value <0.05 was considered to be statistically significant.
Results
TGF-β1 Induces Tubular Epithelial to Myofibroblast Transition in Vitro
To demonstrate tubular epithelial to myofibroblast transition in vitro, we examined the de novo expression of α-SMA, a phenotypic marker for myofibroblast cells, in human renal tubular epithelial cells (HKC). As shown in Figure 1 ▶ , incubation of HKC cells with TGF-β1 induced abundant expression of α-SMA protein. Dose-dependence studies revealed that TGF-β1 was able to induce α-SMA at a concentration as low as 0.1 ng/ml, suggesting that this induction is readily achievable in vivo under pathological conditions. The induction of α-SMA expression in tubular epithelial cells reached a plateau when TGF-β1 was >2 ng/ml.
Figure 1.

TGF-β1 induces de novo expression of α-SMA in tubular epithelial cells. HKC cells treated without (control) or with different amounts of TGF-β1 for 72 hours in serum-free medium. The whole cell lysate was immunoblotted with a specific α-SMA antibody. The same blot was reprobed with β-actin to ensure equal loading of each lane.
Figure 2 ▶ shows the phenotypic conversion of tubular epithelial cells after TGF-β1 treatment. The transformed cells displayed the presence of abundant α-SMA-positive microfilaments in the cytoplasm. Meanwhile, they totally lost the staining of E-cadherin, an epithelial marker that is essential for the structural integrity of renal epithelium (Figure 2) ▶ . We observed dramatic alteration in the organization of actin cytoskeleton. The transformed cells underwent F-actin reorganization to form long stress fibers (Figure 2) ▶ . Consistent with these actin reorganizations that often define cell morphology, the transformed cells lost the typical cobblestone pattern of an epithelial monolayer, and displayed a spindle-shape, fibroblast-like morphology (Figure 2) ▶ . In addition, TGF-β1-treated cells expressed vimentin, a marker of mesenchymal cells, and began to markedly produce fibronectin and collagen I (data not shown). All together, these data suggest that tubular epithelial cells, under appropriate stimulus, undergo a conversion process into myofibroblasts in vitro.
Figure 2.
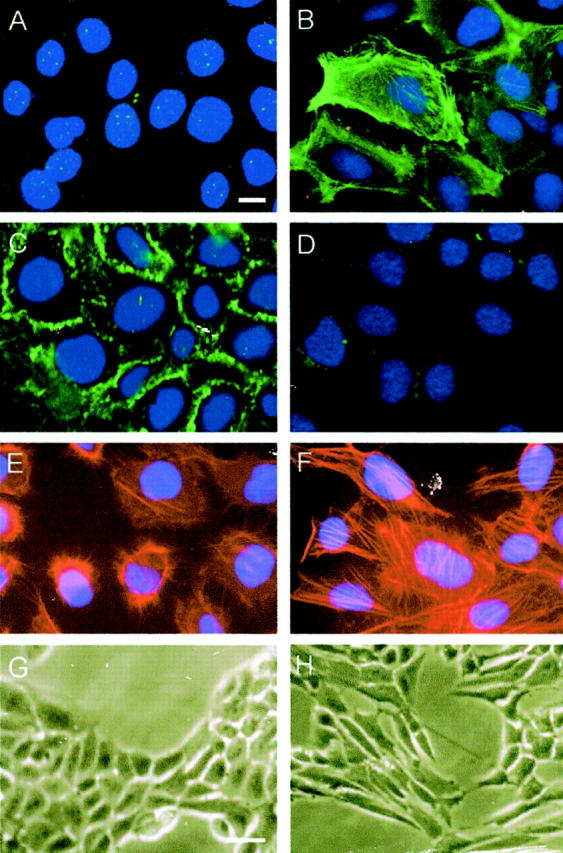
Tubular epithelial to myofibroblast transition in vitro. HKC cells treated without (left column) or with 4 ng/ml of TGF-β1 (right column) for 72 hours in serum-free medium. The myofibroblast or epithelial cell markers were detected by an indirect immunostaining. The transformed cells acquired α-SMA (A, B), lost E-cadherin (C, D), formed stress fiber by actin reorganization (E, F), and displayed myofibroblast morphology (G, H). Scale bars: 10 μm (A–F), 20 μm (G and H).
Loss of E-Cadherin Expression Is an Early Event during Epithelial to Myofibroblast Conversion
To identify the early event essential for epithelial to myofibroblast conversion, we investigated the time course of the gene expression after TGF-β1 treatment. As shown in Figure 3 ▶ , we found that loss of E-cadherin expression was an early event that took place as early as 6 hours after TGF-β1 treatment, whereas induction of de novo expression of α-SMA was a delayed response requiring 36 hours of incubation (Figure 3) ▶ . Other changes in cell phenotype such as induced vimentin and fibronectin expression as well as morphological transformation also required longer periods of persistent incubation with TGF-β1 ranging from 2 to 5 days (data not shown). These results establish that loss of E-cadherin expression probably is an early event, which allows dissociation of structural integrity of renal epithelia and collapse of epithelial polarity.
Figure 3.
