

Abstract
Type XIII collagen is a type II transmembrane protein found at many sites of cell adhesion in tissues. Homologous recombination was used to generate a transgenic mouse line (Col13a1N/N) that expresses N-terminally altered type XIII collagen molecules lacking the short cytosolic and transmembrane domains but retaining the large collagenous ectodomain. The mutant molecules were correctly transported to focal adhesions in cultured fibroblasts derived from the Col13a1N/N mice, but the cells showed decreased adhesion when plated on type IV collagen. These mice were viable and fertile, and in immunofluorescence stainings the mutant protein was located in adhesive tissue structures in the same manner as normal α1(XIII) chains. In immunoelectron microscopy of wild-type mice type XIII collagen was detected at the plasma membrane of skeletal muscle cells whereas in the mutant mice the protein was located in the adjacent extracellular matrix. Affected skeletal muscles showed abnormal myofibers with a fuzzy plasma membrane-basement membrane interphase along the muscle fiber and at the myotendinous junctions, disorganized myofilaments, and streaming of z-disks. The findings were progressive and the phenotype was aggravated by exercise. Thus type XIII collagen seems to participate in the linkage between muscle fiber and basement membrane, a function impaired by lack of the cytosolic and transmembrane domains.
The collagen superfamily of proteins consists of more than 19 types of collagen and several other proteins with collagen-like domains. 1 Type XIII collagen and the hemidesmosomal component type XVII collagen form a subfamily of transmembrane collagens. 2 The genes of human and mouse type XIII collagen are 135 to 138 kb in size, consisting of 42 exons, and they are localized to chromosome 10 in both species. 3-5 The encoded type XIII collagen consists of three collagenous domains (COL1 to COL3) separated and flanked by four noncollagenous domains (NC1 to NC4). 6,7 The precursor RNAs that encode type XIII collagen undergo complex alternative splicing, which is predicted to affect the structures of the COL1, NC2, and COL3 domains of the human and mouse chains. 5,6,8-10 Type XIII collagen produced in insect cells forms α1(XIII) homotrimers, and the three collagenous domains fold into a stable triple-helical conformation. 11 The type XIII collagen molecules have been shown to reside on the plasma membranes of cells in a type II orientation with a short N-terminal cytosolic portion, a transmembrane domain, and an extensive collagenous ectodomain. 12 Sequences that are important for association of the three α1(XIII) chains reside in the N-terminal region, and hence triple helix formation is thought to proceed from the N terminus to the C terminus, in the opposite orientation to that known to occur in the fibrillar collagens. 12 The extracellular ligands of type XIII collagen have not been identified, but recent studies with recombinant protein demonstrate that its ectodomain interacts strongly with the I domain of α1 integrin. 13
In situ hybridization analyses have shown that type XIII collagen mRNAs occur in a wide range of tissues. 9,14 Immunohistochemical studies of mature human and mouse tissues have shown that this collagen is located at many cell-matrix adhesion sites, eg, the myotendinous junctions and at cell-cell adhesion sites. 15 Type XIII collagen is expressed throughout mouse fetal development, with a clear increase toward the time of birth and strongest initial expression in the developing nervous system and heart. As development proceeds, it is found in developing bone, cartilage, intestine, skeletal muscle, lung, and skin, with clear developmental shifts in expression pattern. 16
To understand the biological function of type XIII collagen, we have generated a mouse strain that expresses it in an N-terminally altered form through site-specific Cre-loxP-mediated deletion of exon 1 sequences in embryonic stem (ES) cells. The data suggest a role for the cytosolic and transmembrane domains in skeletal muscle integrity.
Materials and Methods
Construction of the Targeting Vector
A 9.0-kb BamHI fragment containing the promoter, the transcription initiation sequences, the first exon, and part of the first intron of the mouse gene for type XIII collagen was derived from the cosmid clone 19A, originally isolated from a 129svJ genomic library 3 and subcloned into pSP72 (Promega, Madison, WI) (Figure 1) ▶ . The loxP sequence was amplified from the pBSloxP vector using primers flanking the NotI recognition sequences, and the polymerase chain reaction (PCR) product was digested with NotI and inserted into the unique NotI site in the Col13a1 5′-untranslated region. A selection marker gene cassette flanked by loxP sites and containing a novel EcoRI site just upstream of the 5′-loxP sequence was released from the pBSloxP-neor-HSV-tk-loxP plasmid 17 and inserted into the genomic SfiI site 120 bp (bp) downstream of the first exon of Col13a1.
Figure 1.
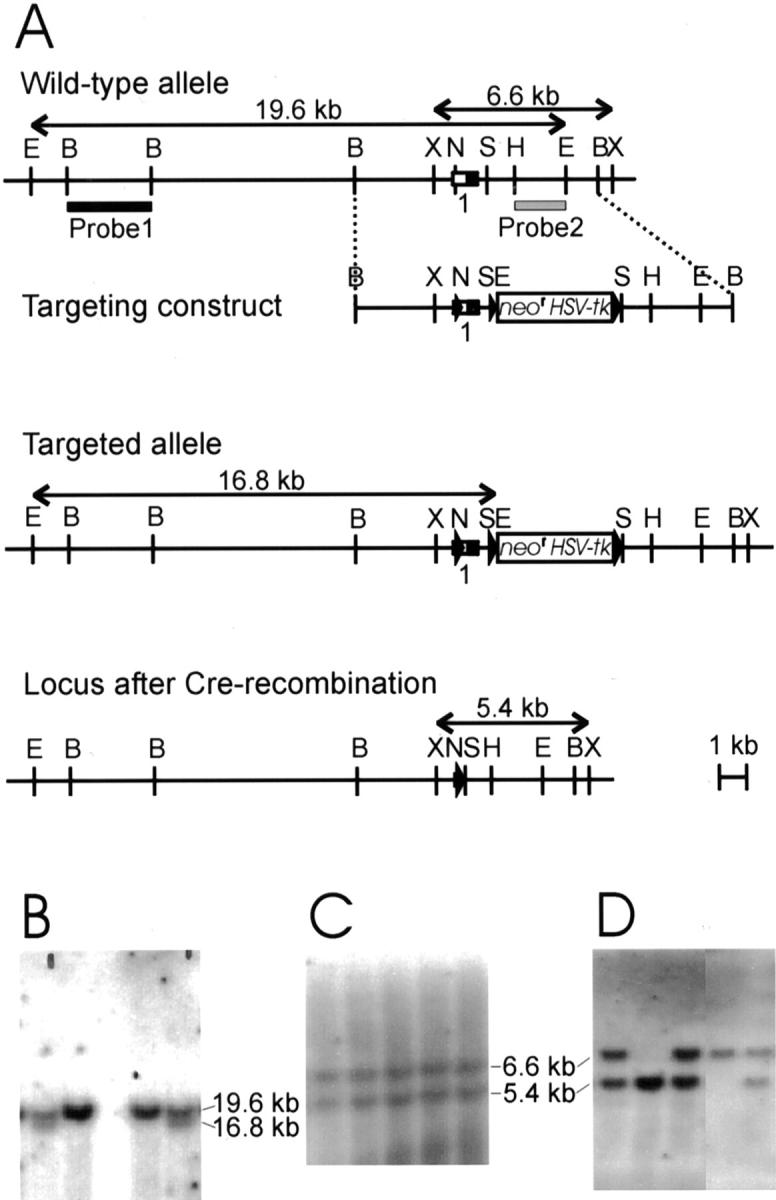
Targeted deletion of exon 1 in Col13a1. A: Targeting strategy and Cre-mediated recombination. Top: Restriction map for the 5′ portion of the Col13a1 gene wild-type allele showing exon 1 and predicted restriction fragments. Untranslated and translated regions of exon 1 are shown as open and filled squares, respectively. A 9-kb BamHI fragment was used to generate a targeting vector with a loxP-neor-HSV-tk-loxP cassette inserted into a SfiI site and a third loxP site inserted into a NotI site. The loxP sites are shown as filled triangles and the bars indicate the two probes used for Southern blot analysis. Middle: Targeted allele. Bottom: Col13a1 allele with exon 1 deleted. B: Genotypes of EcoRI-digested ES cell DNA from wild-type and targeted cell lines. Probe 1 detects a 19.6-kb fragment in the wild-type allele and a 16.8-kb fragment in the targeted allele. C: After Cre-mediated deletion of the targeted allele, probe 2 detects a 5.4-kb XbaI fragment in ES cell DNA instead of the 6.6-kb fragment seen in the wild-type. D: The targeted cells were used to generate mutant mice and a Southern analysis of XbaI-digested DNA with probe 2 reveals wild-type, heterozygous mutant, and homozygous mutant mice in the F2 generation after matings of heterozygous F1 mice. B, BamHI; E, EcoRI; H, HindIII; N, NotI; S, SfiI; X, XbaI.
