

Abstract
Karyotype and fluorescence in situ hybridization analyses have demonstrated the frequent presence of an altered static state of the number of chromosomes (ie, aneuploidy) in lung cancer, but it has not been directly established whether aneuploidy is in fact associated with a persistent increase in the rate of chromosomal losses and gains (ie, chromosome instability, or CIN). The study presented here used a panel of 10 lung cancer cell lines to provide for the first time direct evidence that CIN is a common feature in lung cancer cell lines in association with the presence of significant aneuploidy. In addition, we found that the CIN phenotype correlates well with the presence of p53 mutations. However, human papilloma virus 16-E6-directed inactivation of p53 in a representative non-CIN lung cancer cell line did not result in the induction of CIN, at least up to the 25th generation, suggesting that inactivation of p53 itself is unlikely to directly induce CIN in lung cancer cells. Interestingly, however, significant CIN could be induced in conjunction with the generation of aneuploid populations when the mitotic spindle formation was transiently abrogated in p53-inactivated cells. These results suggest that inactivation of p53 may allow lung cancer cells to go through an inappropriate second division cycle under certain forms of mitotic stresses, which would result in the induction of the CIN phenotype in conjunction with the generation of aneuploidy.
Lung cancer currently claims more than 160,000 lives annually as the number one cause of cancer death in the United States, and it has also become the leading cause in Japan with more than 47,000 deaths annually. 1 Lung cancer cells have been shown to carry frequent structural chromosomal abnormalities such as deletions, which presumably reflect genetic lesions at tumor suppressor loci. 2,3 In addition, previous cytogenetic studies have shown that lung cancer cells frequently exhibit numerical changes in chromosomes. 4 It is conceivable that this form of chromosomal abnormalities may contribute to tumor development and progression by facilitating loss of heterozygosity and the phenotypic manifestation of inactivated tumor suppressor genes, as well as favoring polysomy of chromosomes that harbor growth-promoting genes. 5
Karyotype and fluorescence in situ hybridization (FISH) analyses have demonstrated the frequent occurrence of altered static state of the number of chromosomes (ie, aneuploidy) in lung cancer, but it has not been directly established whether aneuploidy is indeed associated with a persistent increase in the rate of chromosomal losses and gains (ie, chromosome instability, or CIN), or whether it is merely a fingerprint of a few chaotic, nonpersisting chromosomal missegregations. In addition, very few genetic and biochemical data are available at present about how this very common abnormality is acquired in lung cancers. In this connection, Lengauer and his colleagues 6 recently showed that human colon cancer cell lines frequently displayed a CIN phenotype. They also found a consistent association of CIN in colon cancers with dysfunction of the mitotic checkpoint as well as with infrequent mutations in two mitotic checkpoint genes, BUB1 and BUBR1. 7 As for lung cancer, we previously reported that the mitotic checkpoint is impaired in ∼40% of lung cancer cell lines and that the MAD1 mitotic checkpoint gene is infrequently altered in lung cancer, whereas other mitotic checkpoint genes, including MAD2, BUB1, BUBR1, and BUB3, did not show any evidence of mutations in lung cancer. 8-10
The study presented here demonstrates that the CIN phenotype is indeed persistently present in lung cancer cell lines and shows a good concordance with the presence of significant aneuploidy. In addition, our data show that the CIN phenotype correlates well with the presence of p53 mutations. Interestingly, however, inactivation of the p53 function by human papilloma virus (HPV)16-E6 infection did not readily induce the CIN phenotype in a chromosomally stable lung cancer cell line, but rather seemed to permit propagation of aneuploid cells accompanying the CIN phenotype under certain forms of mitotic stresses.
Materials and Methods
Cell Lines
Cells were cultured in RPMI 1640 medium supplemented with 5% fetal calf serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. The cell lines ACC-LC-319, ACC-LC-94, and ACC-LC-176 were established at our laboratory. NCI-H460 and HCT116 were obtained from the American Type Culture Collection (Manassas, VA). Other cell lines were generously provided by Drs. L. J. Old and M. Akiyama (Memorial Sloan-Kettering Cancer Center and Radiation Effect Research Foundation, respectively).
