

Abstract
Chemokine expression is associated with reperfusion of infarcted myocardium in the setting of tissue necrosis, intense inflammation, and inflammatory cytokine release. The specific synthesis of monocyte chemotactic protein (MCP)-1 mRNA by cardiac venules in reperfused infarcts corresponded to the region where leukocytes normally localize. MCP-1 could be induced by exogenous tumor necrosis factor (TNF)-α or by postischemic cardiac lymph containing TNF-α. However, the release of TNF-α during early reperfusion did not explain the venular localization of MCP-1 induction. To better understand the factors mediating MCP-1 induction, we examined the role of ischemia/reperfusion in a model of brief coronary occlusion in which no necrosis or inflammatory response is seen. Adult mongrel dogs were subjected to 15 minutes of coronary occlusion and 5 hours of reperfusion. Ribonuclease protection assay revealed up-regulation of MCP-1 mRNA only in ischemic segments of reperfused canine myocardium. Pretreatment with the reactive oxygen scavenger N-(2-mercaptopropionyl)-glycine completely inhibited MCP-1 induction. In situ hybridization localized MCP-1 message to small venular endothelium in ischemic areas without myocyte necrosis. Gel shift analysis of nuclear extracts from the ischemic area showed enhanced DNA binding of the transcription factors AP-1 and nuclear factor (NF)-κB, crucial for MCP-1 expression, in ischemic myocardial regions. Immunohistochemical staining demonstrated reperfusion-dependent nuclear translocation of c-Jun and NF-κB (p65) in small venular endothelium, only in the ischemic regions of the myocardium, that was inhibited by N-(2-mercaptopropionyl)-glycine. In vitro, treatment of cultured canine jugular vein endothelial cells with the reactive oxygen intermediate H2O2 induced a concentration-dependent increase in MCP-1 mRNA levels, which was inhibited by the antioxidant N-acetyl-l-cysteine, a precursor of glutathione, but not pyrrolidine dithiocarbamate, an inhibitor of NF-κB and activator of AP-1. In contrast to our studies with infarction, incubation of canine jugular vein endothelial cells with postischemic cardiac lymph did not induce MCP-1 mRNA expression suggesting the absence of cytokine-mediated MCP-1 induction after a sublethal ischemic period. These results suggest that reactive oxygen intermediate generation, after a brief ischemic episode, is capable of inducing MCP-1 expression in venular endothelium through AP-1 and NF-κB. Short periods of ischemia/reperfusion, insufficient to produce a myocardial infarction, induce MCP-1 expression, potentially mediating angiogenesis in the ischemic noninfarcted heart.
Monocyte chemotactic protein (MCP)-1 is a member of the CC chemokine family and serves as a chemotactic factor for the recruitment of monocytes and subpopulations of T cells during inflammation. 1 It is induced by the proinflammatory cytokines interleukin (IL)-1β and tumor necrosis factor (TNF)-α, as well as phorbol esters, which regulate its induction at the transcriptional level through the cooperative interaction of the transcription factors AP-1 and nuclear factor (NF)-κB. 2-4 Specifically, in endothelial cells, IL-1β, TNF-α, and phorbol esters mediate human MCP-1 gene expression through the cooperative interaction of the NF-κB binding site at −90 bp (relative to the start site) and the AP-1 binding site at −68 bp. 2,3
Early reperfusion of the ischemic myocardium is a key clinical intervention after myocardial infarction and is attended by a robust inflammatory reaction involving both neutrophils and mononuclear cells. 5,6 It has been shown that late reperfusion, when myocardial salvage is no longer possible, improves myocardial repair and obviates infarct expansion. 7 One of the early events after reperfusion of a myocardial infarct is the dramatic induction of MCP-1 in the cardiac venules. 8 We have demonstrated that this molecule is a chemotactic signal for mononuclear cell migration in the reperfused myocardial infarction 8,9 and others have demonstrated its potential role in angiogenesis. 10 In our previous studies with reperfused myocardial infarcts we demonstrated an important role for TNF-α as an upstream cytokine that was released from a preformed store in mast cells during ischemia 11 and showed that TNF-α could also induce endothelial cell MCP-1 expression. 8 Our studies also showed that cardiac lymph collected after reperfusion induced MCP-1 mRNA. This led us to hypothesize that TNF-α was responsible, at least in part, for MCP-1 induction after reperfusion of the infarcted myocardium; however, the assignment of this role to TNF-α did not explain the highly specific cellular localization of MCP-1 induction to the venular endothelium during early reperfusion. Our studies demonstrated that released TNF-α 11 bound to other cell types throughout the same time period in which localization of MCP-1 mRNA was confined to the venular endothelium. 8 These observations led to the current study that examines the hypothesis that the highly localized induction of MCP-1 is induced by reactive oxygen generated from ischemic myocardium. 12-14 Because both leukocyte transmigration and angiogenic budding occur at the postcapillary venular level, understanding this highly localized induction is an important issue.
At subcytolytic concentrations, reactive oxygen intermediates (ROIs) can induce chemokine and cytokine gene expression. In A549 and HS-24 epithelial cell lines, ROIs were shown to induce chemokines such as interleukin-8 (IL-8) and MCP-1. 15-17 The observation that ROIs may act as transcriptional activators is complicated by the fact that transcriptional regulation of ROI-mediated chemokine genes varies greatly with cell type. 17-19 For example, the ROI H2O2 differentially regulates chemokine expression in a cell-type, gene-type-specific manner in A549 epithelial cells through the transcription factor AP-1, however, it does not induce IL-8 in endothelial cells. 17,18 Similarly, generation of ROIs with xanthine/xanthine oxidase induced IL-8 and MCP-1 production from monocytes, but not U937 cells, and this induction is associated with increased NF-κB DNA binding. 20 Thus the potential for a role of ROIs in induction of MCP-1 in venular endothelium was considered.
In this study, we have elected to use the model of brief ischemia followed by reperfusion that results in the release of ROIs from the myocardium but is not associated with infarction or after reperfusion inflammation (see Results). 12-14 The absence of postinfarction inflammation obviates the complications associated with inflammatory cell adhesion to the postcapillary venular network that might induce MCP-1 in these venules via a paracrine- or adhesion-dependent signal mechanism. The results demonstrate that MCP-1 expression after 15 minutes of ischemia and 5 hours of reperfusion occurs specifically in the small venular endothelium and is inhibited by pretreatment with the anti-oxidant, N-(2-mercaptopropionyl)-glycine (MPG). The oxidative-dependent induction of MCP-1 was attended by histological demonstration of nuclear translocation of NF-κB and c-Jun exclusively in the nuclear proteins of the venules of the reperfused myocardium; gel shift studies of nuclear proteins isolated from the reperfused area correlated with these observations by demonstrating activation of NF-κB and AP-1. In vitro, H2O2 induced MCP-1 mRNA and NF-κB and AP-1 translocation in venular endothelium, however, postischemic cardiac lymph collected during reperfusion after 15 minutes of ischemia did not. The potential significance of MCP-1 induction by ROIs in a noninfarcted ischemia-reperfusion (I/R) model will be discussed.