Targeting Vector Transfections and Southern Blot Analysis of ES Cells
The targeting vector (50 μg) was linearized with ClaI, electroporated into 1.5 × 10 7 R1 embryonal stem (ES) cells 18 and cultured on embryonic fibroblast feeder cells as described earlier. 19 After selection, genomic DNA from G418 (400 μg/ml; Life Technologies, Inc., Rockville, MD)-resistant ES clones was digested with EcoRI and analyzed by Southern blot hybridization with a 5′ external probe (probe 1 in Figure 1 ▶ ). The 360 clones included 4 that were correctly targeted, having the selection marker genes (neor and HSV-tk) in the first intron of the Col13a1 gene and loxP sites flanking the first exon. Targeted ES-cells (6.1 × 107) were electroporated as earlier with 40 μg of supercoiled Cre-plasmid (pIC-Cre, a gift from Dr. W. Müller, Institute for Genetics, University of Cologne, Cologne, Germany), and ganciclovir selection (Syntex, Palo Alto, CA) was started 1 to 4 days afterward, depending on the plate, after which the cells were selected for 5 days. The DNA from the 72 surviving ES clones was digested with XbaI and analyzed by Southern blot hybridization with probe 2 (Figure 1) ▶ . Two ES clones with the mutant Col13a1 allele were injected into blastocysts and implanted in the pseudopregnant mice, and the ensuing mice were bred with C57BL/6J and 129sv females to produce heterozygous F1 mice with mixed and inbred genetic backgrounds, respectively. 19
Genotyping and Computational Analyses
DNA was extracted from the tail, liver, or spleen according to standard protocols 20 and PCR amplifications were performed using three primers in a single reaction: Notinfor [5′-AGATTAGTTTGGGAAGTAGCGCCCG-3′]; Notinrev [5′-TCCCTTCTCTCTTCTGCAGAGTTCTCG-3′]; and Sfiinrev [5′-TTAACTACCTGGGAAGGGAGACTTTTG-3′]. The reactions included 35 cycles (60 seconds at 95°C, 45 seconds at 65°C, 45 seconds at 72°C) after initial activation of AmpliTaq Gold (12 minutes at 96°C) (PE Biosystems, Foster City, CA).
Nucleotide sequences were analyzed using the Chromas (Technelysium Pty. Ltd., Helensvale, Australia) and DNASIS (Amersham Pharmacia Biotech, Uppsala, Sweden) programs, and alignments were performed with the GAP program (Wisconsin Package Version 10.0; Genetics Computer Group, Madison, WI). Statistical analyses were performed with either SigmaStat (Jandel Scientific, SPSS Science, Chicago, IL) or Excel (Microsoft Corp., Redmond, WA).
Isolation of RNA, Reverse Transcriptase (RT)-PCR, Sequencing, and Quantitative RT-PCR
Total RNA was isolated from cultured skin fibroblasts and muscles as described earlier. 21 To analyze the RT-PCR product adjacent to exons 16 to 26, total RNA was isolated from mouse muscles described above and 2.5 μg of this together with 150 ng of random oligohexamers (Life Technologies, Inc., Rockville, MD) were annealed at 70°C for 10 minutes and the RT reaction performed with 200 U of the M-MLV RT enzyme (Life Technologies, Inc.) at 42°C for 50 minutes with subsequent treatment of the products with 2 U of RNaseH (Life Technologies, Inc.) at 37°C for 20 minutes. For the PCR reaction, 2 μl of the RT reaction product was used as a template with the primers 5′-GATGCTGCCATTATAATCCACCATCTC-3′ (complementary to sequence 1808 to 1834) 7 and 5′-CCTAAAGGGGAACAAAGTCAGACTGGC-3′ (corresponding to sequences 1201 to 1207 and 1274 to 1294). 7
For 5′-end analysis, total RNA (6 μg) was transcribed into single-stranded DNA using the type XIII collagen-specific reverse oligonucleotide primer RTex2rev (150 pmol, 5′-CCTGGTGGGCAGTTACATCCT-3′, complementary to nucleotides 811 to 831). 7 The 50-μl reactions contained 30 U of avian myeloblastosis virus RT (Finnzymes Inc., Espoo, Finland), RT buffer, 0.5 mmol/L of each of the four deoxynucleotides, 33 U of RNAsin (Promega) and 0.2 μg/μl of acetylated bovine serum albumin (BSA) (Promega). RT reaction was performed at 50°C for 40 minutes and otherwise as described above. One μl of the RT reaction was used as a template for PCR amplification with the type XIII collagen-specific sense and antisense oligonucleotide primers (5′-GCAGGAGATTAGTTTGGGAAGTAGCG-3′, complementary to nucleotides 4402 to 4427 3 and RTex2rev (see above) (10 pmol each). The 25 μl reaction volume included 0.15 mmol/L deaza-dGTP, 0.05 mmol/L dGTP, 0.2 mmol/L dATP, 0.2 mmol/L dCTP, 0.2 mmol/L dTTP, 2.5 U of AmpliTaq Gold (PE Biosystems) in 10 mmol/L of Tris-HCl pH 8.3, 50 mmol/L of KCl, and 2.0 mmol/L of MgCl2. Amplification included 40 cycles of denaturation (60 seconds at 95°C), annealing (45 seconds at 64°C), and extension (45 seconds at 72°C) after initial activation of AmpliTaq Gold. For the negative control samples the template was omitted and PCR was performed as described above. The PCR products were sequenced using an ABI-Prism DNA sequenator (PE Biosystems).
For quantitative RT-PCR, 200 ng of total RNA from three m. quadriceps femoris (MQF) samples was used as a template for a RT reaction primed by oligohexamers. The RT reaction was performed using the Taqman RT-PCR-Gold kit according to the manufacturer’s protocol (PE Biosystems, reaction: 10 minutes at 25°C, 30 minutes at 48°C, and 5 minutes at 95°C), after which type XIII collagen mRNA levels were measured by quantitative RT-PCR analysis using the forward and reverse primers 5′-ACCTGGACTAGACGCCCCTG-3′ and 5′-TTGTTCCAGCAGCCTTGGACT-3′, respectively (nucleotides 2623 to 2640 and 2688 to 2668). 7 The 66-bp amplicon was detected using the bifunctional fluorogenic probe 5′Fam-CCCGCTGGGAGAAGATGGCTTACC-Tamra-3′ and the collagen type XIII mRNA expression levels were normalized to the levels of 18S mRNA as described. 22 PCR reactions were performed in triplicate on samples of the resulting cDNA using the TaqMan Gold PCR reagent kit (PE Biosystems) (initiation 2 minutes at 50°C followed by 10 minutes at 95°C and 40 cycles of 1 second at 95°C and 1 minute at 60°C). The levels of type XIII collagen expression were compared between the mutant and control muscle samples and the statistical significance of the results analyzed by the Mann-Whitney U test.