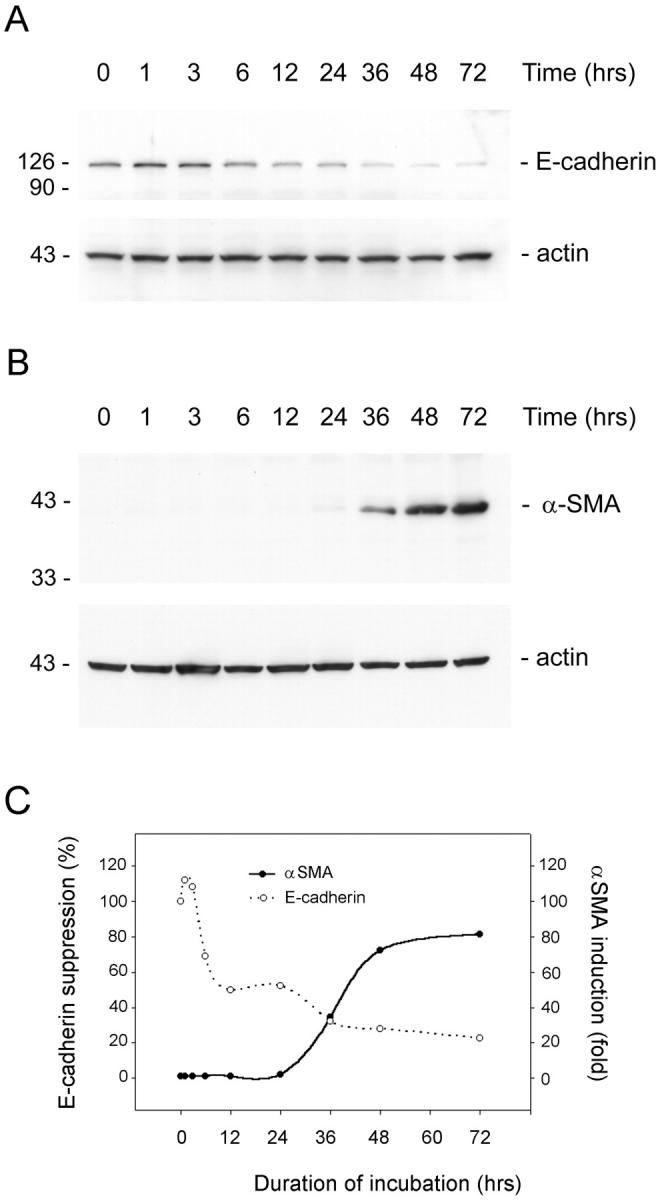
Loss of E-cadherin is an early event during epithelial to myofibroblast transition. HKC cells were treated with TGF-β1 for various periods of time as indicated in serum-free medium. The whole cell lysate was immunoblotted with antibodies against α-SMA, E-cadherin, and β-actin, respectively. A: Time-course of E-cadherin expression. B: Time-dependency of α-SMA expression. C: Graphic presentation of the relative abundance of E-cadherin and α-SMA after TGF-β1-induced cell phenotypic transition.
Up-Regulation of MMP-2 Expression and Its Role in Disrupting Tubular Basement Membrane
To gain insights into the mechanism underlying the destruction of TBM in vivo, we examined the expression pattern of MMPs during TGF-β1-induced EMT. Zymographic analysis of conditioned media exhibited that TGF-β1 induced a marked increase in MMP-2 expression and secretion in a dose-dependent manner (Figure 4A) ▶ . TGF-β1 not only induced pro-MMP-2 abundance, but also stimulated activation of pro-MMP-2, as demonstrated by increased abundance of active MMP-2 in the conditioned media. Time-course studies revealed that this induction was also a delayed response that took place after 48 hours of incubation with TGF-β1 (Figure 4B) ▶ . TGF-β1 also marginally increased MMP-9 activation, as demonstrated by an increase in active MMP-9 abundance in zymographic gels (Figure 4A) ▶ . The induction of MMP-2 expression by TGF-β1 in tubular epithelial cells was independently confirmed by Western blot analyses of the conditioned media (Figure 4) ▶ .
Figure 4.

TGF-β1 induces MMP expression in tubular epithelial cells. A and B: Zymographic analysis of the conditioned media derived from HKC cells treated without (control) or with different amounts of TGF-β1 for various periods of time as indicated. Samples equalized for protein content were separated on a 10% SDS-polyacrylamide gel containing 0.1% gelatin. Proteolytic activity is demonstrated by digestion of gelatin, resulting in the bands of clearing. The locations of bands corresponding to pro- and active MMP-2 as well as pro- and active MMP-9 are indicated. C and D: Western blot analyses of the conditioned media from HKC cells treated without (control) or with various concentration of TGF-β1 for different periods of time in the serum-free medium. The location of a 72-kd band corresponding to MMP-2 is indicated. A and C: HKC cells were incubated with different concentrations of TGF-β1 as indicated for 72 hours. B and D: HKC cells were incubated with 2 ng/ml of TGF-β1 for various periods of time as indicated.