Materials and Methods
I/R Protocol
Mongrel dogs (15 to 25 kg) were surgically instrumented with a hydraulic occluder around the circumflex coronary artery and cardiac lymph duct cannulation as previously described. 11,12,21 After instrumentation, animals were allowed to recover for 72 hours before occlusion. Coronary artery occlusion was initiated by inflating the hydraulic occluder until mean blood flow in the coronary artery was zero as determined by Doppler flow probe. After 15 minutes of occlusion, the coronary occluder was deflated and the heart was reperfused for 5 hours. To investigate the potential role of ROIs as mediators of gene induction, one treatment group of instrumented dogs (n = 4) was infused with the cell soluble antioxidant, MPG 13 (100 mg/kg), administered beginning 15 minutes before onset of occlusion (15 minutes) and reperfusion (5 hours). After reperfusion, the heart was stopped with a rapid infusion of KCl, removed, and sectioned. Myocardial sections were fixed in neutral-buffered formalin, 10% paraformaldehyde, B*5 or frozen in tissue-freezing medium (OCT) for histological studies. Adjacent tissue sections were flash-frozen in liquid N2, then homogenized and processed for either RNA or nuclear protein extraction. Additional sections were used for determination of blood flow with radiolabeled microspheres as previously described. 11,21 Blood flow in regions deemed ischemic had a reduction in blood flow >80% relative to control sections. Blood flow data were collected from 15 animals (n = 7 for 15 minutes occlusion/0 hours reperfusion, and n = 8 for 15 minutes occlusion/5 hours reperfusion, data not shown).
Cell Culture and Treatments
Isolation of canine jugular vein endothelial cells (CJVECs) was performed as previously described. 8 Before agonist administration cell culture groups were washed twice with 1× phosphate-buffered saline (PBS) and incubated for 24 hours in 0.5% growth media (complete media diluted 1/40 with Dulbecco’s modified Eagle’s medium). 17,18,22 CJVECs were treated with H2O2 or TNF-α using a protocol adapted from Lakshminarayanan and colleagues. 17,18 CJVECs were washed twice with 1× PBS and then incubated in serum-free, phenol red-free Dulbecco’s modified Eagle’s medium (Life Technologies, Inc., Gaithersburg, MD) with H2O2 or TNF-α diluted to their appropriate final concentrations.
Immunostaining and Histology
Immunostaining of canine myocardial tissue sections was performed as described by Frangogiannis and colleagues 11 using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA). Tissue sections were incubated with the primary antibodies c-Fos/p62 (K-25), c-Jun/p39 (H-79), NF-κB/p65 (H-286) (Santa Cruz Biotechnology, Santa Cruz, CA), SG8H6 anti-neutrophil, 23 anti-CD31 24 and Mac387 anti-myeloid cell antibody (DAKO, Carpinteria, CA) for 2 hours at room temperature, rinsed three times in 1× PBS and transferred into the corresponding secondary antibody. Tissue sections were then rinsed three times in 1× PBS, and incubated with avidin-biotin-peroxidase solution for 30 minutes. The peroxidase-based reaction was developed with diaminobenzidine.
In Situ Hybridization
All solutions used for in situ hybridization were treated with 0.1% diethyl pyrocarbonate (Sigma Chemical Co., St. Louis, MO) and sterilized, or prepared with diethyl pyrocarbonate-treated water. In situ hybridization was performed as described by Kumar and colleagues. 8 After deparaffinization and rehydration, tissue sections were rinsed in 2× standard saline citrate (SSC) for 5 minutes, incubated in prehybridization buffer (50% deionized formamide, 4× SSC, 1× Denhardt’s reagent, 0.5 mg/ml salmon sperm DNA, 0.25 mg/ml yeast tRNA, 10% dextran sulfate) at room temperature for 1 hour, rinsed in 2× SSC, then incubated with hybridization buffer (prehybridization buffer containing 150 ng of digoxigenin-labeled MCP-1 riboprobe) at 42°C for 24 hours in a humidified incubator. After hybridization, tissue slides were washed twice in 1× SSC at room temperature for 30 minutes each. To reduce nonspecific hybridization, wash stringency was increased by reducing the concentration of SSC to 0.2× and increasing the wash temperature to 55°C. For immunological detection an anti-digoxigenin antibody and nitro blue tetrazolium staining of the alkaline phosphatase reaction were used.
RNA Isolation
All solutions used for RNA isolation were treated with 0.1% diethyl pyrocarbonate and sterilized, or prepared with diethyl pyrocarbonate-treated water. T75 cultures of CJVECs or sections of whole canine myocardium (5 mg) were solubilized in 2 ml or 5 ml of Trizol (Life Technologies, Inc.), respectively. Total RNA was isolated according to the manufacturer’s protocol then lysate was extracted with phenol-chloroform. Samples were mixed vigorously, incubated at room temperature for 2 to 3 minutes, and then spun at ∼12,000 × g for 15 minutes. The aqueous phase was collected and total RNA was precipitated by mixing with equal volumes of isopropyl alcohol and storing at −70°C for 1 hour.
Ribonuclease Protection Assays (RPAs)
The RPA was adapted from previously published protocols. 17,25 The canine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plasmid was a generous gift from Dr. T. Rosol (Ohio State University, Columbus, OH). Canine GAPDH and MCP-1 plasmids were linearized with the restriction enzymes HindIII and SAC I (Promega, Madison, WI), respectively. Radiolabeled cRNA templates were generated by in vitro transcription of canine GAPDH 25 and MCP-1 8 cDNAs in the presence of [32P]-uridine triphosphate (UTP) (Amersham, Pharmacia Biotechnology, Piscataway, NJ) and T7 RNA polymerase (Pharmingen, La Jolla, CA). The radiolabeled templates were combined, precipitated with 3 mol/L ammonium acetate/100% ethanol, and 4 × 10 5 cpm were hybridized to total RNA for 16 hours at 56°C. For negative control and experimental samples, 30 μg of total RNA was used. Because of the robust nature of the lipopolysaccharide controls, only 20 μg of total RNA was used. mRNA probe complexes were treated with RNase A+T1, phenol-chloroform extracted, resolved on a 6% denaturing polyacrylamide-sequencing gel and exposed to radiographic film. Determination of protected complex size was made by linear regression comparison to unprotected probe, and comparison to [32P]-adenosine triphosphate (ATP) end-labeled 1.77-kb RNA ladder (Life Technologies, Inc.). The resulting protected cRNA products for MCP-1 and GAPDH complexes were 282 bp and 191 bp, respectively. Phosphate incorporation analysis was performed using the Storm 860 phosphorimager (Molecular Dynamics, Sunnyvale, CA) and quantitated with ImageQuant software.
DNA-Binding Studies from Nuclear Extracts
Electrophoretic mobility shift assays were performed essentially as described. 18,22 Briefly, nuclear protein extracts (10 μg) were prepared from homogenized canine myocardial sections (5 mg) or CJVECs by a variation of the methods of Muller and colleagues 26 and Roebuck and colleagues 22 and were incubated with 5 × 10 4 cpm (∼0.1 ng) of [32P]-end-labeled oligonucleotide probes for consensus AP-1 or consensus NF-κB sequences (Promega) for 20 to 30 minutes at room temperature in binding buffer containing 12% glycerol, 12 mmol/L HEPES-NaOH (pH 7.9), 60 mmol/L KCl, 5 mmol/L MgCl2, 4 mmol/L Tris-Cl (pH 7.9), 0.6 mmol/L ethylenediaminetetraacetic acid (pH 7.9), 0.6 mmol/L dithiothreitol, and 0.25 μg of poly(dI-dC). To demonstrate binding specificity, a 100-fold molar excess of either specific or nonspecific oligonucleotides was included in the binding reaction. Protein-DNA complexes were resolved in 5% polyacrylamide gels in 0.25× TBE buffer. Gels were dried and exposed to radiographic film with an intensifying screen at −70°C. Gel shifts were performed at least three times using nuclear extracts prepared from different animals or cell culture experiments. Similar results were obtained and a representative gel is shown in each of the figures.