FISH for Detection of Chromosome Instability
Chromosome instability was examined essentially with the methods developed by Lengauer and colleagues. 6 In brief, cells were transfected with pcDNA3 using DMRIE-C (Life Technologies, Inc., Rockville, MD), followed by selection with G418. FISH analysis of methanol/acetic acid (3:1)-fixed nuclei of multiple single-cell-derived clones was performed by using centromeric probes specific for chromosomes 1, 11, 12, and 17 at the 15th and 25th generations after transfection. At least 100 nuclei were evaluated per clone with each chromosome probe. In addition, chromosomal variation in situ was evaluated by means of FISH on small colonies consisting of 100 to 200 cells, which had been grown directly on microscopic slides and fixed with 100% ethanol, essentially in the manner described by Lengauer and colleagues. 6
Centrosome Staining
Exponentially growing cells were plated at a density of 5 × 10 4 cells/well on 24-mm glass coverslips in 12-well plates. After a 48-hour incubation in RPMI 1640 containing 5% fetal calf serum, the cells were washed with phosphate-buffered saline (PBS), sequentially fixed in ice-cold methanol/acetone, preincubated with Tris-buffered saline (0.9% NaCl in 50 mmol/L Tris-HCl, pH 8.0) containing 1% bovine serum albumin and 1% skim milk for 1 hour, and then subjected to incubation with a polyclonal antibody to pericentrin (BAbCO, Richmond, CA) at a dilution of 1:150 for 1 hour. After extensive washing, the slides were incubated with a fluorescein isothiocyanate-conjugated secondary antibody, and examined with a fluorescence microscope after counterstaining with 4′, 6-diamidino-2-phenylindole. To determine centrosome abnormalities in each cell line, the diameter and number of pericentrin spots of at least 100 cells were carefully examined. Centrosomes were considered to be abnormal if they were more than three times larger in diameter than human normal fibroblasts and/or if there were three or more centrosomes per cell. Abnormalities in both numbers and size abnormalities were evaluated together, because it was occasionally difficult to clearly distinguish between one large centrosome and an aggregate of multiple centrosomes under conventional fluorescent microscopy. The criteria used were: ++, >10% of cells had abnormal centrosomes; +, 3 to 10% of cells showed abnormal staining; −, 0 to <3% of cells had abnormal centrosomes. The frequency of abnormal staining in human normal fibroblasts was <3%.
Northern Blot Analysis of STK15/BTAK/aurora2
Northern blot analysis was performed following the standard procedures. A 744-bp cDNA probe of the STK15/BTAK/aurora2 gene was generated by polymerase chain reaction (PCR) amplification with the aid of 5′-AAGCTCAGCTGGAGAAAGCC and 5′-CTCAAGGATTTCTCCCCCTG oligonucleotide primes. Induction of G1 phase arrest in ACC-LC-48 was performed, as described previously. 11
Analysis of Mitotic Delay in Response to Nocodazole
Cells were cultured in the presence of nocodazole, harvested at 12, 18, 24, 32, 40, and 52 hours, and then fixed with 4% formaldehyde in PBS, followed by staining with 4′, 6-diamidino-2-phenylindole. Two hundred nmol/L of nocodazole was used for all cell lines except for QG56, to which 800 nmol/L of nocodazole was added. To measure the mitotic index (percentage of viable cells arrested in mitosis), fluorescence microscopy was used to count at least 300 cells for each measurement as described previously, 8 which was repeated at least twice. Two control cell lines with known mitotic checkpoint defects (SW480 and T47D) 7 were also included. Flow cytometric analysis was also conducted to evaluate the cell-cycle profile of cells harvested at various time points before and after 32 hours incubation with nocodazole. Cells were stained with propidium iodide and analyzed with the aid of FACScan and Cell Fit-DNA software (Becton Dickinson, Bedford, MA). 8
Detection of p53 Mutations and Functional Inactivation of p53 by HPV16-E6 Infection
p53 mutations in each cell line were examined by sequencing as described previously. 12 Functional inactivation of p53 in ACC-LC-176 was accomplished by infecting a replication-defective retrovirus vector (LXSN) carrying the HPV16-E6 gene, followed by selection with G418, as described previously. 13 Infected cells were irradiated with 12-Gy X-ray, lysed in Laemmli’s sample buffer, and examined for induction of the p53 protein by Western blot analysis using an anti-p53 monoclonal antibody (Ab6) (Oncogene Research Products, Cambridge, MA).
Results
Detection of CIN in Lung Cancer Cell Lines
Ten lung cancer cell lines were examined for the presence of persistent changes in the number of chromosomes by FISH analysis of interphase cells with the aid of four centromeric probes. Clones of each of the 10 cell lines were generated and chromosome losses or gains in lung cancer cells within a defined period of time were examined by FISH at the 15th and 25th generations.
As a result, we could divide the 10 cell lines into three groups, similar results being obtained at the 15th and 25th generations for each of the cell lines (Table 1 ▶ , data not shown for the 25th generation). The first group consisted of five cell lines, QG90, QG56, PC-10, Calu1, and SK-MES-1, which showed marked variation in the number of chromosomes within each of the multiple single-cell-derived clones. For example, the QG90 clone a showed that only 64% of cells had a modal number of chromosome 1 (ie, presumably the original number of chromosome 1 in this clone), whereas 24 and 12% of cells showed losses and gains, respectively. FISH analysis using probes specific for chromosomes 11, 12, and 17 similarly revealed dramatic variations in chromosome numbers. Evaluation of the three other clones of QG90 confirmed the reproducibility of these observations. In our study we used an arbitrary instability score, which was based on the following calculation: (number of observations, in which >10% of the cells had other than a modal number of chromosomes)/(chromosomes × clones examined in each cell line). According to this score, all of the five cell lines belonging to this group had an instability score of 1.0 and the rate of CIN could be estimated to be in excess of 10−2 per generation for each chromosome analyzed. These cell lines without exception were found to be markedly aneuploid. In contrast, a near-diploid lung cancer cell line ACC-LC-176 as well as a colon cancer cell line HCT116, which was included as a non-CIN control, were found to have acquired chromosomal losses or gains in <10% of cells in all clones and chromosomes examined, for an instability score of 0. The remaining group, ACC-LC-94, NCI-H460, ACC-LC-319, and PC-1, showed an intermediate phenotype with considerable variation in the instability score, ranging from 0.33 to 0.88.