To directly demonstrate whether the increased expression of MMP-2 is responsible for disrupting the structural and functional integrity of TBM, we examined the ability of conditioned media derived from the transformed cells to degrade Matrigels that essentially reconstitute the native TBM. 24-26 As shown in Figure 5 ▶ , incubation of Matrigels with conditioned media derived from TGF-β1-treated cells markedly disrupted the integrity of basement membrane matrix. Bacterial translocation experiments revealed that there was little resistance for bacteria to translocate from the upper to the bottom compartments of the Boyden chamber across the matrix gels and 3-μm pores on the transwell filters (Figure 5) ▶ , suggesting that the integrity of Matrigels is impaired, presumably because of the degradation by MMP-2 that is rich in the conditioned media. However, under the same conditions, bacterial translocation was primarily blocked when the Matrigels were incubated with conditioned media from control HKC cells (Figure 5) ▶ .
Figure 5.

Functional disruption of the reconstituted TBM by the conditioned media from the transformed cells. Matrigels (15-μm depth) analogous to native TBM matrix were formed on the transwell filters and incubated with the conditioned media from either control or TGF-β1-treated HKC cells for 4 days. The integrity of Matrigels was assessed by bacterial translocation through the gels. A and B: Representative plates show the colonies formed from the bacteria passed through the Matrigels. C: Graphic presentation of the numbers of bacteria translocated via the gels. **, P < 0.01 versus control (n = 3).
Enhanced Motility and Invasive Capacity of the Transformed Cells
To understand how the transformed cells migrate and finally enter the interstitial compartment in vivo, we studied the motility of transformed cells by Boyden chamber assay. As shown in Figure 6 ▶ , incubation of HKC cells with TGF-β1 for 2 days began to induce cell migration across the pores of the transwell filters. Approximately 50% of the pores in transwell filters were filled with cell extensions after 2 days of TGF-β1 treatment, but no cells truly migrated through the pores of filters toward the opposite side of the filters at this stage (Figure 6) ▶ . However, the control HKC cells had barely started to move toward the pores of transwell filters after the same period of incubation. After 5 days of incubation, increased numbers of the cells in TGF-β1-treated groups actually migrated across the pores to the lower surface of the filters, compared to that in control groups (Figure 6) ▶ .
Figure 6.

Enhanced migration of the transformed cells. HKC cells were seeded on the transwell filters of Boyden chamber (pore size, 8 μm) and incubated with or without TGF-β1 for 2 and 5 days, respectively. The cells or cell extensions that passed through the pores of filters were counted after staining. A–C: Most pores of the transwell filters were filled with cell extensions after incubation with TGF-β1 for 2 days, but no cells migrated through the pores to the opposite side of filters. D–F: After incubation with TGF-β1 for 5 days, the transformed cells migrated through the pores to the opposite side of filters. A, B, D, E: Representative micrographs of the transwell filters. C and F: Graphic presentation of the numbers of cells or cell extensions migrated through the pores of the filters after incubation with or without TGF-β1 for 2 and 5 days, respectively. **, P < 0.01 versus control (n = 3).
We next analyzed the invasive capacity of transformed cells by a Matrigel invasion assay. Because the cells were seeded on the top of Matrigel, migration of the transformed cells toward the pores of transwell filters theoretically had to result from both degradation of Matrigel and enhanced motility. As shown in Figure 7 ▶ , incubation of HKC cells with TGF-β1 for 5 days markedly promoted cell invasion into and migration across the Matrigel. Most pores in transwell filters were filled with cell extensions after 5 days of incubation with TGF-β1, which resulted from the cells invading and migrating across the Matrigels (Figure 7) ▶ . Under the same conditions, control HKC cells, without TGF-β1 treatment, displayed a much less invasive property in this assay (Figure 7) ▶ . These data suggest that tubular epithelial cells on stimulation by TGF-β1 acquire new properties that are able to disrupt TBM matrix and invade and migrate through the Matrigel, which presumably allow them to move across the TBM and ultimately to enter the interstitial compartments of the kidney in vivo.
Figure 7.