Statistics
Graphs are presented as the mean (±SEM) of the MCP-1/GAPDH mRNA relative densitometry ratio with significance defined as (P < 0.05). Statistical analysis was performed using a one-way, two-tailed analysis of variance using InStat (GraphPad Software).
Results
Induction of MCP-1 mRNA in Whole Canine Myocardium after Brief I/R
Comparison of acute, ischemia alone (15 minutes/0 hours), brief I/R (15 minutes/5 hours), and lipopolysaccharide myocardium revealed that MCP-1 mRNA was induced in the ischemic region of myocardium subjected to I/R but not in the corresponding acute control (Figure 1A) ▶ . Relative densitometry of MCP-1 mRNA increased significantly in the ischemic regions compared to the acute control (Figure 1B) ▶ . No significant increase was observed in the nonischemic control or ischemic regions of myocardium subjected to ischemia alone, or in the control region of myocardium subjected to I/R, suggesting MCP-1 mRNA induction is dependent on reperfusion of an ischemic bed. When MPG was administered before the I/R protocol, MCP-1 mRNA induction was eliminated suggesting that induction relied on the presence of ROIs (Figure 1, A and B) ▶ .
Figure 1.
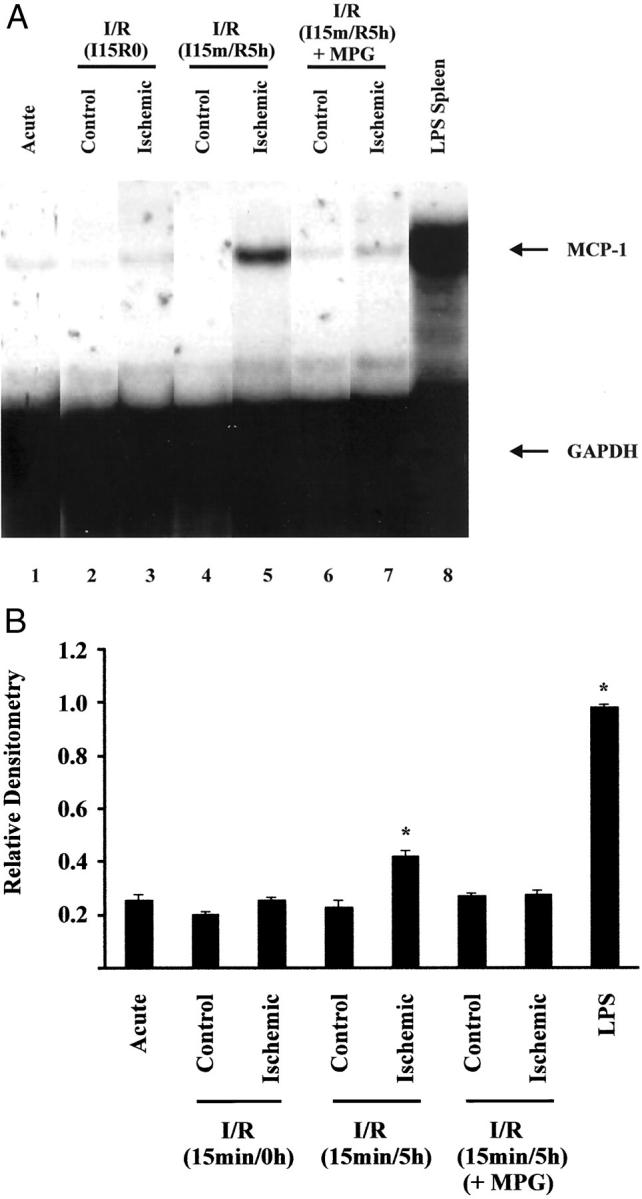
Induction of MCP-1 mRNA in whole canine myocardium by brief ischemia/reperfusion. Total RNA was isolated from control and ischemic sections of canine myocardium after 15 minutes of ischemia alone (lanes 2 and 3), I/R (15 minutes/5 hours) (lanes 4 and 5), I/R (15 minutes/5 hours) + MPG (lanes 6 and 7), or lipopolysaccharide infusion (0.44 mg/kg; 4 hours) (lane 8) and analyzed by RPA. Arrows indicate protected RNA for MCP-1 and GAPDH. A: Autoradiograph. B: Bar graph of relative densitometry of MCP-1 signal normalized to GAPDH signal. Asterisk indicates significant difference (P < 0.05).
In Situ Hybridization of Canine MCP-1 Localizes to Small Venular Endothelium after a Brief Ischemic Insult
In situ hybridization was performed on control and ischemic tissue sections from hearts subjected to brief I/R. Kumar and colleagues 8 previously demonstrated MCP-1 expression specifically in small venular endothelium after I/R (1 hour/3 hours). Similarly, we show brief I/R also induced MCP-1 mRNA expression that was localized to the venular endothelium (Figure 2B) ▶ , but not in hearts subjected to ischemia alone (Figure 2A) ▶ . Serial sections were immunostained with the neutrophil-specific antibody SG8H6, 23 the myeloid cell marker Mac 387 and the anti-CD-31 endothelial-specific antibody, 24 and showed in situ MCP-1 staining was specific for microvascular endothelium in the heart in the absence of neutrophil or monocyte infiltration, indicating that MCP-1 induction was independent of an inflammatory response.
Figure 2.
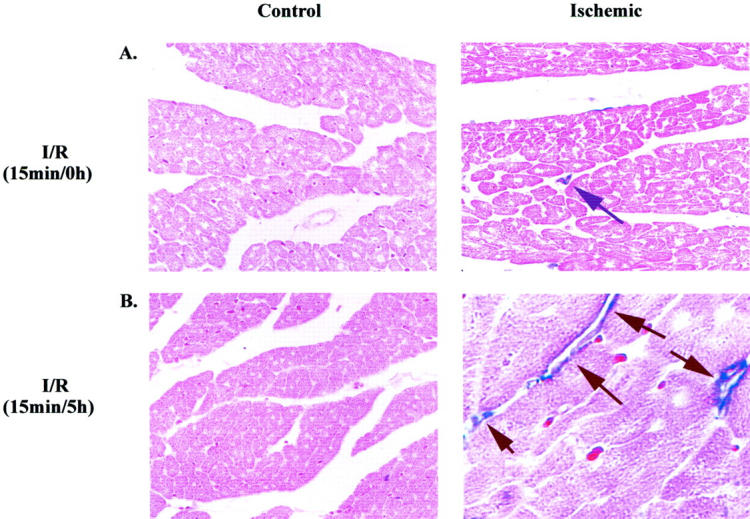
In situ hybridization of canine MCP-1 is localized to small venular endothelium after myocardial stunning. In situ hybridization was performed on paraffin-embedded tissue samples from control and ischemic regions of canine myocardium with the MCP-1 antisense probe. A: Control and ischemic regions of canine myocardium exposed to ischemia alone (15 minutes). B: Control and ischemic regions of canine myocardium exposed to I/R (15 minutes/5 hours). Arrows indicate MCP-1 mRNA expression in tissue section. Experiment presented is representative of four individual experiments.