Table 1.
Variations in Numbers of Chromosomes at 15th Generation in Clones Generated from a Single Cell of Human Lung Cancer Cell Lines
| Cell line | Number of chromosomes | Clone | % with losses or gains | Instability score* | |||
|---|---|---|---|---|---|---|---|
| Ch. 1 | Ch. 11 | Ch. 12 | Ch. 17 | ||||
| QG90 | 84 | a | 36% (4)† | 33% (4) | 29% (4) | 33% (4) | 1.0 (16/16) |
| b | 27% (4) | 20% (4) | 26% (4) | 32% (4) | |||
| c | 41% (4) | 33% (4) | 31% (4) | 32% (4) | |||
| d | 40% (4) | 25% (4) | 35% (4) | 46% (4) | |||
| PC-10 | 102 | a | 32% (5) | 17% (3) | 30% (5) | 37% (4) | 1.0 (16/16) |
| b | 39% (4) | 61% (3) | 43% (5) | 47% (4) | |||
| c | 61% (4) | 40% (3) | 63% (4) | 54% (4) | |||
| d | 55% (5) | 41% (3) | 48% (5) | 57% (3) | |||
| QG56 | 69 | a | 22% (3) | 21% (3) | 26% (3) | 31% (3) | 1.0 (12/12) |
| b | 28% (3) | 23% (3) | 26% (3) | 27% (4) | |||
| c | 18% (3) | 13% (3) | 19% (2) | 23% (4) | |||
| Calu1 | 87 | a | 53% (4) | 18% (3) | 63% (7) | 63% (5) | 1.0 (8/8) |
| b | 66% (4) | 53% (3) | 81% (6) | 73% (5) | |||
| SK-MES-1 | 86 | a | 16% (4) | 16% (3) | 50% (3) | 44% (2) | 1.0 (8/8) |
| b | 19% (4) | 16% (4) | 46% (3) | 40% (2) | |||
| ACC-LC-94 | 50 | a | 13% (2) | 26% (3) | 30% (4) | 12% (2) | 0.88 (7/8) |
| b | 8% (2) | 18% (3) | 58% (4) | 27% (3) | |||
| NCI-H460 | 57 | a | 10% (2) | 1% (2) | 12% (2) | 15% (2) | 0.67 (8/12) |
| b | 33% (4) | 3% (2) | 12% (2) | 19% (2) | |||
| c | 20% (4) | 4% (2) | 16% (2) | 17% (2) | |||
| ACC-LC-319 | 70 | a | 34% (4) | 9% (3) | 17% (4) | 9% (2) | 0.63 (5/8) |
| b | 35% (4) | 11% (2) | 17% (4) | 8% (2) | |||
| PC-1 | 48 | a | 9% (2) | 5% (2) | 4% (2) | 4% (2) | 0.33 (4/12) |
| b | 11% (2) | 13% (2) | 7% (2) | 8% (2) | |||
| c | 14% (2) | 8% (2) | 16% (2) | 9% (2) | |||
| ACC-LC-176 | 44 | a | 6% (2) | 8% (2) | 6% (2) | 4% (2) | 0 (0/16) |
| b | 8% (2) | 8% (2) | 6% (2) | 9% (2) | |||
| c | 6% (2) | 10% (2) | 3% (2) | 10% (2) | |||
| d | 5% (2) | 4% (2) | 3% (2) | 5% (2) | |||
| HCT116 | 45 | a | 7% (2) | 6% (2) | 4% (2) | 7% (2) | 0 (0/8) |
| b | 9% (2) | 9% (2) | 3% (2) | 7% (2) | |||
*(Number of observations of either loss or gain of chromosomes in more than 10% of cells)/(chromosomes × clones examined in each cell line).
†Number in parentheses shows number of modal chromosomes in each clone.
Chromosomal variations in situ were also examined by analyzing small colonies consisting of only 100 to 200 cells, as described by Lengauer and colleagues. 6 Cell lines were plated at low density on microscope slides so that independent clones could be examined after seven to eight generations by FISH without disaggregating them. Examination of five independent colonies of PC-10 with the unstable phenotype showed that 52% (range, 47 to 57%) and 54% (range, 49 to 60%) of the cells had acquired either losses or gains in comparison with the modal numbers of chromosomes 11 and 12, respectively (data not shown for individual colonies). These results were thus consistent with the presence of high rates of change in chromosome numbers. In contrast, ACC-LC-176 with the stable phenotype exhibited little chromosomal variation within each of the three independent colonies, with only 6% (range, 5 to 7%) of the cells having other configurations than two chromosomes 11.