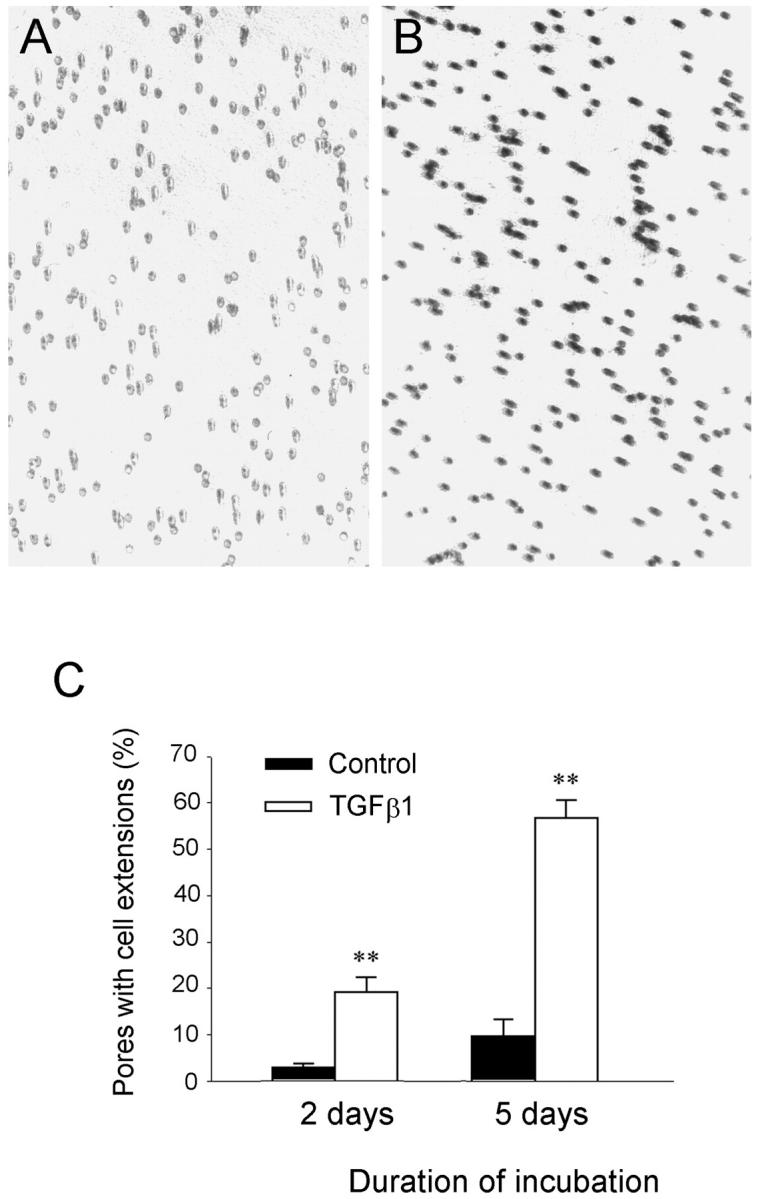
Increased invasive capacity of the transformed cells on Matrigel. HKC cells were seeded on the top of Matrigels in the transwell filters of the Boyden chamber and incubated with or without TGF-β1 for 2 and 5 days, respectively. The cell extensions that migrated across the Matrigels and passed through the pores of filters were counted after staining. A and B: Representative micrographs show the pores of the transwell filters filled with cell extensions after incubation without or with TGF-β1 for 5 days. C: Graphic presentation of the numbers of pores filled with cell extensions after incubation with or without TGF-β1 for 2 and 5 days, respectively. **, P < 0.01 versus control (n = 3).
Tubular Epithelial to Myofibroblast Transition in Vivo
To demonstrate tubular epithelial to myofibroblast conversion in vivo under pathological conditions, we examined the kidneys of mice with chronic renal disease induced by UUO. As shown in Figure 8, a ▶ rapid and marked induction of both TGF-β1 and its type I receptor (TβR-I) was observed in the obstructed kidneys as early as 1 day after UUO, as demonstrated quantitatively by enzyme-linked immunosorbent assay and Western blot analyses. Immunohistochemical localization studies revealed that the expression of TGF-β1 and TβR-I was specifically induced in renal tubular epithelia of the obstructed kidneys (Figure 8) ▶ . This rapid, tubule-specific induction of TGF-β1 axis clearly precedes the phenotypic changes observed in the obstructed kidneys in this model (see below), suggesting that hyperactive TGF-β1 signaling in vivo specifically targets renal tubules and its induction is early enough for initiating epithelial to myofibroblast transition under pathological conditions.
Figure 8.
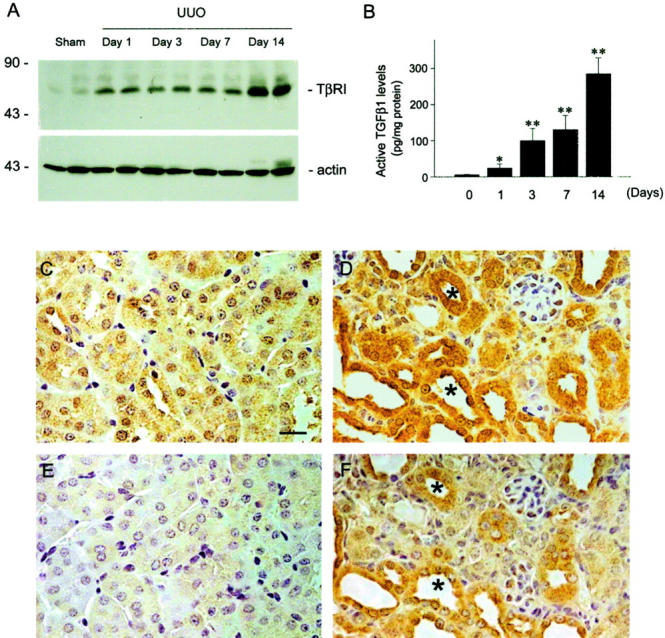
Expression of both TGF-β1 and its type I receptor increases rapidly and specifically in renal tubular epithelia in mouse model of renal interstitial fibrosis induced by UUO. A: Western blot demonstrates an early induction of TGF-β type I receptor expression in the obstructed kidneys after UUO. Representative picture shows the immunoblotting results of two animals per time point. Marked TβR-I elevation was observed as early as 1 day after UUO. B: Quantitative determination of TGF-β1 protein by enzyme-linked immunosorbent assay reveals a time-dependent induction of TGF-β1 in the obstructed kidneys. Data are presented as mean ± SE from four animals per group. *, P < 0.05; **, P < 0.01 versus sham. Representative micrographs show the localization of TGF-β type I receptor (C and D) and TGF-β1 (E and F) in the kidneys at 7 days after UUO (D and F) or sham operation (C and E). Both TGF-β1 and TβR-I are specifically induced in renal tubular epithelia after UUO (F and D). Asterisk indicates positively stained tubules for both TGF-β1 and TβR-I in serial sections. Scale bar, 20 μm.