Brief I/R Induces c-Jun and NF-κB, but Not c-Fos, that Are Localized in the Nuclei of Small Venular Endothelium
To further characterize the expression of AP-1 and NF-κB after brief I/R, control and ischemic tissue sections from hearts subjected to ischemia alone or brief I/R were immunohistochemically stained with antibodies specific for the DNA-binding factors NF-κB, c-Jun, and c-Fos. Ischemic tissue sections from brief I/R myocardium, but not nonischemic control myocardium, showed significant positive nuclear staining for the DNA binding factor NF-κB and the AP-1 component c-Jun, but not c-Fos (Figure 3; A to C ▶ ). Staining serial sections of the control and ischemic tissue with the endothelial-specific antibody CD-31 confirmed that NF-κB and c-Jun protein expression were specifically found in venular endothelium (Figure 3D) ▶ . Tissue sections from animals that received MPG before the I/R protocol did not show nuclear translocation of c-Jun and NF-κB in the venular endothelium. No AP-1 or NF-κB nuclear translocation occurred in ischemic, nonreperfused samples.
Figure 3.
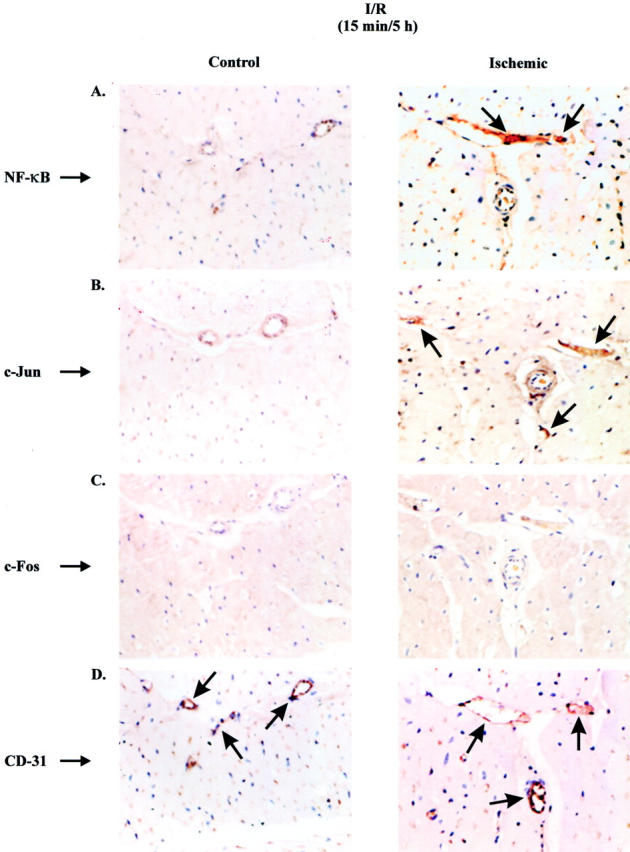
Brief ischemia/reperfusion induces c-Jun and NF-κB, but not c-Fos, that are localized in nuclei of small venular endothelium. Paraformaldehyde-fixed tissues from canine myocardium were immunostained (diaminobenzidine, black) with monoclonal antibodies against NF-κB (A), c-Jun (B), c-Fos (C), or CD-31 (D) and counterstained with eosin. Control (left) and ischemic (right) tissue sections of canine myocardium exposed to 15 minutes of ischemia and 5 hours of reperfusion. Arrows indicate localization of antibody signal in small venular endothelium for NF-κB (A), c-Jun (B), and CD-31 (D). Experiment presented is representative of four individual experiments.
Brief I/R Induces Both AP-1- and NF-κB-Binding Activity in Whole Myocardium
Two of the transcription factors intricately involved in the regulation of MCP-1 are AP-1 and NF-κB. 3,4 Because AP-1 and NF-κB are redox-sensitive transcription factors, 27,28 we investigated their role in activation of MCP-1 gene expression associated with brief I/R using gel shift analysis of nuclear extracts prepared from ischemic and control canine myocardium subjected to ischemia alone or brief I/R. In hearts subjected to brief I/R, enhanced DNA binding to consensus AP-1 and NF-κB oligonucleotides was observed in nuclear extracts of ischemic tissue sections (Figure 4, A and B ▶ ; lane 5) relative to the nonischemic control sections (Figure 4, A and B ▶ ; lane 4). Minimal DNA binding was apparent in nuclear extracts from the control and ischemic tissue sections from hearts subjected to ischemia alone, without reperfusion (Figure 4, A and B ▶ ; lanes 2 and 3). Competition with 100-fold molar excess of nonlabeled oligonucleotides to AP-1 and NF-κB abrogated DNA binding with respect to each probe, whereas the nonspecific oligonucleotide OCT-1 did not inhibit binding, showing specificity of the DNA binding reaction. May-Newman and colleagues 29 demonstrated that canine myocardium contains a high vascular endothelial density, comprising a potential source for the enhanced AP-1 and NF-κB DNA binding induced by ROIs in myocardial nuclear extracts.
Figure 4.

Brief I/R (15 minutes/5 hours) induces both AP-1- and NF-κB-binding activity in nuclear extracts. Nuclear protein extracts prepared from acute (lane 1), control 15 minutes/0 hours (lane 2), ischemic 15 minutes/0 hours (lane 3), control 15 minutes/5 hours (lane 4), ischemic 15 minutes/5 hours (lane 5), or lipopolysaccharide infusion (lanes 6 to 8) tissue extracts were incubated with [32P] end-labeled oligonucleotides. A: Consensus AP-1-binding site. B: Consensus NF-κB-binding site. Arrow indicates induced DNA-binding complex. Sequences for oligonucleotides are as follows: consensus AP-1: 5′-CGCTTGATGAGTCAGCCGGAA-3′; consensus NF-κB: 5′-AGTTGAGGGGACTTTCCCAGGC-3′; consensus OCT-1: 5′TGTCGAATGCAAATCACTAGAA-3′.
MCP-1 mRNA Induction by H2O2 in CJVECs
MCP-1 is an oxy-sensitive chemokine induced by H2O2 in endothelial cells. 30 To further examine the potential mechanism of oxidant induction of MCP-1 in venular endothelium, we used CJVECs as an in vitro endothelial cell culture model. ROI scavenger experiments by Bolli and colleagues 12 used a glucose/glucose oxidase reaction to generate H2O2 and measured its rate of production to be 11 μmol/L/min. Experiments by Wung and colleagues 31 induced MCP-1 expression in bovine artery endothelial cells with exogenous H2O2 at a concentration of 100 μmol/L. Therefore, we stimulated CJVECs with equivalent concentrations of H2O2. CJVECs were isolated as previously described 8 and stimulated with H2O2 (10 to 200 μmol/L) or TNF-α (100 U/ml) for 3 hours, and MCP-1 induction was measured by RPA. Figure 5A ▶ shows MCP-1 message increased in a dose-dependent manner with maximal expression at 100 to 200 μmol/L H2O2. Phosphorimager analysis (Figure 5B) ▶ indicates 100 μmol/L H2O2 induced MCP-1 mRNA ∼2.8-fold relative to unstimulated CJVECs. To address the question of a stable soluble agonist (eg, cytokines) secreted into the cardiac lymph stimulating MCP-1 expression, canine cardiac lymph was isolated at 1-hour intervals of reperfusion after brief ischemia and used to stimulate CJVECs. Previous studies from Kumar and colleagues 8 showed postischemic canine cardiac lymph (1 hour/3 hours) was capable of stimulating MCP-1 message in CJVECs. In contrast, postischemic canine cardiac lymph after brief I/R was unable to induce MCP-1 mRNA in CJVECs (data not shown). Thus, in the absence of infarction, there is no apparent cytokine-stimulated MCP-1 induction; this contrasts with the study by Kumar and colleagues 8 (see Introduction).
Figure 5.