Cell Cultures
Fibroblast cultures were established from 8-week-old mouse skin biopsies and 13.5 days post coitus embryos from both control 129sv and homozygous mutant mice. The fibroblasts were grown in Dulbecco’s modified Eagle’s medium (Biochrom KG Seromed, Berlin, Germany) supplemented with 10 to 15% heat-inactivated fetal bovine serum (Autogen Bioclear UK Ltd., Calne Wiltshire, UK), 2 mmol/L l-glutamine (Life Technologies, Inc., Rockville, MD), 50 μg/ml l(+)-ascorbic acid sodium salt (Fluka, Buchs, Switzerland), nonessential amino acids (Life Technologies, Inc.), 100 U/ml penicillin (Life Technologies, Inc.), 100 μg/ml streptomycin (Life Technologies, Inc.). and 250 ng/ml Amphotericin B (JRH Biosciences, Hampshire, UK). For embryonal fibroblast cultures 1 mmol/L of sodium pyruvate (Life Technologies, Inc.) was added to the medium.
Adhesion Studies
Fibroblast adhesion to uncoated 96-well plates (Becton Dickinson Labware, Franklin Lakes, NY) was studied first by seeding 0.2 ml of embryonal fibroblast cells (150,000 cells/ml) per well and allowing them to attach for 20, 40, or 60 minutes. Seven to 11 replicate measurements were performed at each time point. After the attachment period the wells were emptied by suction, the plate was washed and the DNA content was assayed using the CyQuant Cell proliferation assay kit (Molecular Probes, Eugene, OR). The amount of bound DNA was measured with the Victor enzyme-linked immunosorbent assay plate reader (Wallac, Turku, Finland).
Adhesion to specific substrates was then tested by coating Maxisorp 96-well plates (Nunc Brand Products, Roskilde, Denmark) with 2 μg/cm 2 of mouse laminin, human fibronectin, mouse type IV collagen (all from Collaborative Biochemical Products, Bedford, MA), or BSA (heat-inactivated at 60°C for 10 minutes; Boehringer Mannheim, Mannheim, Germany) in phosphate-buffered saline (PBS) and incubating them overnight at 4°C. The wells were washed with PBS and nonspecific protein binding was blocked with 10 mg/ml heat-inactivated BSA incubated for 1 hour at room temperature followed by washing with PBS and distilled water. Subconfluent embryonal fibroblasts were washed twice with PBS, detached with trypsin, treated with soybean trypsin inhibitor (1 mg/1.8 mg trypsin, Sigma, St. Louis, MO) and washed with serum-free medium. The same amounts of cells as described above were allowed to attach for 5, 10, 20, or 40 minutes (five replicate measurements at each time point), after which the wells were emptied by suction, washed with PBS, and the DNA content assayed.
Immunofluorescence Staining of Fibroblasts
For the immunofluorescence stainings, mutant and wild-type skin fibroblasts were cultured on sterilized glass coverslips for 1, 2, 3, 4, 9, and 12 hours. The cells were fixed for 5 minutes in precooled methanol at −20°C and incubated in 1% BSA-PBS, pH 7.2, for 30 minutes to reduce nonspecific staining. Rabbit anti-human type XIII collagen XIII/NC3, 7 mouse anti-talin (Chemicon International, Inc., Temecula, CA) and mouse anti-vinculin antibodies were applied at the appropriate dilutions and incubated for 30 minutes at room temperature, followed by extensive washing with PBS. Porcine anti-rabbit-fluorescein isothiocyanate and rabbit anti-mouse-tetramethylrhodamine B isothiocyanate secondary antibodies were diluted according to the manufacturer’s instructions (DAKO A/S, Glostrup, Denmark) and allowed to bind to the specimens for 1 hour at room temperature. After washing with PBS, the coverslips were mounted on microscope slides with Immu-Mount (Shandon Inc., Pittsburgh, PA), viewed, and photographed on a Leica Aristoplan microscope with the appropriate filter units (Leica Inc., Deerfield, IL).
Light Microscopy and Immunofluorescence Staining of Tissues
Heart, brain, muscle, lung, skin, liver, spleen, kidney, and testis samples from 17-week-old wild-type and mutant mice were embedded in TissueTec medium (Sakura Finetek U.S.A., Inc., Torrance, CA) and frozen immediately in liquid nitrogen. Either 5 μm or 10 μm sections were cut and stained with the rabbit anti-human XIII/NC3 antibody 7 and hematoxylin and eosin (H&E), respectively. In addition, m. gastrocnemius and MQF samples were prepared from 8-, 17-, and 43-week-old mice, mounted in embedding medium and frozen immediately in isopentane precooled in liquid nitrogen. The muscle samples were stained with H&E, rabbit anti-collagen IV (Chemicon International, Inc., Temecula, CA), rabbit anti-laminin-2 (merosin) (ProGen, Tustin, CA), mouse anti-vinculin (Sigma), rat anti-tenascin-C (Sigma), rat anti-α5-integrin (Pharmingen, San Diego, CA), mouse anti-desmin (Sigma), mouse anti-proliferating cell nuclear antigen (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rat anti-PECAM (CD31) (Pharmingen), and rabbit anti-human XIII/NC3 antibodies. 7 Nonspecific staining was reduced by incubating the slides for an hour in 1% BSA-PBS, pH 7.2, after which they were exposed to the primary antibodies in a moist chamber overnight at 4°C. After several washes in PBS, the secondary antibodies goat anti-mouse-fluorescein isothiocyanate (DAKO A/S), goat anti-rat-CY3 (Jackson ImmunoResearch, Laboratories Inc., West Grove, PA) and goat anti-rabbit-CY3 (Jackson ImmunoResearch) were applied and incubated in a dark moist chamber at room temperature for an additional half hour. For mouse monoclonal antibodies, 2% goat serum and a 1:50 dilution of goat anti-mouse IgG (DAKO A/S) were added to the blocking solution.
Electron Microscopy
For electron microscopy, the muscles described above were fixed in 2% glutaraldehyde and 0.1 mol/L phosphate buffer, postfixed in 1% osmium tetroxide, dehydrated in acetone, and embedded in Epon LX112. Thin sections were cut with a Reichert Ultracut E-ultramicrotome (Reichert-Jung, Vienna, Austria) and examined in a Philips CM100 transmission electron microscope (Philips Export B.V., Eindhoven, Netherlands) at an acceleration voltage of 80 kV. Density measurements were performed on the plasma membrane-basement membrane region using a charge-coupled device camera and an Electron Microscopy Menu version 2.1 from Tietz Video and Image Processing Systems GmbH (Gaunting, Germany). Density values were measured for three wild-type and three mutant animals, five to eight cells from each individual, totaling 19 mutant and 17 control cells. All of the values measured for each cell were divided by the plasma membrane density value for the same cell to normalize the results and facilitate comparison. Moving averages of 20 data points were used to reduce fluctuations in the measurements and show the trend more clearly.
Polyclonal Antibodies against Mouse Type XIII Collagen
A synthetic peptide (DYNGSINEALQEIRTL) corresponding to the mouse NC3 domain 7 was used as an antigen to generate a rabbit polyclonal antibody (Innovagen, Sweden). The mouse NC3 sequence differs from the human counterpart by one residue. 7 The ensuing anti-mouse XIII/NC3 antibody was affinity purified using the above peptide and tested for specificity by Western blotting of insect cell-derived extracts containing recombinant type XIII collagen as previously described (data not shown). 15 The anti-mouse XIII/NC3 antibody was used for immunoelectron microscopy.
Immunoelectron Microscopy
Samples of m. gastrocnemius obtained from 24-week-old wild-type and mutant mice were fixed in 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4, containing 2.5% sucrose for 2 hours. Small tissue pieces were immersed in 2.3 mol/L of sucrose in PBS overnight and frozen in liquid nitrogen. Thin cryosections were cut with Leica Ultracut UCT microtome. For the immunolabeling, the sections were first incubated in 5% BSA with 0.1% cold water fish skin (CWFS) gelatin (Aurion, Wageningen, The Netherlands) in PBS. Antibodies and gold conjugate were diluted in 0.1% BSA-C (Aurion) in PBS. All washings were performed in 0.1% BSA-C in PBS. Sections were then incubated with the polyclonal anti-human XIII/NC3 7 and anti-mouse XIII/NC3 antibodies described above for 60 minutes followed by protein A-gold complex (size 10 nm) for 30 minutes. 23 The controls were prepared by performing the labeling procedure without primary antibody. The sections were embedded in methylcellulose and examined in Philips CM100 transmission electron microscope.