We observed dramatic changes in renal tubular epithelial cell phenotypes after UUO, with complete loss of E-cadherin expression and de novo expression of α-SMA (Figure 9) ▶ . There was a reciprocally inverse correlation between the loss of E-cadherin and de novo expression of α-SMA in the obstructed kidneys. These alterations in cell phenotypes after UUO were coinciding with an increase in TBM-degrading enzymes in these kidneys (Figure 10) ▶ . Zymographic analysis of whole kidney lysates revealed that MMP-2 and MMP-9 expressions were dramatically increased in a time-dependent manner (Figure 10) ▶ . All together, these data essentially recapitulate our in vitro findings and suggest that tubular epithelial to myofibroblast conversion occurs in vivo as well, and presumably plays an important role in the pathogenesis of renal interstitial fibrosis.
Figure 9.

Alterations of protein expression pattern in the obstructed kidneys recapitulate key events in tubular epithelial to myofibroblast transition. Kidney sections were prepared from mice at 7 days after either UUO or sham operation and stained with various specific antibodies. Representative micrographs show the alterations in protein expression pattern after UUO. A and C: Sham-operated mice. B and D: UUO mice. A and B: E-cadherin. C and D: α-SMA. Scale bar, 20 μm. Arrowheads indicate positively stained cells. E: Western blot demonstrates a time-dependent alteration in the expression of E-cadherin and α-SMA in the obstructed kidneys after UUO. Representative pictures show the results of two animals per time point.
Figure 10.

Induction of renal MMP-2 and -9 expressions in vivo. Kidney tissues were collected from mice receiving UUO at different time points as indicated. A: Zymographic analysis of whole kidney lysates indicates that renal MMP-2 and -9 expressions were markedly induced in a time-dependent manner. B: Graphic presentation of the relative abundance (fold induction) of MMP-2 and MMP-9 in the kidneys at different time points after UUO.
Discussion
Although tubular epithelial to myofibroblast conversion may require a sequential activation and repression of the expression of many sets of genes in a coordinated way, we hypothesized that, at cellular level, several key events (or steps) could be both necessary and sufficient for the completion of the entire EMT process in vivo. In this report, using cultured tubular epithelial cells as a model system, we have identified the four key steps that are crucial in the process of tubular epithelial to myofibroblast transition. As illustrated in Figure 11 ▶ , these four key events include: 1) loss of epithelial adhesion properties, 2) de novo expression of α-SMA and actin reorganization, 3) disruption of the TBM, and 4) enhanced cell migration and invasion. TGF-β1, a well-characterized pro-fibrogenic cytokine, 27,28 is capable of inducing tubular epithelial cells to undergo all four steps, and thereby, leads to the completion of the entire EMT course. Our data establish that EMT is an orchestrated, step-wise process that depends on hyperactive TGF-β1 signaling and provide novel insights into the mechanism underlying the myofibroblast activation under pathological conditions.
Figure 11.

Schematic illustration depicting four key events during tubular epithelial to myofibroblast transition. The diagram illustrates the four key events essential for the completion of entire EMT course at cellular level, which include: 1) loss of epithelial adhesion properties, 2) de novo expression of α-SMA and actin reorganization, 3) disruption of the TBM, and 4) enhanced migration and invasive capacity of the transformed cells. TGF-β1 as a sole factor is capable of inducing tubular epithelial cells to undergo all four steps.