MCP-1 mRNA induction by H2O2 in canine venular endothelial cells. CJVECs were treated with 10, 50, 100, and 200 μmol/L of H2O2 (lanes 2 to 5) or 100 U/ml of TNF-α (lane 6) for 3 hours. Total RNA was isolated and analyzed by RPA. A: Autoradiograph. B: Bar graph of relative densitometry of MCP-1 signal normalized to GAPDH signal. Asterisk indicates significant difference (P < 0.05).
H2O2 Induces AP-1- and NF-κB-Binding Activity in CJVECs
To further understand the role of ROI-mediated AP-1 and NF-κB DNA binding, nuclear extracts were prepared from CJVECs stimulated with H2O2 (100 μmol/L) or TNF-α (100 U/ml), then incubated with consensus oligonucleotides against AP-1 and NF-κB. As shown in Figure 6, A and B ▶ , nuclear extracts from H2O2-stimulated CJVECs increased AP-1 and NF-κB binding compared to unstimulated CJVECs. The increase in DNA binding activity in H2O2-stimulated CJVECs correlated with the enhanced binding seen in the nuclear extracts prepared from ischemic myocardial tissue sections from brief I/R experiments (Figure 3, A and B) ▶ and the increased c-Jun and NF-κB nuclear translocation (Figure 4B) ▶ seen in histological sections from brief I/R hearts.
Figure 6.
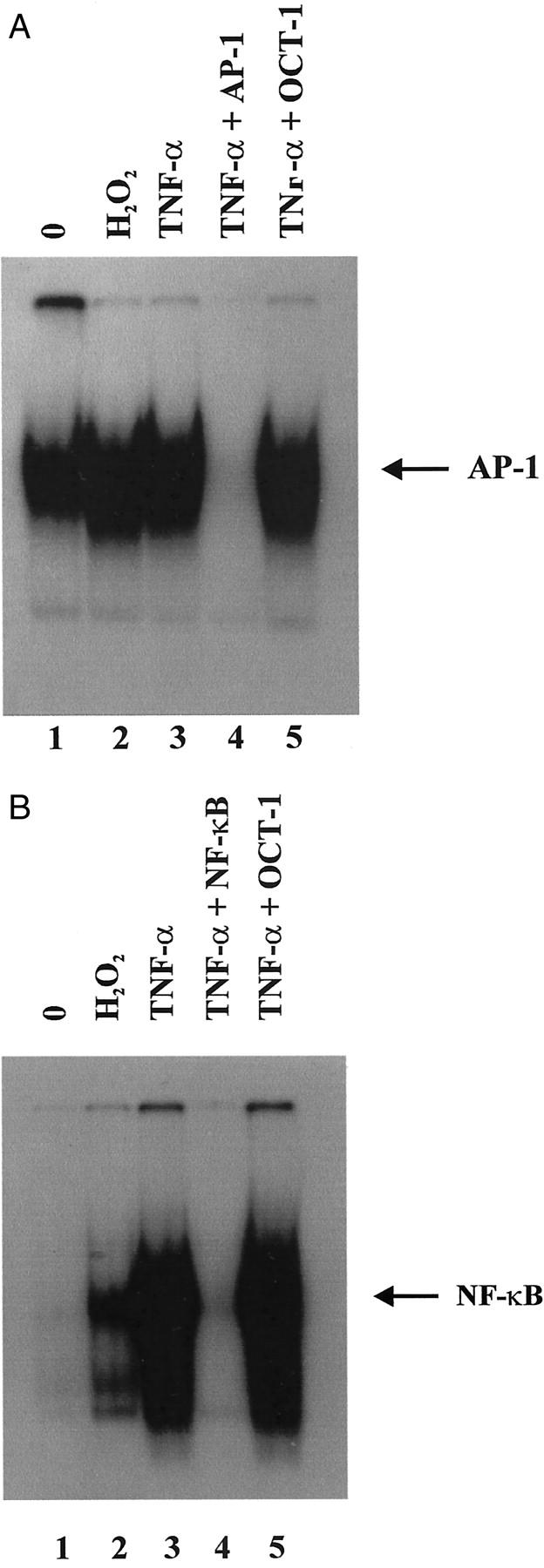
H2O2 induces AP-1- and NF-κB-binding activity in CJVECs. Nuclear protein extracts from CJVECs stimulated with 100 μmol/L of H2O2 (lane 2) or 100 U/ml of TNF-α (lanes 3 to 5) for 1 hour were incubated with [32P] end-labeled oligonucleotides. A: Consensus AP-1-binding site. B: Consensus NF-κB-binding site.
Effects of N-Acetyl-l-Cysteine (NAC) and Pyrrolidine Dithiocarbamate (PDTC) on H2O2 Induction of MCP-1 mRNA in CJVECs
Because oxidant-induced MCP-1 expression in CJVECs is potentially mediated by AP-1 and NF-κB, we investigated the induction of MCP-1 mRNA in the presence of the antioxidants NAC, a precursor of glutathione (GSH), and PDTC, an anti-oxidant, which has been shown to inhibit NF-κB binding. 32-34 CJVECs were pretreated with NAC (5 mmol/L) or PDTC (1 mmol/L) for 1 hour before stimulation with H2O2 (100 μmol/L) for 3 hours (Figure 7A) ▶ . These concentrations of NAC (5 mmol/L) and PDTC (1 mmol/L) were previously shown to inhibit the oxidant-induced expression of ICAM-1 in human umbilical vein endothelial cells. 22 Total RNA was isolated and analyzed for MCP-1 mRNA by RPA from H2O2-stimulated CJVECs. Densitometry indicated H2O2 induced MCP-1 mRNA approximately twofold relative to unstimulated CJVECs (Figure 7B ▶ , lane 4). Pretreatment with NAC abrogated the H2O2 induction of MCP-1 (lane 5) but had no effect on MCP-1 mRNA in untreated controls (lane 2). This effect is similar to the ICAM-1 studies. 22 In contrast, PDTC alone (lane 3) increased MCP-1 message 2.6-fold in the absence of H2O2 treatment, and pretreatment with PDTC further enhanced the H2O2 induction of MCP-1 mRNA (lane 6). These studies are in contrast to those described for ICAM-1. 22 To examine these observations further, we investigated the effects of NAC and PDTC on the binding activity of AP-1 and NF-κB.
Figure 7.