Presence of Mitotic Checkpoint Defects in a Fraction of Cell Lines with CIN
The mitotic checkpoint is thought to be essential for ensuring accurate chromosome segregation by implementing mitotic delay in response to a spindle defect. 14,15 The panel of lung cancer cell lines used in our study was therefore examined for the presence of mitotic checkpoint impairment in relation to the presence of CIN (Table 2) ▶ . Two cell lines each of the unstable and intermediate phenotype groups showed a significant reduction (mean, 21%; range, 17 to 25%) of cells arrested at mitosis. This reduction was comparable to the mitotic indices of the control cell lines (SW480 and T47D) with known mitotic checkpoint defects (25 and 21%, respectively), 7 when treated with a microtubule poison, nocodazole. In contrast, the stable lung cancer cell line ACC-LC-176 and the non-CIN control HCT116 exhibited normal responses (peak mitotic indices, 48 and 57%, respectively).
Table 2.
Abnormalities in Mitotic Delay, Centrosome, STK15/BTAK/aurora2, and p53 Status in Relation to the Presence of CIN
| CIN phenotype* | Cell line | Mitotic delay† | Centrosome abnormality‡ | STK15/BTAK/aurora2§ | p53 Gene status |
|---|---|---|---|---|---|
| Unstable | QG90 | Normal | + | ++ | Pro to Ser (codon 249) |
| PC-10 | Abnormal | ++ | ++ | Gly to Cys (codon 245) | |
| QG56 | Normal | − | ++ | Pro to Ser (codon 249) | |
| Calu1 | Abnormal | ++ | ++ | Homozygous deletion | |
| SK-MES-1 | Normal | ++ | ++ | Glu to Stop (codon 298) | |
| Intermediate | ACC-LC-94 | Normal | + | ++ | Asp to Gly (codon 281) |
| NCI-H460 | Normal | − | ++ | Wild-type | |
| ACC-LC-319 | Abnormal | ++ | ++ | 1-bp deletion (codon 71) | |
| PC-1 | Abnormal | − | ++ | Wild-type | |
| Stable | ACC-LC-176 | Normal | − | ++ | Wild-type |
| Control | HCT116 | Normal | − | N.D. | Wild-type |
*Unstable, instability score 1.0; intermediate, instability scores between 0 and 1.0; stable, 0.
†Data for QG90, PC-10, QG56, NCI-H460, ACC-LC-319, PC-1, and ACC-LC-176 from our previous study (Takahashi et al 8 ) were used.
‡Centrosomes were considered to be abnormal if they were more than three times larger in diameter than human normal fibroblasts and/or if there were three or more centrosomes per cell. ++, >10% of cells had abnormal centrosomes; +, 3 to 10% of cells showed abnormal staining, −, 0 to <3% of cells had abnormal centrosomes. The frequency of abnormal staining in human normal fibroblasts was less than 3% both in number and size in this assay.
§STK15/BTAK/aurora2 expression was classified as follows. ++, significantly higher expression than that in normal lung; +, expression comparable to that in normal lung; −, no expression.
Association of CIN with the Presence of Centrosome Abnormalities
It has been suggested that abnormality in centrosomes, which play a vital role in organizing mitotic spindle during cell division, may be involved in the acquisition of CIN. 16,17 The panel of lung cancer cell lines was used to study centrosome abnormalities in relation to CIN by using an antibody against pericentrin, a component of the centrosome matrix protein involved in microtubule nucleation (Table 2 ▶ and Figure 1 ▶ ). 18 Four of the five unstable cell lines (80%) exhibited abnormal centrosome staining (mean, 13%; range, 4 to 21%), whereas three cell lines showed abnormality in >10% of the cells with a frequency of up to 21%. Two of the four cell lines with an intermediate phenotype also showed abnormal centrosome staining. Both ACC-LC-176 and HCT116 without CIN exhibited abnormal centrosome staining in <3% of the cells.
Figure 1.

Representative abnormal centrosome staining in SK-MES-1 with significant CIN. Centrosomes were considered abnormal if there were three or more signals and/or the diameter was more than three times larger than that of human normal fibroblasts. Representative SK-MES-1 cells exhibiting normal centrosome staining (arrow in A, enlarged figure in B) and abnormalities in size (open arrowhead in A, enlarged figure in C) or numbers (solid arrowhead in A, enlarged figure in D). Representative centrosome staining of normal fibroblasts is shown at the same magnification in the inset of B.
Ubiquitous High Expression Levels of STK15/BTAK/aurora2
Northern blot analysis was performed to examine the expression of STK15/BTAK/aurora2 centrosome kinase. All lung cancer cell lines examined showed significantly higher expression levels than normal lung tissues, regardless of the presence or absence of CIN (Figure 2) ▶ . Interestingly, a high STK15/BTAK/aurora2 expression level was also detectable in normal lung epithelial cell lines, whereas G1-arrested lung cancer cell lines expressed it at a very low level, comparable to that in normal lung tissues.