Running Exercise
Fourteen mutant and 12 wild-type male mice aged 8 to 10 weeks were made to run on a motor-driven treadmill with 6° uphill tracks at a speed of 8.5 m × min−1 for 6 hours. After 2 and 4 hours of running there was a 20-minute resting period during which the mice had free access to pelleted food and water. Forty-eight hours after the cessation of exercise the animals were sacrificed together with unexercised control and mutant mice (wild type, n = 8; mutants, n = 7). The proximal part of the MQF was excised from the left hind limb, frozen in liquid nitrogen, and β-glucuronidase activity was measured as described. 24 For histological analysis, H&E and immunostained sections of the MQF and m. gastrocnemius from the contralateral leg were frozen in liquid nitrogen-cooled isopentane and oriented under a microscope.
Results
Generation of a Mouse Strain Lacking Exon 1 of the Type XIII Collagen Gene
The mouse type XIII collagen gene is 135 kb in size and contains 42 exons. 3 Our aim was to generate null mice for this collagen type by deleting exon 1, which encodes the 96 extreme N-terminal amino acid residues of the α1(XIII) chain, including the cytosolic and transmembrane domains. A knockout targeting vector was constructed in which one loxP site was inserted into the NotI site of exon 1 and a selection marker cassette (neor and HSV-tk) flanked by loxP sites was inserted into the SfiI site of the first intron of the mouse genomic sequences (Figure 1A) ▶ . Homologous recombination was identified in several ES cell clones by Southern blot analysis of EcoRI-digested genomic DNA using probe 1 (Figure 1B) ▶ . Two correctly targeted clones were exposed to Cre treatment, and ES cell clones surviving gancyclovir selection were subjected to Southern blot analysis of XbaI digested DNA using probe 2 (Figure 1C) ▶ . Several alleles with a deletion encompassing the first exon and adjacent noncoding sequences were obtained, but no conditional recombinants were achieved (Figure 1C) ▶ . One ES cell clone with a Col13a1 allele lacking exon 1 was used to generate chimeric males that transmitted the mutant allele to their progeny (Figure 1D) ▶ .
Homologous Recombination Results in N-Terminally Altered Type XIII Collagen
To confirm that the mutation leads to a loss of type XIII collagen function, mRNA and protein expression were studied. Surprisingly, immunofluorescence stainings of mice that were homozygous for the altered allele revealed clear staining for type XIII collagen (Figure 4D) ▶ . Furthermore, RT-PCR analysis with primers adjacent to exons 16 and 26 revealed clear expression of the α1(XIII) mRNA (data not shown). This prompted us to investigate the 5′ sequences of the type XIII collagen transcripts in the homozygous mutant mice by RT-PCR using primers flanking the NotI site in the 5′-untranslated region and exon 2. Sequencing of the ensuing 365-bp RT-PCR product revealed that the transcript of the targeted allele had acquired a new 5′ end that included the loxP site and extended 155 bp to the first intron, after which the sequence continued into exon 2. Sequence analysis showed the first intron to contain a cryptic splice site TCCCAGgtagtta (nucleotides shown with capital letters being included in the RT-PCR product) that serves as a donor site for splicing to the second exon (Figure 2) ▶ .
Figure 4.

H&E and immunofluorescence stainings of muscles from wild-type mice (A, C, and E) and Col13a1N/N mice (B, D, and F). The muscle fibers of the mutant mice have an uneven appearance in the H&E stainings, with rough edges (B). Staining with anti-XIII/NC3 antibodies, which detect the ectodomain of type XIII collagen, reveal clear expression of this collagen type in the Col13a1N/N mice (D), but the myofiber structure is uneven compared with that in the wild-type mice (C). Smooth, linear staining of longitudinal muscle sections with anti-type IV collagen antibodies is seen in the wild-type (E), whereas the basement membranes between the muscle fibers are highly uneven and fuzzy in appearance in the mutant case (F). Scale bars, 20 μm.
Figure 2.

Schematic presentation of the wild-type and mutant forms of exon 1 and the deduced amino acid sequences. The coding regions of the wild-type exons 1 and 2 are marked as filled squares and the 5′-untranslated region as an open square. The cryptic donor splice site used in the targeted allele is indicated by an asterisk. The intron 1-derived sequence found in the ensuing mutant mRNA is marked as a striped box in the altered exon 1, and the filled triangle indicates a loxP sequence. The amino acid sequences encoded by the wild-type and altered forms of exon 1 and the first three residues encoded by exon 2 are indicated. Potential initiation methionines are marked with shaded circles, the transmembrane domain encoded by the wild-type exon 1 is underlined, and the 20-kb intron 1 is shown.
Assessment of the new 5′ sequences of the mutant type XIII collagen transcript indicated that there is an ATG codon in the loxP sequence and another in the intron 1 sequence that are retained in the transcript. Both ATG codons are in-frame with exon 2-derived sequences. Thus the transcripts derived from the targeted allele can be predicted to result in synthesis of N-terminally altered type XIII collagen molecules in which the 96 amino acid residues encoded by the first exon are replaced by a new sequence that is either 65 or 11 residues in length, depending on which of the two potential initiation codons is used (Figure 2) ▶ . As a result, the N-terminally altered type XIII collagen molecules lack the cytosolic and transmembrane domains and the first 37 residues of the ectodomain, and these are replaced by unique sequences. The consensus sequence for the initiation of translation is gcca/gccAUGg, 25 and the corresponding sequences for the longer and shorter mutants were uaauguAUGc and gaaaggAUGc, respectively. Sequences downstream of both methionines contain some hydrophobic amino acid residues, but neither of these is predicted to serve as a transmembrane domain or signal peptide (Figure 2) ▶ . The mutant allele was designated as Col13a1N.
Viability and Breeding
Mice that were homozygous for the altered Col13a1N allele were normal in appearance and were indistinguishable from their wild-type littermates. Southern blot genotyping of 381 offspring from heterozygous intercrosses showed that 22% were of the wild type, 51% were heterozygous, and 27% were homozygous for the Col13a1N allele. Thus Mendelian transmission of the Col13a1N allele to the offspring of heterozygous crosses indicates that the absence of exon 1 sequences does not lead to loss of essential functions during development. No gross phenotypic abnormalities were detected on inspection of homozygous mutant embryos or new born mutant mice. Furthermore, mice that were homozygous for the allelic change showed no changes in their growth, behavior, or reproductive capacity compared with normal littermates.
N-Terminally Altered Type XIII Collagen Results in Decreased Fibroblast Adhesion
Type XIII collagen can be detected at focal adhesions of cultured fibroblasts, and it has been predicted to play a role in cellular adhesion. 15 To explore the properties of the N-terminally altered type XIII collagen molecules, we established skin fibroblast cultures from 8-week-old homozygous and control mice. Surprisingly, immunofluorescence stainings of adhering cells and mature spread cells with an anti-human XIII/NC3 antibody recognizing the NC3 domain of human and mouse α1(XIII) chains resulted in similar staining patterns in both the mutant and control cell lines (Figure 3, A and C) ▶ . The type XIII collagen signal co-localized with that of the classical focal adhesion marker vinculin in both cell lines (Figure 3, B and D) ▶ . Thus, despite the lack of the cytosolic and transmembrane domains, the N-terminally altered type XIII collagen molecules are correctly transported to the focal adhesions and located there.
Figure 3.