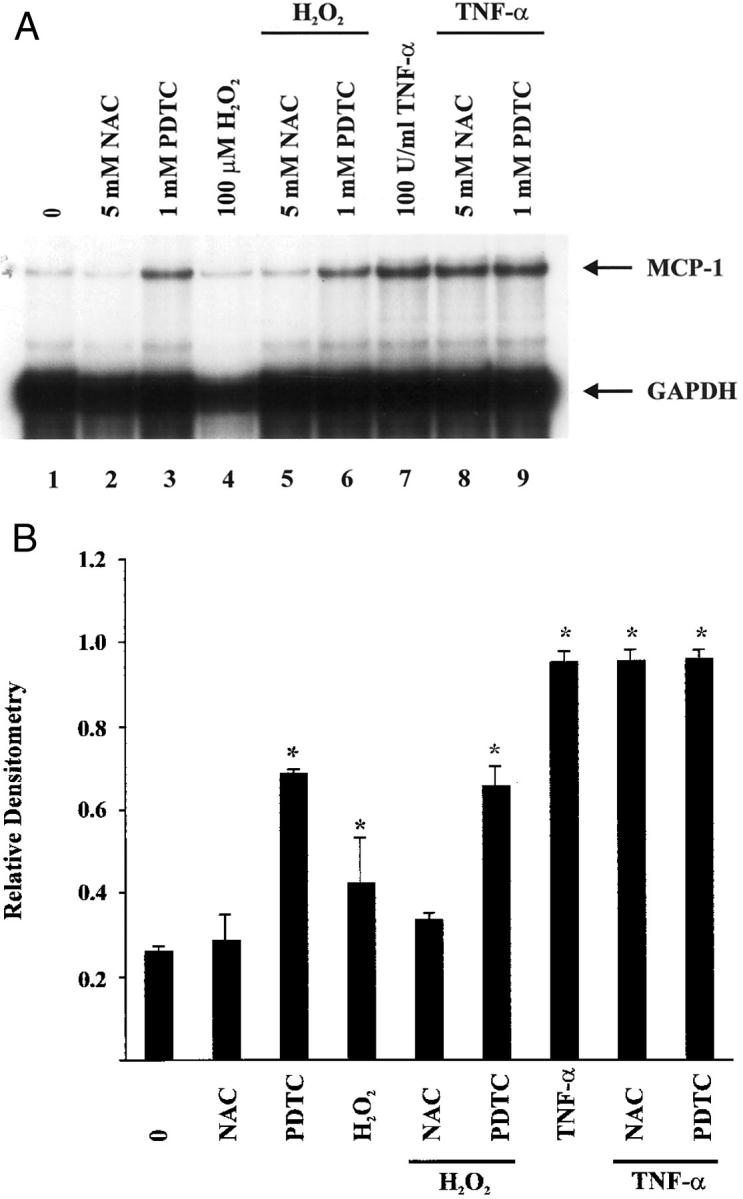
Effects of NAC and PDTC on H2O2 induction of MCP-1 mRNA in CJVECs. CJVECs were pretreated with NAC (5 mmol/L) (lanes 5 and 8) or PDTC (1 mmol/L) (lanes 6 and 9) for 1 hour and then stimulated with H2O2 (100 μmol/L) or TNF-α (100 U/ml) for 3 hours. Total RNA was isolated and analyzed by RPA. A: Autoradiograph. B: Bar graph of relative densitometry of MCP-1 signal normalized to GAPDH signal. Asterisk indicates significant difference (P < 0.05).
Effects of NAC and PDTC on Oxidant-Induced AP-1 and NF-κB Binding
Previous work from Munoz and colleagues 35 showed PDTC alone, or in concert with TNF-α, enhanced AP-1 DNA-binding activity while inhibiting TNF-α-mediated NF-κB activity in human umbilical vein endothelial cells. Because of the oxy-sensitive nature of the transcription factors AP-1 and NF-κB 28 and their potential role in mediating the induction of MCP-1 by oxidant stress, 31 we investigated the effects of anti-oxidants NAC and PDTC on oxidant-induced AP-1 and NF-κB DNA binding (Figure 8) ▶ . Nuclear proteins isolated from CJVECs pretreated with NAC (5 and 10 mmol/L) or PDTC (1 and 10 mmol/L) for 1 hour before stimulation with H2O2 (100 μmol/L) were incubated with consensus oligonucleotides against AP-1 (Figure 8A) ▶ and NF-κB (Figure 8B) ▶ . In Figure 8A ▶ , NAC alone (lanes 2 and 3) failed to induce AP-1 DNA binding relative to unstimulated CJVECs (lane 1). In contrast, PDTC (lanes 4 and 5) and H2O2 (lane 6) enhanced AP-1 DNA binding. Pretreatment with 10 mmol/L NAC significantly reduced H2O2-mediated AP-1-binding activity (lane 8) whereas 1 mmol/L PDTC pretreatment enhanced H2O2-induced AP-1 binding (lane 9). In Figure 8B ▶ , H2O2 induced NF-κB DNA binding (lane 6) that was inhibited by NAC (5 and 10 mmol/L) (lanes 7 and 8) as well as PDTC (1 and 10 mmol/L) (lanes 9 and 10) pretreatment. The inhibitory effect of PDTC on oxidant-induced NF-κB binding was more pronounced at higher concentrations, reducing NF-κB binding at 1 mmol/L and abrogating the response at 10 mmol/L (Figure 8B ▶ , lanes 9 and 10). Taken as a whole, these studies show that PDTC effectively inhibited ROI-mediated NF-κB DNA-binding activity while activating AP-1.
Figure 8.

Effects of NAC and PDTC on oxidant-induced AP-1 and NF-κB binding. CJVECs were pretreated with NAC (5, 10, or 100 mmol/L) (lanes 7, 8, 12, and 13) or PDTC (0.1, 1, or 10 mmol/L) (lanes 9, 10, 14,and 15) for 1 hour and then stimulated with H2O2 (100 μmol/L) or TNF-α (100 U/ml) for 1 hour. Nuclear protein extracts were isolated and incubated with [32P] end-labeled oligonucleotides and analyzed by gel shift analysis. A: Consensus AP-1-binding site. B: Consensus NF-κB-binding site. Arrow indicates induced DNA-binding complex. Experiment presented is representative of three individual experiments.
Discussion
Our previous experiments using a reperfused myocardial infarction model demonstrated that MCP-1 mRNA expression was confined to the postcapillary venular endothelial cells that are the site of leukocyte transendothelial migration. 8,9 The presence of TNF-α in the reperfused infarction during early reperfusion and the ability of both postischemic cardiac lymph and TNF-α to induce MCP-1 identified TNF-α as a major candidate for MCP-1 induction. 8 However, cellular distribution of TNF-α is far more extensive than merely the cardiac venules and would not completely explain such an exquisite localization 9 of MCP-1 induction to cardiac venules. 8
Extensive evidence supports a role for reactive oxygen in chemokine induction chemokines in various cell types; however, this observation is confounded by significant variability of response to ROI with regard to cell type and chemokine species. 17,19 Nevertheless, the possibility that reactive oxygen might be responsible for the induction of MCP-1 localized in the venules was considered because: 1) H2O2 is capable of inducing MCP-1 mRNA in endothelial cells, 1,36 2) ROI formation is induced during early reperfusion of the myocardium, 12,14,37 and 3) ROIs from the reperfused myocardium egress to the capillary vascular bed.
We also considered the fact that the postcapillary venular endothelium represented the site to which leukocytes adhere and through which leukocytes transmigrate. Because leukocyte adhesion and transmigration might induce genes in the endothelium either through paracrine cytokine secretion mechanisms or through direct stimulation associated with adhesion, we chose to examine MCP-1 induction in a model using brief, nonlethal ischemia followed by reperfusion that has been well characterized in the past as inducing ROI formation and secretion from myocytes associated with dysfunction but no cell death. 5,12-14 We have also confirmed that there is no significant leukocyte infiltration associated with this protocol.
Spin-trapping techniques have allowed quantitation of ROI generation in models of stunned myocardium, which are characterized by reperfusion of ischemic noninfarcted hearts. Administration of α-phenyl-N-tert-butylintrone and analysis of coronary vessel blood by electron paramagnetic resonance has been used to assess the time course of ROI generation during brief ischemia (15 minutes) in a canine model, 14,38 demonstrating a release of ROIs at the onset of reperfusion after ischemia, peaking within the first 2 to 3 minutes, but sustained for 3 hours of reperfusion. 38 Thus, in this model of stunned myocardium and reversible cardiac dysfunction, ROIs are potential candidates to induce expression of oxy-sensitive chemokines such as MCP-1.