Figure 2.

Ubiquitous high expression levels of STK15/BTAK/aurora2 in lung cancer cell lines. Note that the G1-arrested ACC-LC-48 lung cancer cell line shows a low STK15/BTAK/aurora2 expression level comparable to that in normal lung tissues.
Association of CIN with the Presence of p53 Mutations
It has been reported that embryonic fibroblasts of p53 knockout mouse exhibit mitotic checkpoint impairment and centrosome abnormality, leading to an increase in CIN. 19,20 All of the five cell lines with marked CIN as well as two of the four cell lines with an intermediate phenotype were found to carry p53 mutations. Notably, all cell lines with centrosome abnormalities carried p53 alterations including both missense and null mutations. On the other hand, all but one cell line with mutant p53 exhibited centrosome abnormalities. Three of the four cell lines with impaired response to nocodazole carried altered p53.
Indirect Role of p53 Inactivation in the Induction of Aneuploid Cells with CIN
We also investigated whether functional inactivation of p53 can directly induce CIN in association with mitotic checkpoint impairment and/or centrosome abnormality. A non-CIN lung cancer cell line ACC-LC-176 was infected with a retrovirus carrying HPV16-E6. Abrogation of p53 induction in response to irradiation or nocodazole treatment was confirmed by Western blot analysis of HPV16-E6-infected clones (Figure 3A ▶ , data not shown for nocodazole). FISH analysis of five independent HPV16-E6-infected clones at both the 15th and 25th generation revealed that p53 inactivation by itself did not appreciably induce CIN, mitotic checkpoint impairment or centrosome abnormality (Figure 3B ▶ and Table 3 ▶ ; data not shown for the 15th generation).
Figure 3.
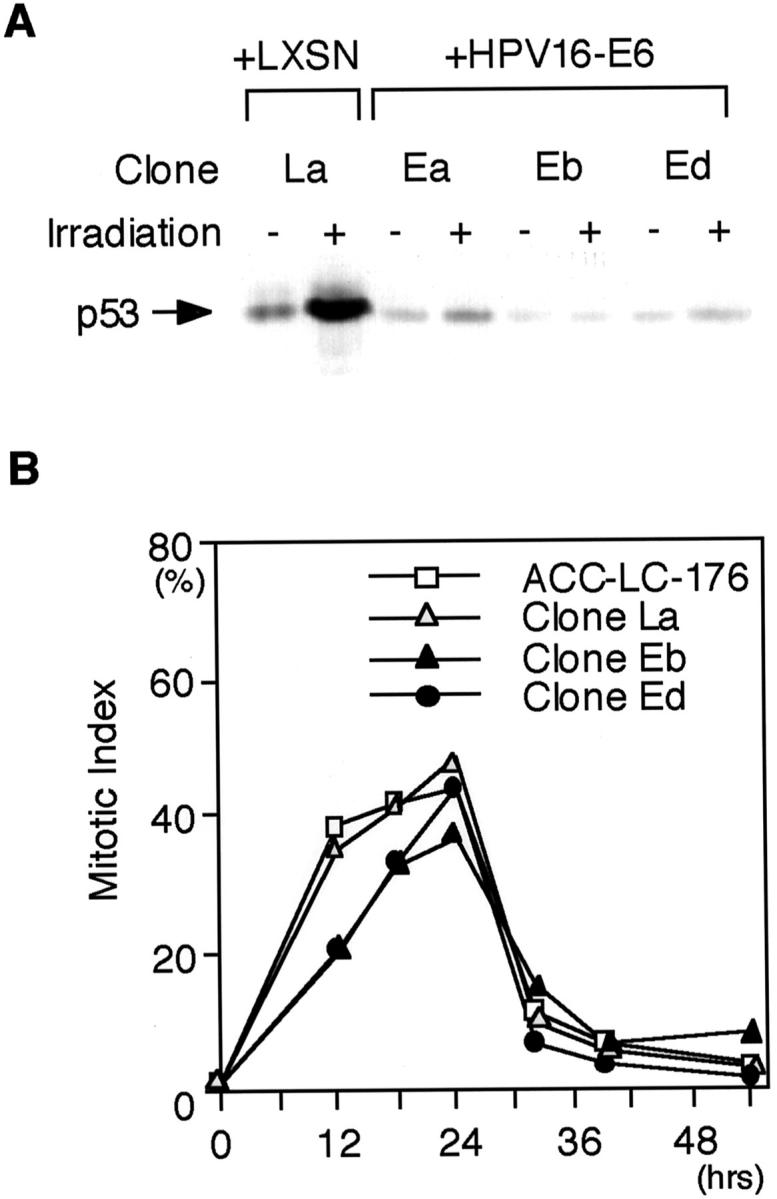
Functional p53 inactivation by HPV-E6 infection in ACC-LC-176. A: Western blot analysis of p53 in HPV16-E6-infected clones. Induction of p53 in response to irradiation, which is clearly seen in the control clone (La), is abrogated in HPV16-E6-infected clones (Ea, Eb, and Ed). B: Mitotic indices of HPV16-E6-infected clones of ACC-LC-176 in the presence of nocodazole. Transient accumulation of cells at the metaphase and subsequent adaptation is seen within 32 hours regardless of the p53 status.