Alterations in the expression of many genes have been identified during epithelial to mesenchymal conversion. 29,30 For instance, the transformed cells acquire de novo expression of α-SMA and Fsp1. 13,14 However, it remains to be one of the greatest challenges to distinguish those changes playing a key role in EMT from those just associated with it. Because TGF-β1 rapidly suppresses E-cadherin expression in cultured tubular epithelial cells, it is conceivable that loss of epithelial adhesion properties is an early important step that precedes all other major events such as induction of α-SMA and MMP-2 expression. Because E-cadherin, the well-characterized adhesion receptor found within adherens-type junctions, plays an essential role in maintaining the structural integrity of renal epithelia and in epithelial polarization, 31,32 its loss consequently allows destabilization of the structural integrity of renal epithelium and makes cells dissociate from their neighbors and lose polarity. E-cadherin is linked to the actin filament network by catenins, a family of intracellular adhesive junction proteins. The importance of E-cadherin for development of normal epithelium has been established by knock-out of its gene in mice 33 and by its role in embryonic epitheliogenesis during early nephrogenesis. 34 Recent results further demonstrate that suppression of E-cadherin expression alone, by the transcription factor Snail, induces EMT in carcinoma cells. 35,36
Because tubular epithelial cells and myofibroblasts locate in separate tissue compartments in vivo, disruption of TBM will be of fundamental importance in clearing the path for transformed cells to migrate toward interstitium. In light of the fact that MMP-2 specifically cleave native type IV collagen and laminin, 24,37 the principal proteins found in the TBM, our results on induction of MMP-2 during TGF-β1-initiated EMT provide significant insights into the mechanism underlying the destruction of TBM in vivo (Figures 4 and 5) ▶ ▶ . In accordance with this, incubation of Matrigel, which essentially reconstitutes TBM as shown by the similarity in its structure, composition, physical property, and ability to retain functional characteristics typical of TBMs in vivo, 20 with conditioned media from the transformed cells results in drastic destruction of its structural and functional integrity as evidenced by the bacterial translocation assay (Figure 5) ▶ . Of note, induction of MMP-2 expression occurs at 48 hours after TGF-β1 incubation in vitro (Figure 4) ▶ and at 3 days after UUO in vivo (Figure 10) ▶ , suggesting that destruction of TBM is a delayed event that follows loss of epithelial adhesion as well as α-SMA de novo expression during the entire course of tubular epithelial to myofibroblast transition. It is of interest to note that because TGF-β1 does not increase MMP-2 abundance in cultured rat renal interstitial fibroblast (NRK-49F) cells (data not shown), this induction of MMP-2 expressions in vivo is contributed, at least in a large part, by tubular epithelial cells under pathological conditions.
Because the transformed cells have to enter interstitial compartments, it is essential for them to acquire the motile and invasive capacity to eventually migrate into peritubular interstitium. The reorganization of actin cytoskeleton, and induction of α-SMA, may provide a structural foundation not only for defining the morphology of the transformed cells, 38,39 but also for them to migrate, invade, and even acquire the capacity for contractility. The observation that the transformed cells are more motile suggests that the enhanced motility could readily allow them to migrate through the TBM, which is already destructed by elevated MMP-2 as discussed above, toward the interstitial compartment. The transformed cells could, in reality, combine the efforts of simultaneous destruction of TBM and migration. Such a notion is experimentally confirmed by the Matrigel invasion assay, in which transformed cells grown on top of Matrigel gels have the ability to destroy and migrate through a reconstituted TBM matrix. Of note, TGF-β1 promotion of HKC cell motility and invasion is also a late event that occurs after long periods of incubation. This suggests that enhanced cell motility could be a consequence resulting from the tubular epithelial to myofibroblast conversion. In addition, myofibroblasts are morphologically intermediate between fibroblasts and smooth muscle cells. 4-6 Like fibroblasts, they produce interstitial matrix components including collagens I and III and fibronectin; and like smooth muscle cells, they retain α-SMA expression and have the ability to contract. 40,41 The possibility of these transformed cells to attain contractility, as evidenced by the well-assembled α-SMA microfilament fibers, implies that contraction could potentially be another powerful force leading the transformed cells toward the interstitium.
Major key events during EMT, such as loss of epithelial adhesion, de novo expression of α-SMA, and induction of TBM-degrading enzymes, are recapitulated in the diseased kidneys in an animal model of renal interstitial fibrosis. The fact that rapid, tubule-specific induction of TGF-β1 axis in the obstructed kidneys suggests that, similar to in vitro situation, TGF-β1 is also responsible for initiating epithelial to myofibroblast transition in vivo. However, suppression of E-cadherin expression in vivo does not significantly precede other alterations (Figure 9) ▶ . This discrepancy between in vitro and in vivo studies is probably because of the nature of heterogeneity in cell population in the diseased kidneys. Unlike cultured epithelial cells with homogeneous population, the response of tubular cells in vivo is more complex so that loss of E-cadherin at an early time point in a small percentage of the cell population may not be readily detected by Western blot analyses of whole kidney lysate. Consistent with this notion, loss of E-cadherin staining is observed in renal epithelia in the areas that otherwise are relatively normal (Figure 9) ▶ , suggesting that disruption of epithelial adhesion perhaps is an early event in vivo as well. Nonetheless, we cannot exclude the possibility that in vivo once tubular epithelial cells are initiated to undergo EMT, they may be programmed to progress by simultaneously inducing suppression of E-cadherin, α-SMA expression, and TBM destruction.
TGF-β1, as a sole factor, initiates and completes the entire EMT course that consists of four key steps. This extraordinary ability of TGF-β1 leads one to re-think its roles and mechanisms in progressive renal fibrosis. TGF-β1 is widely considered as a key modulator of organ fibrosis after a wide variety of tissue injuries. 27,28,42 Although TGF-β1 stimulation of fibroblasts to become activated is well documented, little is known about the effects of TGF-β1 on tubular epithelial cells in renal fibrogenesis. Ironically, it is in the tubular epithelium where TGF-β1 receptors are rapidly and specifically up-regulated in diseased kidneys 43 (Figure 8) ▶ , suggesting that tubular epithelial cells are the natural targets of TGF-β1 under pathological conditions in vivo. Our current results indicate that the pro-fibrogenic role of TGF-β1 is mediated, at least in part, by promoting myofibroblast activation via inducing tubular epithelial to myofibroblast transition.