In this report, we demonstrate that MCP-1 mRNA is expressed exclusively in the postcapillary venules of the ischemic myocardium during reperfusion after brief sublethal periods of ischemia (15 minutes). In situ hybridization (Figure 2) ▶ showed reperfusion-dependent-specific localization of MCP-1 mRNA expression in the endothelium of small venules, similar to that described in the infarction protocol. 8 In contrast to the infarction model however, the 15 minutes/5 hours protocol demonstrated no significant inflammatory infiltrate, as determined by immunostaining with the anti-neutrophil-specific monoclonal antibody SG8H6 23 or Mac 387, which identifies infiltrating neutrophils and monocytes. 24 MCP-1 mRNA induction was accompanied by nuclear translocation of NF-κB and c-Jun in the same postcapillary venules, and nuclei isolated from ischemic myocardium demonstrated both NF-κB and AP-1 activation by gel retardation assays. A brief ischemic insult induced prolonged expression of NF-κB and c-Jun, as previously described in the isolated perfused rat heart. 39 Finally, MCP-1 mRNA induction and venular nuclear translocation of NF-κB and c-Jun were not seen in animals in whom MPG infusion preceded the I/R protocol. Taken as a whole the data suggest that ROI production is necessary for transcriptional activation of the MCP-1 gene in the absence of inflammation or infarction. The methods used in vivo do not allow dissection of other factors in the extracellular fluid that might contribute to this induction. To examine whether ROI was sufficient for MCP-1 induction and activation of NF-κB and AP-1, we examined an in vitro model of venular endothelial cultures in crystalloid solution.
In the in vitro study, we used two antioxidants (NAC and PDTC) with distinct mechanisms of inhibition to investigate the up-regulation of MCP-1 transcription by H2O2. NAC readily diffuses into cells and serves as a precursor to GSH. 34 Pretreatment of CJVECs with 5 mmol/L NAC abrogated induction of MCP-1 mRNA by H2O2 (Figure 7) ▶ . The anti-oxidant PDTC specifically inhibits NF-κB activation by preventing I-κB separation from the active NF-κB cytoplasmic domain, thereby precluding nuclear translocation and regulation of gene transcription. 33 In contrast to the inhibitory effects by NAC on H2O2-induced MCP-1 mRNA, PDTC was ineffective as an inhibitor of MCP-1 mRNA (Figure 7) ▶ . In fact, PDTC alone (Figure 7A ▶ , lane 3) and in combination with H2O2 (Figure 7A ▶ , lane 6) induced MCP-1 message relative to H2O2 alone (Figure 7A ▶ , lane 4). This apparent discrepancy was reconciled by the observation that PDTC also strongly enhanced DNA binding to the consensus AP-1 oligonucleotide (Figure 8A ▶ , lanes 4 and 5). This suggests that PDTC may induce AP-1 gene expression in addition to inhibiting NF-κB in endothelial cells, which has been well described by others. 35 Meyer and colleagues 40 showed PDTC had strikingly different effects on NF-κB and AP-1 DNA binding under oxidative stress. Similarly, in our in vitro system PDTC inhibited oxidant-induced NF-κB activity and had the secondary effect of activating AP-1 in CJVECs. Thus, it seems that AP-1 activation is sufficient to support MCP-1 induction in cultured adult endothelial cells, which is in contrast to observations on ICAM-1 stimulation. 22 The in vivo demonstration in our experiments of concomitant nuclear translocation of NF-κB suggests that, at least in cardiac venules, the oxidant response involves both factors.
To our knowledge, this represents the first demonstration of reperfusion-dependent chemokine expression induced by ROI release in vivo after an acute ischemic episode in the absence of injury. It is interesting to speculate what the role of MCP-1 induction might be in a physiological circumstance in which it is induced in the absence of injury. Ito and co-workers 10 demonstrated that injection of MCP-1 into ischemic skeletal muscle beds results in angiogenesis and collateral blood flow. Because angiogenesis begins with endothelial budding at the venular level, locally generated venular MCP-1 might be involved in an angiogenic response to reversible ischemia. The mechanism by which MCP-1 can induce angiogenesis remains unclear, but recent reports demonstrate the presence of a CCR2 receptor in endothelial cells. 41 In addition, the recent experiments 42 demonstrated that repeated brief occlusions of the coronary artery resulted in the formation of collateral circulation without inducing myocardial injury. It is interesting to speculate whether similar mechanisms might result from repeated bouts of myocardial ischemia distal to a critical coronary lesion and result in collateral blood flow formation. It is possible that ROIs generated by these reversible ischemic episodes induce MCP-1 in venules facilitating the angiogenic response.
Acknowledgments
We thank Leonardo Mendoza, Peggy Jackson, and Lisa Thurmon for their technical assistance.
Footnotes
Address reprint requests to Mark L. Entman M.D., Department of Medicine, Section of Cardiovascular Sciences, Baylor College of Medicine, One Baylor Plaza, M. S. F-602, Houston, Texas 77030-3498. E-mail: mentman@bcm.tmc.edu.
Supported by National Institutes of Health grant HL-42550 and a grant from the Methodist Hospital Foundation (to N. G. F.).
References
- 1.Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B: Monocyte chemotactic proteins MCP-1, MCP-2, and MCP-3 are major attractants for human CD4+ and CD8+ T lymphocytes. FASEB J 1994, 8:1055-1060 [DOI] [PubMed] [Google Scholar]
- 2.Sica A, Wang JM, Colotta F, DeJana E, Mantovani A, Oppenheim JJ, Larsen CG, Zachariae CO, Matsushima K: Monocyte chemotactic and activating factor gene expression induced in endothelial cells by IL-1 and tumor necrosis factor. J Immunol 1990, 144:3034-3038 [PubMed] [Google Scholar]
- 3.Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR: Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol 1997, 27:1091-1097 [DOI] [PubMed] [Google Scholar]
- 4.Shyy JY, Lin MC, Han J, Lu Y, Petrime M, Chien S: The cis-acting phorbol ester “12-O-tetradecanoylphorbol 13-acetate”-responsive element is involved in shear stress-induced monocyte chemotactic protein 1 gene expression. Proc Natl Acad Sci USA 1995, 92:8069-8073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dreyer WJ, Michael LH, West MS, Smith CW, Rothlein R, Rossen RD, Anderson DC, Entman ML: Neutrophil accumulation in ischemic canine myocardium. Insights into time course, distribution, and mechanism of localization during early reperfusion. Circulation 1991, 84:400-411 [DOI] [PubMed] [Google Scholar]
- 6.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, LaRosa GJ, Hawkins HK, Smith CW, Michael LH, Entman ML, Rossen RD: Complement C5a, TGF-beta 1, and MCP-1, in sequence, induce migration of monocytes into ischemic canine myocardium within the first one to five hours after reperfusion. Circulation 1997, 95:684-692 [DOI] [PubMed] [Google Scholar]
- 7.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML: A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol 2000, 278:H1049-H1055 [DOI] [PubMed] [Google Scholar]
- 8.Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, MacKay CR, LaRosa GJ, Rossen RD, Smith CW, Entman ML: Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation 1997, 95:693-700 [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, Michael LH, Ballantyne CM, Smith CW, Entman ML: Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol 1998, 30:2567-2576 [DOI] [PubMed] [Google Scholar]