Table 3.
Lack of Direct Induction of CIN by HPV16-E6-Directed Inactivation of p53 in ACC-LC-176 Cells
| Infection | Clone | % with losses or gains | Instability score* | Mitotic delay | Centrosome abnormality† | |
|---|---|---|---|---|---|---|
| Ch. 11 | Ch. 12 | |||||
| LXSN | La | 6% (2)‡ | 5% (2) | 0 (0/10) | Normal | — |
| Lb | 5% (2) | 1% (2) | Normal | — | ||
| Lc | 8% (2) | 8% (2) | N.D. | — | ||
| Ld | 5% (2) | 9% (2) | N.D. | — | ||
| Le | 6% (2) | 8% (2) | N.D. | — | ||
| HPV16-E6 | Ea | 6% (2) | 3% (2) | 0.1 (1/10) | N.D. | — |
| Eb | 5% (2) | 1% (2) | Normal | — | ||
| Ec | 2% (2) | 5% (2) | N.D. | — | ||
| Ed | 12% (2) | 7% (2) | Normal | — | ||
| Ee | 4% (2) | 7% (2) | N.D. | — | ||
*Fraction of events (i.e., losses or gains compared with modal number of chromosomes) observed in the two infections.
†Determined as described in the footnote of Table 2 ▶ .
‡Numbers in parentheses show modal numbers of each chromosome.
Because chemotherapeutic agents affecting the mitotic spindle, such as vinorelbine, have been introduced to clinical practice, we here investigated whether transient abrogation of mitotic spindle formation, a condition mimicking chemotherapy, may lead to the generation of aneuploid cells and whether such cells exhibit CIN. Two HPV16-E6-infected clones, Eb and Ed, as well as a control clone, La, were incubated with nocodazole for 32 hours, followed by thorough washing with fresh medium to remove the agent. A small fraction of the treated cells, which had survived the treatment, could subsequently be propagated (Figure 3B) ▶ . Fluorescence-activated cell sorting analysis at various time points up to 12 days revealed that, after the transient nocodazole treatment, cells with a content of >2N DNA could survive in p53-inactivated clones (Eb and Ed), whereas predominance of cells with a content of 2N DNA gradually became apparent in the surviving control clone (La) (Figure 4) ▶ . Consistent with this finding, FISH analysis using a probe specific for chromosome 11 showed that, in contrast to the predominance of diploid colonies in clone La, the vast majority of the colonies of clones Eb and Ed were aneuploid. Interestingly, these colonies were found to exhibit marked CIN during the analysis of chromosomal variations in situ, resulting in either losses or gains of chromosome 11 in 33% (range, 24 to 44%) of the cells in clone Eb and in 36% (range, 27 to 44%) of those in clone Ed.
Figure 4.

FACS and FISH analyses after transient nocodazole treatment of HPV16-E6-infected ACC-LC-176 clones. Preferential propagation of aneuploid populations was observed in p53-inactivated clones (Eb and Ed) but not in a clone with normal p53 (La).
Discussion
In the study presented here, we have provided for the first time direct evidence that CIN is a common feature in lung cancer cell lines in consistent association with the presence of significant aneuploidy and p53 mutations. However, our results also indicate that it is unlikely that inactivation of p53 directly induces CIN in lung cancer cells, because HPV16-E6-directed inactivation of p53 in a representative non-CIN lung cancer cell line did not result in the induction of CIN at least up to the 25th generation. Importantly, however, significant CIN could be induced in conjunction with the generation of aneuploid populations, when mitotic spindle was transiently eliminated in p53-inactivated cells. Although it has been shown that p53 prohibits subsequent endoreduplication in the prolonged presence of nocodazole, 21-23 our study suggests that p53 may play a role in the imposition of sustained arrest in a G1-like state associated with 4N DNA content in response to transient disruption of mitotic spindles. Thus, inactivation of p53 seems to allow lung cancer cells to go through an inappropriate second division cycle under certain types of mitotic stresses, resulting in the induction of the CIN phenotype by means of a presently unclear mechanism. In addition to artificial disruption of mitotic spindles by nocodazole, mitotic spindle-affecting chemotherapeutic agents as well as alterations in the mitotic apparatus and checkpoint might also have similar consequences, leading to the generation of aneuploid populations with CIN in lung cancers carrying mutant p53. In fact, transient treatment with 5 nmol/L vinorelbine, which is an active agent for the treatment of lung cancer, similarly induced an aneuploid population in the p53-inactivated clones (Eb and Ed) but not in clone La with the wild-type p53 function (data not shown). It is possible that induction of aneuploid populations may in turn increase the chances for surviving lung cancer cells to acquire selective growth advantage after chemotherapy, as it has been suggested that CIN may contribute to tumor heterogeneity and progression. 5
It is conceivable that once CIN is acquired, lung cancers are increasingly likely to generate karyotypes altered to most suitable for genetic lesions, which are distinct in each case. Selective growth advantage may be acquired by the loss of the wild-type allele of tumor suppressor genes as a result of loss of heterozygosity and an increase in the gene dosage of growth-promoting genes by polysomy as well as by changes in the expression profiles of thousands of genes on the affected chromosomes. In this connection, the presence of allele-specific imbalance in various chromosomes in lung cancer, which has been reported by us and others, 24,25 might also reflect such a CIN-directed selection of chromosomes containing genes with allele-specific expression.