Type XIII collagen and vinculin expression by wild-type and Col13a1N/N fibroblasts and analysis of their adhesion properties. Skin fibroblasts derived from wild-type (A and B) and Col13a1N/N (C and D) mice were grown on glass coverslips for 9 hours and subjected to double-immunofluorescence staining with anti-type XIII collagen antibodies (A and C) and anti-vinculin antibodies (B and D). The cells that were homozygous for the N-terminally altered type XIII collagen (C and D) were the same in overall morphology and appearance as the control cells (A and B). E and F: Wild-type (shaded bars) and Col13a1N/N embryonal fibroblasts (open bars) plated (30,000 cells/well) on uncoated wells in a serum-containing medium (E) or on type IV collagen-coated wells in a serum-free medium (F). The unattached cells were removed after 20, 40, or 60 minutes (n = 54) (E) or 5, 10, 20, or 40 minutes (n = 5) (F), the adhering cells were lysed, and the amount of DNA in the wells was measured. The binding results are expressed relative to the last time point for the wild-type cells. Significant differences between the Col13a1N/N and wild-type fibroblasts (P < 0.01) are indicated by two asterisks. Scale bars, 10 μm.
The fibroblasts derived from the mutant mice cannot be described as entirely normal, however. When the adhesion of fibroblasts derived from 13.5-day-old embryos plated in the presence of a serum-containing medium was assayed 20, 40, and 60 minutes after subculturing, the cells expressing altered type XIII collagen demonstrated an 8 to 15% lower adherence to the culture plates than the controls, a difference that was statistically significant (Figure 3E) ▶ . When cellular adhesion was studied by plating the fibroblasts on plates coated with type IV collagen, the difference was even more dramatic (Figure 3F) ▶ . The other substrates tested, namely fibronectin, laminin, and BSA, did not give measurable differences (data not shown). The lowered adhesion properties of the fibroblasts expressing altered type XIII collagen were also observed after centrifugation of the cells, when it was consistently found that cell pellets derived from affected mice detached from the tubes more readily than wild-type cell pellets.
Morphological Changes in Skeletal Muscle
Systematic examination of the gross anatomy of a number of tissues from 17-week-old homozygous mice and histological examination by light microscopy revealed changes only in the skeletal muscle. Some of the muscle fibers appeared uneven in size in H&E stainings, and the fibers had a wavy sarcolemma that could not be attributed to problems of sample preparation (Figure 4, A and B) ▶ . The diameter of the muscle fibers appeared smaller in mutants compared with controls (Figure 4; A to D ▶ ). Immunofluorescence staining with antibodies detecting type XIII collagen revealed a clear staining in the Col13a1N/N mice that was comparable in intensity and location to that in the controls (Figure 4, C and D) ▶ , except that the staining along the muscle fibers was somewhat uneven and adjacent fibers appeared to be more loosely attached to each other. These findings suggested that the N-terminally altered type XIII collagen chains are expressed at a comparable level to intact molecules but cause disturbances in skeletal muscle integrity. Quantitative RT-PCR analysis using RNA isolated from muscles subsequently confirmed equal levels of expression of the type XIII collagen alleles in the wild-type and mutant mice (data not shown).
The skeletal muscle tissues of the Col13a1N/N mice were further characterized by immunofluorescence staining with antibodies against type IV collagen, a component of all basement membranes. The staining pattern was suggestive of an infirm, fuzzy basement membrane in the muscle expressing altered type XIII collagen (Figure 4, E and F) ▶ . These findings were focal but rather common, ie, they could be found easily in each set of muscle sections. Immunofluorescence staining of the ligand for the dystrophin-dystroglycan receptor complex laminin-2 revealed the same staining pattern as for type IV collagen, and the signal intensity was comparable to that in the controls (data not shown).
Ultrastructural Abnormalities in Skeletal Muscle
A more detailed analysis was performed by electron microscopy of m. gastrocnemius and m. quadriceps femori (MQF) samples from 8-, 17-, and 43-week-old wild-type and Col13a1N/N mice. The mutant skeletal muscle showed vacuolization and disorganization of myofilaments and z-bands when compared with wild-type mice (Figure 5, A and B) ▶ . Accumulation and enlargement of mitochondria were often detected (data not shown). The sarcolemma and the adjacent basement membrane showed a disorganized, fuzzy structure compared with that seen in the corresponding wild-type mice, and this was particularly evident at the myotendinous junctions (Figure 5, C and D) ▶ . These abnormalities were detected more frequently and were more pronounced in the older mice, suggesting a progressive condition.
Figure 5.
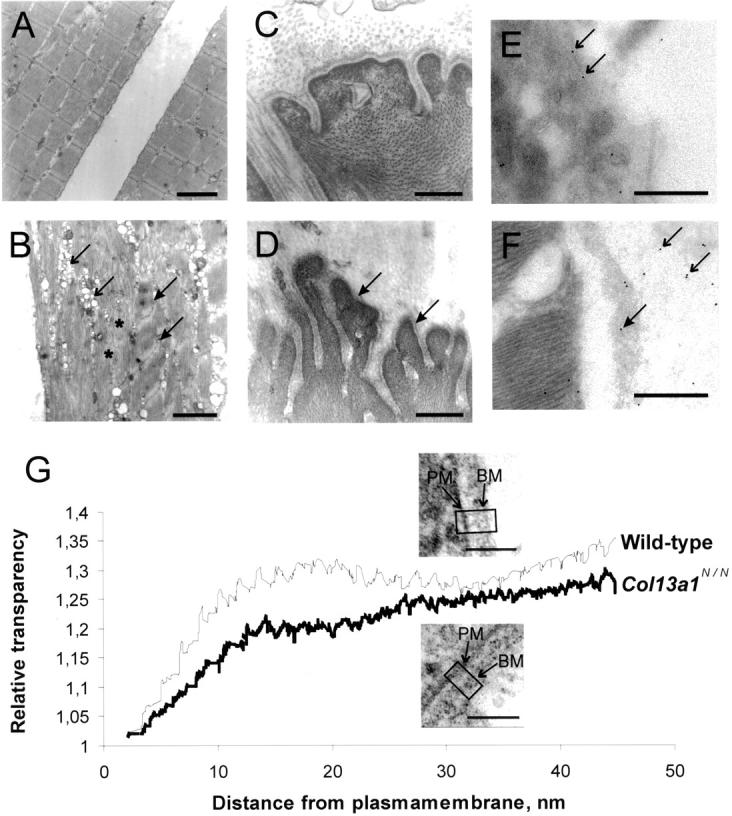
Transmission electron microscopy and immunoelectron microscopy of wild-type and Col13a1N/N skeletal muscle. Samples of the MQF from 43-week-old wild-type (A and C) and Col13a1N/N mice were prepared for electron microscopy (B and D). In the mutant case the myofibrils (asterisk) are disorganized and there is z-band streaming (filled arrows) and vacuolization (arrows) indicating muscle cell degeneration (B). The basement membranes attached to the muscle fibers are abnormal, this being most clearly visible at the myotendinous junctions (filled arrows in D). Samples of the m. gastrocnemius from 24-week-old wild-type (E) and Col13a1N/N (F) mice were used for immunoelectron microscopy with the antibody anti-mouse XIII/NC3. In wild-type samples most of the staining is localized to the plasma membrane (arrows in E) whereas in the mutant most of the staining can be seen in the detached basement membrane (filled arrow in F) and adjacent extracellular space (arrows in F). In G the relative transparency is plotted against the distance from the plasma membrane toward the extracellular matrix. The values are normalized as indicated in Materials and Methods. Insets: Wild-type (top) and lower Col13a1N/N (bottom) demonstrate areas used for the density measurements. The curves summarize the results for a total of 19 mutant and 17 control cells. PM and BM indicate plasma membranes and basement membranes, respectively. Scale bars: 2 μm (A and B), 400 nm (C–F), 100 nm (G).