- 10.Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W: Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res 1997, 80:829-837 [DOI] [PubMed] [Google Scholar]
- 11.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML: Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98:699-710 [DOI] [PubMed] [Google Scholar]
- 12.Bolli R, Zhu WX, Hartley CJ, Michael LH, Repine JE, Hess ML, Kukreja RC, Roberts R: Attenuation of dysfunction in the postischemic ‘stunned’ myocardium by dimethylthiourea. Circulation 1987, 76:458-468 [DOI] [PubMed] [Google Scholar]
- 13.Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, McCay PB: Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ Res 1989, 65:607-622 [DOI] [PubMed] [Google Scholar]
- 14.Bolli R, Marban E: Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999, 79:609-634 [DOI] [PubMed] [Google Scholar]
- 15.DeForge LE, Preston AM, Takeuchi E, Kenney J, Boxer LA, Remick DG: Regulation of interleukin 8 gene expression by oxidant stress. J Biol Chem 1993, 268:25568-25576 [PubMed] [Google Scholar]
- 16.Yoshida Y, Maruyama M, Fujita T, Arai N, Hayashi R, Araya J, Matsui S, Yamashita N, Sugiyama E, Kobayashi M: Reactive oxygen intermediates stimulate interleukin-6 production in human bronchial epithelial cells. Am J Physiol 1999, 276:L900-L908 [DOI] [PubMed] [Google Scholar]
- 17.Lakshminarayanan V, Beno DW, Costa RH, Roebuck KA: Differential regulation of interleukin-8 and intercellular adhesion molecule-1 by H2O2 and tumor necrosis factor-alpha in endothelial and epithelial cells. J Biol Chem 1997, 272:32910-32918 [DOI] [PubMed] [Google Scholar]
- 18.Lakshminarayanan V, Drab-Weiss EA, Roebuck KA: H2O2 and tumor necrosis factor-alpha induce differential binding of the redox-responsive transcription factors and NF-kappaB to the interleukin-8 promoter in endothelial and epithelial cells. J Biol Chem 1998, 273:32670-32678 [DOI] [PubMed] [Google Scholar]
- 19.Roebuck KA, Carpenter LR, Lakshminarayanan V, Page SM, Moy JN, Thomas LL: Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors and NF-kappaB. J Leukoc Biol 1999, 65:291-298 [DOI] [PubMed] [Google Scholar]
- 20.Lee JS, Kahlon SS, Culbreth R, Cooper AD, Jr: Modulation of monocyte chemokine production and nuclear factor kappa B activity by oxidants. J Interferon Cytokine Res 1999, 19:761-767 [DOI] [PubMed] [Google Scholar]
- 21.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML: Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 1999, 99:546-551 [DOI] [PubMed] [Google Scholar]
- 22.Roebuck KA, Rahman A, Lakshminarayanan V, Janakidevi K, Malik AB: H2O2 and tumor necrosis factor-alpha activate intercellular adhesion molecule 1 (ICAM-1) gene transcription through distinct cis-regulatory elements within the ICAM-1 promoter. J Biol Chem 1995, 270:18966-18974 [DOI] [PubMed] [Google Scholar]
- 23.Hawkins HK, Entman ML, Zhu JY, Youker KA, Berens K, Dore M, Smith CW: Acute inflammatory reaction after myocardial ischemic injury and reperfusion. Development and use of a neutrophil-specific antibody. Am J Pathol 1996, 148:1957-1969 [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrer L, Fondevila D, Rabanal RM, Vilafranca M: Immunohistochemical detection of CD31 antigen in normal and neoplastic canine endothelial cells. J Comp Pathol 1995, 112:319-326 [DOI] [PubMed] [Google Scholar]
- 25.Grone A, Weckmann MT, Capen CC, Rosol TJ: Canine glyceraldehyde-3-phosphate dehydrogenase complementary DNA: polymerase chain reaction amplification, cloning, partial sequence analysis, and use as loading control in ribonuclease protection assays. Am J Vet Res 1996, 57:254-257 [PubMed] [Google Scholar]
- 26.Muller FU, Boknik P, Horst A, Knapp J, Linck B, Schmitz W, Vahlensieck U, Bohm M, Deng MC, Scheld HH: cAMP response element binding protein is expressed and phosphorylated in the human heart. Circulation 1995, 92:2041-2043 [DOI] [PubMed] [Google Scholar]
- 27.Chandrasekar B, Freeman GL: Induction of nuclear factor kappaB and activation protein 1 in postischemic myocardium. FEBS Lett 1997, 401:30-34 [DOI] [PubMed] [Google Scholar]
- 28.Muller JM, Rupec RA, Baeuerle PA: Study of gene regulation by NF-kappa B and AP-1 in response to reactive oxygen intermediates. Methods 1997, 11:301-312 [DOI] [PubMed] [Google Scholar]
- 29.May-Newman K, Mathieu-Costello O, Omens JH, Klumb K, McCulloch AD: Transmural distribution of capillary morphology as a function of coronary perfusion pressure in the resting canine heart. Microvasc Res 1995, 50:381-396 [DOI] [PubMed] [Google Scholar]
- 30.Kilgore KS, Imlay MM, Szaflarski JP, Silverstein FS, Malani AN, Evans VM, Warren JS: Neutrophils and reactive oxygen intermediates mediate glucan-induced pulmonary granuloma formation through the local induction of monocyte chemoattractant protein-1. Lab Invest 1997, 76:191-201 [PubMed] [Google Scholar]
- 31.Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL: Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ Res 1997, 81:1-7 [DOI] [PubMed] [Google Scholar]
- 32.Moellering D, McAndrew J, Jo H, Darley-Usmar VM: Effects of pyrrolidine dithiocarbamate on endothelial cells: protection against oxidative stress. Free Radic Biol Med 1999, 26:1138-1145 [DOI] [PubMed] [Google Scholar]
- 33.Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA: Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med 1992, 175:1181-1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toledano MB, Leonard WJ: Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci USA 1991, 88:4328-4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz C, Pascual-Salcedo D, Castellanos MC, Alfranca A, Aragones J, Vara A, Redondo MJ, de Landazuri MO: Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8, and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: modulation of NF-kappa B and AP-1 transcription factors activity. Blood 1996, 88:3482-3490 [PubMed] [Google Scholar]
- 36.Seino Y, Ikeda U, Sekiguchi H, Morita M, Konishi K, Kasahara T, Shimada K: Expression of leukocyte chemotactic cytokines in myocardial tissue. Cytokine 1995, 7:301-304 [DOI] [PubMed] [Google Scholar]
- 37.Bolli R: Oxygen-derived free radicals and postischemic myocardial dysfunction (“stunned myocardium”). J Am Coll Cardiol 1988, 12:239-249 [DOI] [PubMed] [Google Scholar]
- 38.Bolli R, McCay PB: Use of spin traps in intact animals undergoing myocardial ischemia/reperfusion: a new approach to assessing the role of oxygen radicals in myocardial “stunning.” Free Radic Res Commun 1990, 9:169-180 [DOI] [PubMed] [Google Scholar]
- 39.Maulik N, Goswami S, Galang N, Das DK: Differential regulation of Bcl-2, AP-1 and NF-kappaB on cardiomyocyte apoptosis during myocardial ischemic stress adaptation. FEBS Lett 1999, 443:331-336 [DOI] [PubMed] [Google Scholar]
- 40.Meyer M, Schreck R, Baeuerle PA: H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J 1993, 12:2005-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richard V, Murry CE, Reimer KA: Healing of myocardial infarcts in dogs. Effects of late reperfusion. Circulation 1995, 92:1891-1901 [DOI] [PubMed] [Google Scholar]
- 42.Weihrauch D, Tessmer J, Warltier DC, Chilian WM: Repetitive coronary artery occlusions induce release of growth factors into the myocardial interstitium. Am J Physiol 1998, 275:H969-H976 [DOI] [PubMed] [Google Scholar]