We note that our results are markedly different from those for colon cancer, because it has been reported that cell fusions of two non-CIN colon cancer cell lines, 6 hence artificially creating aneuploid cells, did not result in the induction of CIN. It is possible that fundamental differences in the ways aneuploid cells were created (ie, transient nocodazole treatment versus cell fusion) or distinct genetic backgrounds of the cell lines used might account for this difference. CIN in lung cancer appears to be less closely related to the presence of mitotic checkpoint impairment than that in colon cancer, because all colon cancer cell lines with a CIN phenotype reportedly had mitotic checkpoint impairment. 7 This finding also points to the possibility that cancer type-specific differences may exist in the major underlying causes of CIN of these two common cancers of adults. Our results, however, do not preclude a role for mitotic checkpoint impairment in the acquisition of CIN in lung cancer. In fact, we previously reported that the MAD1 mitotic checkpoint gene is indeed mutated in lung cancer in vivo, although with low frequency. 9
Although there is increasing evidence of a direct link between centrosome dysfunction and chromosome missegregation during tumorigenesis, 26,27 inactivation of p53 has been reported to induce abnormal centrosome amplification, resulting in unequal segregation of chromosomes. 19 Although the present study suggests a potential connection between the presence of centrosome abnormalities and p53 mutations in the acquisition of CIN, we did not observe significant abnormalities in the numbers or size of centrosomes in p53-inactivated clones and identical p53 mutation was present in two cell lines, one with and the other without centrosome abnormalities. Exogenous overexpression of a human serine/threonine kinase STK15/BTAK/aurora2 was previously claimed to cause abnormal centrosome amplification, leading to aneuploidy and transformation, 28 but the ubiquitous high expression levels observed in our study, regardless of the presence or absence of CIN, suggests that the functional significance of a high STK15/BTAK/aurora2 expression level needs further clarification in terms of the induction of CIN in lung cancer cells.
As to possible other mechanisms that might make lung cancer cells susceptible to the acquisition of CIN, there is a large number of potential targets, alterations of which can theoretically give rise to CIN. These include genes involved in chromosome condensation, cohesion, kinetochore structure and function, centrosome/microtubule formation, and various checkpoints such as cytokinesis checkpoint. 5 In this connection, our preliminary examination of 39 lung cancer cell lines, including all 10 cell lines examined here, did not find any alterations in the human EB1 gene, a yeast homologue of which has been implicated in cytokinesis checkpoint, 29 whereas 44 primary lung cancer specimens did not carry any mutations in the human securin gene (unpublished observation), which is thought to play a role in sister chromatid separation. 30
Evidence to date indicates that further studies are needed to identify and clarify the underlying mechanisms directly responsible for the induction of CIN in lung cancer. In this regard, human homologues, which, when altered, can result in CIN phenotypes in yeast, would be excellent candidates for such genetic lesions. Clarification of the molecular bases and biological significance of CIN should provide important clues for the development of new therapeutic approaches for this fatal cancer.
Acknowledgments
We would like to thank Y. Inoue and W. Kaufmann for their helpful suggestions and stimulating discussions.
Footnotes
Address reprint requests to Dr. Takashi Takahashi, Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan. E-mail: tak@aichi-cc.pref.aichi.jp.
Supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture, Japan; and by a Grant-in-Aid for the Second Term Comprehensive Ten-Year Strategy for Cancer Control from the Ministry of Health and Welfare, Japan.