In summary, the results of our study suggest that tubular epithelial to myofibroblast transition is an orchestrated, highly regulated, step-wise process that depends on hyperactive TGF-β1 signaling. Hence, disruption of any of these key steps could potentially offer unique opportunities to block the EMT process and, thereby, to inhibit myofibroblast activation and prevent renal interstitial fibrogenesis.
Acknowledgments
We thank Dr. Aaron Bell for critical review of this manuscript.
Footnotes
Address reprint requests to Youhua Liu, Ph.D., Department of Pathology, University of Pittsburgh School of Medicine, S-405 Biomedical Science Tower, 200 Lothrop St., Pittsburgh, PA 15261. E-mail: liuy@msx.upmc.edu.
Supported by the National Institutes of Health (grants R01 DK-54922 and K02 DK-02611 to Y. L.) and in part by a Pathology Postdoctoral Research Training Grant from the Department of Pathology at the University of Pittsburgh School of Medicine (to J. Y.).
References
- 1.Port FK, Fenton SSA, Mazzuchi N: ESRD throughout the world: morbidity, mortality and quality of life. Kidney Int 2000, 57:S1-S2 [Google Scholar]
- 2.Eddy AA: Molecular insights into renal interstitial fibrosis. J Am Soc Nephrol 1996, 7:2495-2508 [DOI] [PubMed] [Google Scholar]
- 3.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 1998, 339:1448-1456 [DOI] [PubMed] [Google Scholar]
- 4.Roberts IS, Burrows C, Shanks JH, Venning M, McWilliam LJ: Interstitial myofibroblasts: predictors of progression in membranous nephropathy. J Clin Pathol 1997, 50:123-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Essawy M, Soylemezoglu O, Muchaneta-Kubara EC, Shortland J, Brown CB, el Nahas AM: Myofibroblasts and the progression of diabetic nephropathy. Nephrol Dial Transplant 1997, 12:43-50 [DOI] [PubMed] [Google Scholar]
- 6.Tang WW, Ulich TR, Lacey DL, Hill DC, Qi M, Kaufman SA, Van GY, Tarpley JE, Yee JS: Platelet-derived growth factor-BB induces renal interstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol 1996, 148:1169-1180 [PMC free article] [PubMed] [Google Scholar]
- 7.Ong AC, Fine LG: Loss of glomerular function and tubulointerstitial fibrosis: cause or effect? Kidney Int 1994, 45:345-351 [DOI] [PubMed] [Google Scholar]
- 8.Bohle A, Strutz F, Muller GA: On the pathogenesis of chronic renal failure in primary glomerulopathies: a view from the interstitium. Exp Nephrol 1994, 2:205-210 [PubMed] [Google Scholar]
- 9.Kliem V, Johnson RJ, Alpers CE, Yoshimura A, Couser WG, Koch KM, Floege J: Mechanisms involved in the pathogenesis of tubulointerstitial fibrosis in 5/6-nephrectomized rats. Kidney Int 1996, 49:666-678 [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Rajur K, Tolbert E, Dworkin LD: Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney Int 2000, 58:2028-2043 [DOI] [PubMed] [Google Scholar]
- 11.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG: Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 1995, 130:393-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng YY, Huang TP, Yang WC, Chen ZP, Yang AH, Mu W, Nikolic-Paterson DJ, Atkins RC, Lan HY: Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int 1998, 54:864-876 [DOI] [PubMed] [Google Scholar]
- 13.Okada H, Danoff TM, Kalluri R, Neilson EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997, 273:F563-F574 [DOI] [PubMed] [Google Scholar]
- 14.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan YH: Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 1999, 56:1455-1467 [DOI] [PubMed] [Google Scholar]
- 15.Racusen LC, Monteil C, Sgrignoli A, Lucskay M, Marouillat S, Rhim JG, Morin JP: Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med 1997, 129:318-329 [DOI] [PubMed] [Google Scholar]
- 16.Liu Y: Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol 1999, 277:F624-F633 [DOI] [PubMed] [Google Scholar]
- 17.Hammad FT, Wheatley AM, Davis G: Long-term renal effects of unilateral ureteral obstruction and the role of endothelin. Kidney Int 2000, 58:242-250 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Tolbert EM, Lin L, Thursby MA, Sun AM, Nakamura T, Dworkin LD: Up-regulation of hepatocyte growth factor receptor: an amplification and targeting mechanism for hepatocyte growth factor action in acute renal failure. Kidney Int 1999, 55:442-453 [DOI] [PubMed] [Google Scholar]
- 19.Kim TH, Mars WM, Stolz DB, Michalopoulos GK: Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology 2000, 31:75-82 [DOI] [PubMed] [Google Scholar]