To analyze the sarcolemma-matrix region further, we measured the optical density of the extracellular matrix immediately adjacent to the sarcolemma. A transparent interphase of 10 to 20 nm was observed between the plasma membrane and the basement membrane in the controls, whereas in the Col13a1N/N mice this space was opaque (Figure 5G) ▶ .
Immunoelectron Microscopy
To obtain a clearly detectable signal in immunoelectron microscopy, a rabbit polyclonal antibody against a synthetic peptide corresponding to the mouse NC3 domain was generated and affinity purified. The specificity of the anti-mouse XIII/NC3 antibody was confirmed by Western blotting of cellular extracts derived from insect cells expressing recombinant human type XIII collagen and the key enzyme of collagen synthesis, prolyl 4-hydroxylase. The anti-mouse XIII/NC3 antibody recognized the same type XIII collagen bands (data not shown) as the previously characterized anti-human XIII/NC3 antibody 7 produced against the human sequence that differs from the mouse NC3 by one residue. No bands could be detected with the anti-mouse XIII/NC3 antibody in insect cells infected only with the virus encoding human prolyl 4-hydroxylase (data not shown). 15
Both antibodies gave similar staining patterns in immunoelectron microscopy, but with the anti-mouse XIII/NC3 resulting in more intense signals. In skeletal muscle from wild-type mice gold particles were most prominently seen in close contact with plasma membranes as is expected for molecules anchored to the cell membranes (Figure 5E) ▶ . Some intracellular staining was visible as well as staining in the extracellular matrix (Figure 5E) ▶ , which is not surprising because furin-type endoproteases have been shown to cleave the ectodomain of type XIII collagen. 12 The proportions of membrane-bound and shed forms of type XIII collagen in tissues is not known, but the immunogold staining suggests that the membrane-bound forms represent the majority of the molecules in wild-type muscle. In contrast to the wild-type muscle, staining of Col13a1N/N muscle located most of the immunogold particles in the extracellular space and only some gold particles were detected at the plasma membrane or inside the cell (Figure 5F) ▶ . Furthermore, in the mutant muscles there were areas where the basement membrane was detached and contained some gold particles (Figure 5F) ▶ . The immunoelectron microscopy confirms that the N-terminally mutant type XIII collagen molecules are transported outside the cell and that they are located in the matrix near the vicinity of the plasma membrane but not embedded to these structures.
Exercise-Induced Muscle Damage
The histopathological signs of a muscular disorder led us to study the possibility that the expression of N-terminally mutant type XIII collagen may confer susceptibility to exercise-induced damage. A single episode of excessive physical exercise is known to cause acute damage to the skeletal muscle and result in focal necrosis and regeneration lesions in the damaged muscle areas. 26,27 When male Col13a1N/N mice and age- and sex-matched wild-type mice were subjected to running on a motor-driven treadmill, the wild-type mice maintained the 6-hour exercise protocol well, but some of the Col13a1N/N mice began to show signs of exhaustion during the last 2 hours, so that the experiment had to be stopped somewhat prematurely. The mice were sacrificed 2 days after the running session and the severity of muscle cell damage was estimated from H&E and immunostained sections of the MQF and gastrocnemius muscles, β-glucuronidase activity in the MQF being used as a quantitative measure of injury, as previously described. 28 This latter activity (μmol s−1 kg−1 protein) was significantly higher in the Col13a1N/N runners than in the wild-type runners, with absolute values of 2.76 ± 0.91 versus 1.39 ± 0.32 (P < 0.001, mean ± SD) (Figure 6A) ▶ . The increase in β-glucuronidase activity between the exercised and unexercised Col13a1N/N mice was 311%, compared with 190% between the exercised and unexercised control mice.
Figure 6.

Increased muscle injury in 8- to 10-week-old mutant mice after acute exercise. A: β-glucuronidase activity in the MQF of unexercised (U) and exercised (E) wild-type and Col13a1N/N mice 48 hours after cessation of running exercise. The difference between the exercised mutant and wild-type mice was statistically significant, P < 0.001 (n = 14 and 12), as also was that between the exercised and unexercised groups of control and mutant mice (P < 0.001, n = 8 and 7), but the control and mutant mice in the unexercised group did not differ from each other (n = 7). B–E: Immunofluorescence staining of the MQF with anti-tenascin antibodies before (B and C) and after (D and E) exercise. Identical staining patterns are seen in the wild-type (B) and mutant (C) mice not subjected to exercise, but stronger staining is seen in the mutant individuals (E) than in the wild-type (D) after exercise. Scale bars, 100 μm.
Comparison of MQF and gastrocnemius sections from mutant and control runners indicated that the muscle damage varied between animals, but that the mutant runners in general had more numerous fibers undergoing degeneration and more intensive inflammation. Immunostaining for tenascin-C, used as a marker of inflammation 29,30 revealed stronger signals in the exercised mutant mice than in the exercised wild-type mice (Figure 6; B to E ▶ ). Thus both the β-glucuronidase assays and the histopathological findings indicated that the Col13a1N/N mice were more sensitive to muscle damage during the running protocol.
Discussion
Homologous targeting of the type XIII collagen gene in ES cells followed by Cre-induced deletion of exon 1 and the adjacent noncoding sequences resulted in synthesis of N-terminally altered type XIII collagen molecules. These molecules differ from normal ones in that the cytosolic domain and transmembrane domain are replaced with short unrelated sequences. Thus the loxP and intron 1-derived sequences served as a template for producing a new N-terminal end for the type XIII collagen molecule instead of the expected inactivation of the gene. The promoter and 5′-flanking sequences of Col13a1 were retained in the mutant allele, and thus apparently served to drive expression of the mutant α1(XIII) chains. The new N-terminal sequence is either 65 or 11 residues in length, depending on which of the two potential initiation methionines is used, and these sequences are devoid of any known functional motifs. Because the 11-residue nucleotide sequence surrounding the downstream ATG shows somewhat better resemblance to the consensus sequence for the initiation of translation, 31 this is a more likely candidate for the new N-terminus. In light of the nondistinctive nature of the new sequence and the correct cellular and tissue location of the altered type XIII collagen molecules, the results obtained with the mutant mouse line can probably be attributed to the lack of the 96 extreme N-terminal residues of type XIII collagen rather than to the gaining of new functions attributable to the new N-terminus. Contrary to our predictions, the mice that were homozygous for this allelic change were viable and fertile.
Type XIII collagen is a type II transmembrane protein that lacks a signal sequence and is inserted into the membrane of the endoplasmic reticulum through a single transmembrane domain, whereas the bulk of the molecule, consisting mainly of collagenous sequences, projects into the lumen of the endoplasmic reticulum and forms the ectodomain of molecules transported to the plasma membrane. 7,12 The N-terminally altered type XIII collagen was correctly deposited in focal adhesions in the cultured fibroblasts and occurred at the expected locations in tissues of Col13a1N/N mice, showing that the cytosolic and transmembrane domains of type XIII collagen are not essential for the development and viability of the mice, and surprisingly, not even for the localization of the mutant molecules. Thus the altered molecules seem to be secreted despite the lack of an apparent mechanism for translocation into the endoplasmic reticulum. In fact, earlier studies have shown that insect cell expression of human type XIII collagen chains lacking the 83 extreme N-terminal residues leads to at least some of the molecules being translocated to the endoplasmic reticulum even though they lack a typical signal sequence. 11 Like the mutant mouse chains, these human α1(XIII) chains lacked the cytosolic and transmembrane domains. Another example of a protein that is secreted despite the lack of a signal peptide is provided by basic fibroblast growth factor. 32
Because type XIII collagen molecules lacking the cytosolic and transmembrane domains are correctly transported to focal adhesions in the plasma membrane, we hypothesize that the altered molecules associate with some of the other focal adhesion components in the endoplasmic reticulum, the Golgi network or the subsequent secretion vehicles and are transported to the plasma membrane via such complexes. Immunoelectron microscopy of skeletal muscle revealed the N-terminally mutant molecules in the extracellular matrix adjacent to the sarcolemma whereas normal molecules were mainly found at the plasma membrane. Thus the analysis of the affected tissue also supports secretion of the mutant molecules. Furthermore, using light microscopy the mutant type XIII collagen location is comparable with authentic type XIII collagen, but the ultrastructural analysis highlights the loss of plasma membrane anchorage of the mutant molecules.