N. H. and T. H. contributed equally to this study.
References
- 1.Statistics and Information Department: Vital Statistics of Japan, vol. 3. Ministry of Health and Welfare, Tokyo, 1998, pp 384–411
- 2.Minna JD, Sekido Y, Fong KM, Gazdar AF: Cancer of the lung. De Vita VT Hellman S Rosenberg SA eds. Cancer Principles and Practice of Oncology. 1997, :pp 849-857 Lippincott-Raven, Philadelphia [Google Scholar]
- 3.Gazdar AF: Cell biology and molecular biology of small cell and non-small cell lung cancer. Curr Opin Oncol 1990, 2:321-327 [DOI] [PubMed] [Google Scholar]
- 4.Testa JR: Chromosome alterations in human lung cancer. Pass HI Mitchell JB Johnson DH Turrisi AT eds. Lung Cancer: Principles and Practice. 1996, :pp 55-71 Lippincott-Raven, Philadelphia [Google Scholar]
- 5.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 6.Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancers. Nature 1997, 386:623-627 [DOI] [PubMed] [Google Scholar]
- 7.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392:300-303 [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, Haruki N, Nomoto S, Masuda A, Saji S, Osada H, Takahashi T: Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 1999, 18:4295-4300 [DOI] [PubMed] [Google Scholar]
- 9.Nomoto S, Haruki N, Takahashi T, Masuda A, Koshikawa T, Fujii Y, Osada H, Takahashi T: Search for in vivo somatic mutations in the mitotic checkpoint gene, hMAD1, in human lung cancers. Oncogene 1999, 18:7180-7183 [DOI] [PubMed] [Google Scholar]
- 10.Haruki N, Saito H, Harano T, Nomoto S, Takahashi T, Osada H, Fujii Y, Takahashi T: Molecular analysis of the mitotic checkpoint genes BUB1, BUBR1 and BUB3 in human lung cancers. Cancer Lett 2001, 162:201-205 [DOI] [PubMed] [Google Scholar]
- 11.Masuda A, Osada H, Yatabe Y, Kozaki K, Tatematsu Y, Takahashi T, Hida T, Takahashi T, Takahashi T: Protective function of p27KIP1 against apoptosis in small cell lung cancer cells in unfavorable microenvironments. Am J Pathol 2001, 158:87-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi T, Takahashi T, Suzuki H, Hida T, Sekido Y, Ariyoshi Y, Ueda R: The p53 gene is very frequently mutated in small-cell lung cancer with a distinct nucleotide substitution pattern. Oncogene 1991, 6:1775-1778 [PubMed] [Google Scholar]
- 13.Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ: Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396:84-88 [DOI] [PubMed] [Google Scholar]
- 14.Rudner AD, Murray AW: The spindle assembly checkpoint. Curr Opin Cell Biol 1996, 8:773-780 [DOI] [PubMed] [Google Scholar]
- 15.Elledge SJ: Mitotic arrest: Mad2 prevents sleepy from waking up the APC. Science 1998, 279:999-1000 [DOI] [PubMed] [Google Scholar]
- 16.Doxsey S: The centrosome—a tiny organelle with big potential. Nat Genet 1998, 20:104-106 [DOI] [PubMed] [Google Scholar]
- 17.Pihan GA, Doxsey SJ: The mitotic machinery as a source of genetic instability in cancer. Semin Cancer Biol 1999, 9:289-302 [DOI] [PubMed] [Google Scholar]
- 18.Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M: Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 1994, 76:639-650 [DOI] [PubMed] [Google Scholar]
- 19.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF: Abnormal centrosome amplification in the absence of p53. Science 1996, 271:1744-1747 [DOI] [PubMed] [Google Scholar]
- 20.Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ: A p53-dependent mouse spindle checkpoint. Science 1995, 267:1353-1356 [DOI] [PubMed] [Google Scholar]
- 21.Lanni JS, Jacks T: Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol 1998, 18:1055-1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notterman D, Young S, Wainger B, Levine AJ: Prevention of mammalian DNA reduplication, following the release from the mitotic spindle checkpoint, requires p53 protein, but not p53-mediated transcriptional activity. Oncogene 1998, 17:2743-2751 [DOI] [PubMed] [Google Scholar]
- 23.Khan SH, Wahl GM: p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res 1998, 58:396-401 [PubMed] [Google Scholar]
- 24.Kondo M, Matsuoka S, Uchida K, Osada H, Nagatake M, Takagi K, Harper JW, Takahashi T, Elledge SJ, Takahashi T: Selective maternal-allele loss in human lung cancers of the maternally expressed p57KIP2 gene at 11p15.5. Oncogene 1996, 12:1365-1368 [PubMed] [Google Scholar]
- 25.Hung J, Kishimoto Y, Sugio K, Virmani A, McIntire DD, Minna JD, Gazdar AF: Allele-specific chromosome 3p deletions occur at an early stage in the pathogenesis of lung carcinoma. JAMA 1995, 273:558-563 [PubMed] [Google Scholar]
- 26.Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, Doxsey SJ: Centrosome defects and genetic instability in malignant tumors. Cancer Res 1998, 58:3974-3985 [PubMed] [Google Scholar]
- 27.Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL: Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci USA 1998, 95:2950-2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S: Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998, 20:189-193 [DOI] [PubMed] [Google Scholar]
- 29.Muhua L, Adames NR, Murphy MD, Shields CR, Cooper JA: A cytokinesis checkpoint requiring the yeast homologue of an APC-binding protein. Nature 1998, 393:487-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zou H, McGarry TJ, Bernal T, Kirschner MW: Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 1999, 285:418-422 [DOI] [PubMed] [Google Scholar]