- 20.Vukicevic S, Somogyi L, Martinovic I, Zic R, Kleinman HK, Marusic M: Reconstituted basement membrane (Matrigel) promotes the survival and influences the growth of murine tumors. Int J Cancer 1992, 50:791-795 [DOI] [PubMed] [Google Scholar]
- 21.Tisher CC, Madsen KM: Anatomy of the kidney. ed 5 Brenner BM eds. The Kidney, 1996, :pp 3-71 W.B. Saunders Co., Philadelphia [Google Scholar]
- 22.Lech K, Brent R: Escherichia coli, plasmids and bacteriophages. Ausubel FM Brent R Kingston RE Moore DD Seidman JG Smith JA Struhl K eds. Current Protocols in Molecular Biology. 1995, :pp 1.0.3-1.2.2 John Wiley & Sons, Inc., New York [Google Scholar]
- 23.Liu Y, Centracchio JN, Lin L, Sun AM, Dworkin LD: Constitutive expression of HGF modulates renal epithelial cell phenotype and induces c-met and fibronectin expression. Exp Cell Res 1998, 242:174-185 [DOI] [PubMed] [Google Scholar]
- 24.Birkedal-Hansen H: Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol 1995, 7:728-735 [DOI] [PubMed] [Google Scholar]
- 25.Borden P, Heller RA: Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit Rev Eukaryot Gene Exp 1997, 7:159-178 [DOI] [PubMed] [Google Scholar]
- 26.Martin J, Steadman R, Knowlden J, Williams J, Davies M: Differential regulation of matrix metalloproteinases and their inhibitors in human glomerular epithelial cells in vitro. J Am Soc Nephrol 1998, 9:1629-1637 [DOI] [PubMed] [Google Scholar]
- 27.Sharma K, Ziyadeh FN: The emerging role of transforming growth factor-β in kidney diseases. Am J Physiol 1994, 266:F829-F842 [DOI] [PubMed] [Google Scholar]
- 28.Border WA, Noble NA: Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 1998, 31:181-188 [DOI] [PubMed] [Google Scholar]
- 29.Hay ED, Zuk A: Transformations between epithelium and mesenchyme: normal, pathological, and experimentally induced. Am J Kidney Dis 1995, 26:678-690 [DOI] [PubMed] [Google Scholar]
- 30.Arias AM: Epithelial mesenchymal interactions in cancer and development. Cell 2001, 105:425-431 [DOI] [PubMed] [Google Scholar]
- 31.Steinberg MS, McNutt PM: Cadherin and their connections: adhesion junctions have broader functions. Curr Opin Cell Biol 1999, 11:554-560 [DOI] [PubMed] [Google Scholar]
- 32.Bush KT, Tsukamoto T, Nigam SK: Selective degradation of E-cadherin and dissolution of E-cadherin-catenin complexes in epithelial ischemia. Am J Physiol 2000, 278:F847-F852 [DOI] [PubMed] [Google Scholar]
- 33.Riethmacher D, Brinkmann V, Birchmeier C: A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci USA 1995, 92:855-859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horster MF, Braun GS, Huber SM: Embryonic renal epithelia: induction, nephrogenesis, and cell differentiation. Physiol Rev 1999, 79:1157-1191 [DOI] [PubMed] [Google Scholar]
- 35.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA: The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000, 2:76-83 [DOI] [PubMed] [Google Scholar]
- 36.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A: The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat Cell Biol 2000, 2:84-89 [DOI] [PubMed] [Google Scholar]
- 37.Lenz O, Elliot SJ, Stetler-Stevenson WG: Matrix metalloproteinases in renal development and disease. J Am Soc Nephrol 2000, 11:574-581 [DOI] [PubMed] [Google Scholar]
- 38.Bar-Sagi D, Hall A: Ras and Rho GTPases: a family reunion. Cell 2000, 103:227-238 [DOI] [PubMed] [Google Scholar]
- 39.Krizbai IA, Bauer H, Amberger A, Hennig B, Szabo H, Fuchs R, Baue HC: Growth factor-induced morphological, physiological and molecular characteristics in cerebral endothelial cells. Eur J Cell Biol 2000, 79:594-600 [DOI] [PubMed] [Google Scholar]
- 40.Powell DW, Miffin RC, Valentich JD, Crowe SE, Saada JI, West AB: Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol 1999, 277:C1-C19 [DOI] [PubMed] [Google Scholar]
- 41.Kropp BP, Zhang Y, Tomasek JJ, Cowan R, Furness PD, Vaughan MB, Parizi M, Cheng EY: Characterization of cultured bladder smooth muscle cells: assessment of in vitro contractility. J Urol 1999, 162:1779-1784 [PubMed] [Google Scholar]
- 42.Yang SP, Worlf AS, Yuan HT, Scott RJ, Risdon RA, O’Hare MJ, Winyard PJD: Potential biological role of transforming growth factor-beta1 in human congenital kidney malformations. Am J Pathol 2000, 157:1633-1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sutaria PM, Ohebshalom M, McCaffrey TA, Vaughan ED, Jr, Felsen D: Transforming growth factor-β receptor type I and II are expressed in renal tubules and are increased after chronic unilateral ureteral obstruction. Life Sci 1998, 62:1965-1972 [DOI] [PubMed] [Google Scholar]