The fact that the lack of the 96 N-terminal residues reduced the adhesion properties of the cultured fibroblasts derived from the Col13a1N/N mice indicates that these residues are of functional importance. In particular, the mutant fibroblasts showed a marked decrease in adhesion to type IV collagen compared with wild-type cells. This may have been because of impaired direct interaction between collagens of types XIII and IV. Fibroblasts express α1β1 integrin, a subtype known to have the highest affinity for type IV collagen in the various collagen-binding integrins. 33,34 It is of interest that the ectodomain of recombinant type XIII collagen associates closely with the I domain of the α1 integrin subunit, 13 so that another possibility would be that the mutant type XIII collagen molecules that are not bound to the cell membrane are in effect soluble molecules capable of blocking α1β1 integrin-type IV collagen interaction.
Detailed analyses of the affected mice revealed a fuzzy plasma membrane-basement membrane interphase in the skeletal muscle, and disruption of this linkage under contraction could explain several of the pathological findings, including the basement membrane detachment seen in older animals, streaming of z-bands, and disorganization of myofilaments. Consistent with the observed ultrastructural abnormalities, the muscle fibers were smaller in diameter in the mutant than in the wild-type mice, as is often seen in other myopathies. 35 The skeletal muscle of the Col13a1N/N mice did not show any clear signs of necrosis or regeneration of the abnormal fibers. All in all, the changes were focal, because all cases involved many muscle fibers that were normal in appearance, but the condition was progressive with age. A combination of only some of the muscle fibers being affected, a relatively short life span, and a sedentary lifestyle could prevent the development of severe myopathy. However, exercise induced more prominent muscle damage in the mutant mice, as indicated by a failure to sustain the running protocol and greater changes in β-glucuronidase activity and histological features than in the exercised control mice. These differences were observed in mutant mice only at 8 to 10 weeks of age, and in view of the progressive nature of the muscular disorder, it is likely that older animals would show even more severe changes.
Laminin-2 and its receptor the dystrophin-dystroglycan complex play a major role in linking the muscle cell cytoskeleton and the extracellular matrix. 35 This is exemplified by the muscular dystrophies, which are characterized by progressive muscle wasting and weakness because of mutations in the laminin-2-dystrophin complex. 35-37 Several members of the integrin family of cell-matrix receptors are also expressed in muscle and mediate muscle cell attachment. 38 Absence of the α7 integrin subunit causes a progressive muscular dystrophy starting soon after birth, with a histopathological picture characterized by marked changes in the myotendinous junctions. 39 A partial lack of the integrin α5 subunit in chimeric mice resulted in muscle dystrophy characterized by fiber size variability, central nuclei, degeneration of fibers, and mitochondrial proliferation. 40 Bethlem myopathy is an autosomal-dominant muscle disease caused by mutations in the type VI collagen α1(VI) and α2(VI) chains. 41 A knock-out mouse model for the α1(VI) gene showed histological features of myopathy such as fiber necrosis and phagocytosis and pronounced variation in the fiber diameter. 42 Our findings indicate that type XIII collagen is also a molecule implicated in mediating the linkage between the muscle fiber and the extracellular matrix. In vitro experiments indicate that the ectodomain of recombinant type XIII collagen binds with the basement membrane components nidogen-2 and perlecan (H Tu, University of Oulu, Oulu, Finland, personal communication). Thus the observed microscopic changes speak for a need for type XIII collagen to bind with some basement membrane components, eg, nidogen-2 and perlecan, and that its anchorage to the plasma membrane is necessary for viable muscle cell-matrix linkage. The findings caused by mutations in the genes for type VI collagen are similar to those described for the type XIII collagen mutation in this study excluding fiber necrosis. 41,42 Whether these two myopathies have some common denominator remains to be studied.
In view of the Col13a1N/N mouse phenotype it is of interest to search for diseases located in the vicinity of this gene on chromosome 10. A subtype of Miyoshi-type distal muscular dystrophy has been located in a 23-cM region on chromosome 10 43 and facioscapulohumeral muscular dystrophy at 10qter. 44 Both disease loci are some distance away from the type XIII collagen gene locus, and thus the myopathy observed in Col13a1N/N mice is not represented by the muscle disorders so far linked to chromosome 10.
We demonstrate in this report that the expression of N-terminally mutant type XIII collagen causes a new form of progressive muscular disorder, and postulate that this collagen type participates in linkage between the muscle fiber and the basement membrane. We conclude that mutations in type XIII collagen may cause certain inherited muscular myopathies for which the cause has not yet been identified.
Acknowledgments
We thank Aila Jokinen, Sirkka Vilmi, Jaana Väisänen, Maija Seppänen, Anna-Liisa Oikarainen, and Sirpa Kellokumpu for their technical assistance.
Footnotes
Address reprint requests to Prof. Taina Pihlajaniemi, Department of Medical Biochemistry, University of Oulu, P.O. Box 5000, FIN-90014, Oulu, Finland. E-mail: taina.pihlajaniemi@oulu.fi.
Supported by grants from the Finnish Centre of Excellence Programme (2000–2005) of the Academy of Finland (no. 44843), FibroGen Inc. (South San Francisco, CA), the Emil Aaltonen Foundation, the Jenny and Antti Wihuri Foundation, the Research and Science Foundation of Farmos, and the Sigrid Juselius Foundation.
References
- 1.Myllyharju J, Kivirikko KI: Collagens and collagen-related diseases. Ann Med 2001, 33:7-21 [DOI] [PubMed] [Google Scholar]
- 2.Pihlajaniemi T, Rehn M: Two new collagen subgroups: membrane associated collagens and types XV and XVIII. Prog Nucleic Acid Res Mol Biol 1995, 50:225-262 [DOI] [PubMed] [Google Scholar]
- 3.Kvist AP, Latvanlehto A, Sund M, Horelli-Kuitunen N, Rehn M, Palotie A, Beier D, Pihlajaniemi T: Complete exon-intron organization and chromosomal location of the gene for mouse type XIII collagen (col13a1) and comparison with its human homologue. Matrix Biol 1999, 18:261-274 [DOI] [PubMed] [Google Scholar]
- 4.Shows TB, Tikka L, Byers MG, Eddy RL, Haley LL, Henry WM, Prockop DJ, Tryggvason K: Assignment of the human collagen α1(XIII) chain gene (COL13A1) to the q22 region of chromosome 10. Genomics 1989, 5:128-133 [DOI] [PubMed] [Google Scholar]
- 5.Tikka L, Elomaa O, Pihlajaniemi T, Tryggvason K: Human α1(XIII) collagen gene. Multiple forms of the gene transcripts are generated through complex alternative splicing of several short exons. J Biol Chem 1991, 266:17713-17719 [PubMed] [Google Scholar]
- 6.Pihlajaniemi T, Tamminen M: The α1 chain of type XIII collagen consists of three collagenous and four noncollagenous domains, and its primary transcript undergoes complex alternative splicing. J Biol Chem 1990, 265:16922-16928 [PubMed] [Google Scholar]
- 7.Hägg P, Rehn M, Huhtala P, Väisänen T, Tamminen M, Pihlajaniemi T: Type XIII collagen is identified as a plasma membrane protein. J Biol Chem 1998, 273:15590-15597 [DOI] [PubMed] [Google Scholar]
- 8.Juvonen M, Sandberg M, Pihlajaniemi T: Patterns of expression of the six alternatively spliced exons affecting the structures of the COL1 and NC2 domains of the α1(XIII) collagen chain in human tissues and cell lines. J Biol Chem 1992, 267:24700-24707 [PubMed] [Google Scholar]
- 9.Juvonen M, Pihlajaniemi T, Autio-Harmainen H: Location and alternative splicing of type XIII collagen RNA in the early human placenta. Lab Invest 1993, 69:541-551 [PubMed] [Google Scholar]
- 10.Peltonen S, Rehn M, Pihlajaniemi T: Alternative splicing of mouse α1(XIII) collagen RNAs results in at least 17 different transcripts, predicting α1(XIII) collagen chains with length varying between 651 and 710 amino acid residues. DNA Cell Biol 1997, 16:227-234 [DOI] [PubMed] [Google Scholar]
- 11.Snellman A, Keränen MR, Hägg PO, Lamberg A, Hiltunen JK, Kivirikko KI, Pihlajaniemi T: Type XIII collagen forms homotrimers with three triple helical collagenous domains and its association into disulfide-bonded trimers is enhanced by prolyl 4-hydroxylase. J Biol Chem 2000, 275:8936-8944 [DOI] [PubMed] [Google Scholar]
- 12.Snellman A, Tu H, Väisänen T, Kvist AP, Huhtala P, Pihlajaniemi T: A short sequence in the N-terminal region is required for the trimerization of type XIII collagen and is conserved in other collagenous transmembrane proteins. EMBO J 2000, 19:5051-5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nykvist P, Tu H, Ivaska J, Käpylä J, Pihlajaniemi T, Heino J: Distinct recognition of collagen subtypes by α1β1 and α2β1 integrins. α1β1 mediates cell adhesion to type XIII collagen. J Biol Chem 2000, 275:8255-8261 [DOI] [PubMed] [Google Scholar]
- 14.Sandberg M, Tamminen M, Hirvonen H, Vuorio E, Pihlajaniemi T: Expression of mRNAs coding for the α1 chain of type XIII collagen in human fetal tissues: comparison with expression of mRNAs for collagen types I, II, and III. J Cell Biol 1989, 109:1371-1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hägg P, Väisänen T, Tuomisto A, Rehn M, Tu H, Huhtala P, Eskelinen S, Pihlajaniemi T: Type XIII collagen: a novel cell adhesion component present in a range of cell-matrix adhesions and in the intercalated discs between cardiac muscle cells. Matrix Biol 2001, 19:727-742 [DOI] [PubMed] [Google Scholar]
- 16.Sund M, Väisänen T, Kaukinen S, Ilves M, Tu H, Autio-Harmainen H, Rauvala H, Pihlajaniemi T: Distinct expression of type XIII collagen in neuronal structures and other tissues during mouse development. Matrix Biol 2001, 20:215-231 [DOI] [PubMed] [Google Scholar]
- 17.Potocnik AJ, Brackebusch C, Fässler R: Fetal and adult hematopoietic stem cells require β1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 2000, 12:653-663 [DOI] [PubMed] [Google Scholar]
- 18.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC: Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA 1993, 90:8424-8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fässler R, Meyer M: Consequences of lack of β1 integrin gene expression in mice. Genes Dev 1995, 9:1896-1908 [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989. NY, Cold Spring Harbor Laboratory Press, Cold Spring Harbor
- 21.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 22.Majalahti-Palviainen T, Hirvinen M, Tervonen V, Ilves M, Ruskoaho H, Vuolteenaho O: Gene structure of a new cardiac peptide hormone: a model for heart-specific gene expression. Endocrinology 2000, 141:731-740 [DOI] [PubMed] [Google Scholar]
- 23.Slot JW, Geuze HJ: A new method of preparing gold probes for multiple-labeling cytochemistry. Eur J Cell Biol 1985, 38:87-93 [PubMed] [Google Scholar]
- 24.Barrett AJ: Lysosomal enzymes. Dingle JT eds. Lysosomes, A Laboratory Handbook. 1972, :pp 46-126 North-Holland, Amsterdam [Google Scholar]
- 25.Kozak M: At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J Mol Biol 1987, 196:947-950 [DOI] [PubMed] [Google Scholar]
- 26.Ebbeling CB, Clarkson PM: Exercise-induced muscle damage and adaptation. Sports Med 1989, 7:207-234 [DOI] [PubMed] [Google Scholar]
- 27.Komulainen J, Vihko V: The course of exercise-induced skeletal muscle fibre injury. Reznick AZ Packer L Sen CK Holloszy O Jackson MJ eds. Oxidative Stress in Skeletal Muscle. 1998, :- Birkhäuser Verlag Basel, Switzerland [Google Scholar]
- 28.Salminen A, Kihlström M: Lysosomal changes in mouse skeletal muscle during the repair of exercise injuries. Muscle Nerve 1985, 8:269-279 [DOI] [PubMed] [Google Scholar]
- 29.Irintchev A, Salvini TF, Faissner A, Wernig A: Differential expression of tenascin after denervation, damage or paralysis of mouse soleus muscle. J Neurocytol 1993, 22:955-965 [DOI] [PubMed] [Google Scholar]
- 30.Settles DL, Cihak RA, Erickson HP: Tenascin-C expression in dystrophin-related muscular dystrophy. Muscle Nerve 1996, 19:147-154 [DOI] [PubMed] [Google Scholar]
- 31.Kozak M: An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 1987, 15:8125-8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mignatti P, Morimoto T, Rifkin DB: Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol 1992, 151:81-93 [DOI] [PubMed] [Google Scholar]
- 33.Kern A, Eble J, Golbik R, Kuhn K: Interaction of type IV collagen with the isolated integrins α1β1 and α2β1. Eur J Biochem 1993, 215:151-159 [DOI] [PubMed] [Google Scholar]
- 34.Kern A, Marcantonio EE: Role of the I-domain in collagen binding specificity and activation of the integrins alpha1beta1 and alpha2beta1. J Cell Physiol 1998, 176:634-641 [DOI] [PubMed] [Google Scholar]
- 35.Campbell KP: Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80:675-679 [DOI] [PubMed] [Google Scholar]
- 36.Bulfield G, Siller WG, Wight PA, Moore KJ: X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA 1984, 81:1189-1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forrest SM, Cross GS, Speer A, Gardner-Medwin D, Burn J, Davies KE: Preferential deletion of exons in Duchenne and Becker muscular dystrophies. Nature 1987, 329:638-640 [DOI] [PubMed] [Google Scholar]
- 38.Hynes RO: Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69:11-25 [DOI] [PubMed] [Google Scholar]
- 39.Mayer U, Saher G, Fässler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Poschl E, von der Mark K: Absence of integrin α7 causes a novel form of muscular dystrophy. Nat Genet 1997, 17:318-323 [DOI] [PubMed] [Google Scholar]
- 40.Taverna D, Disatnik MH, Rayburn H, Bronson RT, Yang J, Rando TA, Hynes RO: Dystrophic muscle in mice chimeric for expression of α5 integrin. J Cell Biol 1998, 143:849-859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jöbsis GJ, Keizers H, Vreijling JP, de Visser M, Speer MC, Wolterman RA, Baas F, Bolhuis PA: Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat Genet 1996, 14:113-115 [DOI] [PubMed] [Google Scholar]
- 42.Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM: Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 1998, 7:2135-2140 [DOI] [PubMed] [Google Scholar]
- 43.Linssen WH, de Visser M, Notermans NC, Vreyling JP, Van Doorn PA, Wokke JH, Baas F, Bolhuis PA: Genetic heterogeneity in Miyoshi-type distal muscular dystrophy. Neuromuscul Disord 1998, 8:317-320 [DOI] [PubMed] [Google Scholar]
- 44.Cacurri S, Piazzo N, Deidda G, Vigneti E, Galluzzi G, Colantoni L, Merico B, Ricci E, Felicetti L: Sequence homology between 4qter and 10qter loci facilitates the instability of subtelomeric KpnI repeat units implicated in facioscapulohumeral muscular dystrophy. Am J Hum Genet 1998, 63:181-190 [DOI] [PMC free article] [PubMed] [Google Scholar]