

Abstract
Chronic chagasic cardiomyopathy (CChC) is characterized by an inflammatory reaction which may eventually lead to heart enlargement, arrythmia, and death. As described herein, interleukin-4-deficient mice mount increased specific T helper (Th) 1 immune responses when infected with Trypanosoma cruzi, as compared to wild-type mice. Interestingly, these mice had reduced parasitism and mortality and exacerbated inflammation in their hearts, demonstrating a clear dissociation between inflammation and parasite load. The modulation of these phenomena so as to maximize host and parasite survivals may depend on a fine balance between Th responses, in which a Th1 response will, on one hand, control parasitism and, on the other hand, enhance heart inflammation throughout the course of the infection.
Perhaps the most intriguing feature of Chagas’ disease is the fact that only about 25% of Trypanosoma cruzi-infected individuals develop chronic chagasic cardiomyopathy (CChC), a debilitating disease which is the leading cause of heart failure in several Latin American countries. 1 The factors determining the progression from asymptomatic individuals to patients with chagasic cardiopathy are unknown. Moreover, no currently available treatment prevents disease development. CChC occurs in a time point when tissue parasitism is very scarce or virtually absent. 2,3 This observation has prompted many authors to consider the participation of autoallergic phenomena in the pathogenesis of the disease, 4-9 in opposition to the alternative hypothesis that CChC is totally due to T. cruzi-specific immune responses. 10,11
The DTH-like aspect of the inflammatory lesions in CChC suggests the participation of T helper (Th) 1 (Th1) cells in its pathogenesis, as this lymphocyte subset strongly promotes the activation of macrophages by the secretion of cytokines such as interferon-γ (IFN-γ). The expansion and function of Th1 cells are regulated by cytokines produced by Th2 cells, such as interleukin (IL) -4, -10 and -13, potent promoters of humoral immune responses. 12 In several disease models, it has been shown that cross-regulations between these two T-cell subsets are critical for the determination of disease outcome. In fact, certain pathogens can induce the preferential expansion of one Th subset, while suppressing the other. 13 T. cruzi infection, however, induces a non-polarized response: in different murine experimental models, both resistant and susceptible mice had a mixed pattern of response to the parasite. 14,15 On the other hand, an association between progression to severe CChC and high IFN-γ levels has been demonstrated in human beings. 16,17 If Th1 responses indeed mediate disease in T. cruzi-infected individuals, cytokines such as IL-4 could play a beneficial, anti-pathogenic role during T. cruzi infection. Thus, we investigated a possible modulating role of IL-4 in a CChC model in which IL-4-deficient mice are infected with T. cruzi, as described below.
Materials and Methods
Mice
Specific-pathogen-free, 6- to 8-week-old female or male wild-type, IL-4−/− and nu/nu BALB/c mice were raised and maintained at the animal facilities at the Gonçalo Moniz Research Center-FIOCRUZ, and provided with rodent diet and water ad libitum.
Parasites, Infection and T. cruzi Antigen Preparation
Trypomastigotes of Colombian-strain 18 T. cruzi were obtained by infection of LCC-MK2 cell line. Infection was performed by inoculation of 100 trypomastigotes by intraperitoneal route. Parasitemia was evaluated at different time points after infection by counting the number of trypomastigotes in peripheral blood aliquots contained between a glass slide and a coverslip, with a determined blood volume per microscopic field. 19 Epimastigotes of Colombian and PF strain 20 were obtained by axenic culture in liver infusion tryptase medium. T. cruzi antigen was prepared by subjecting epimastigotes to five cycles of freezing and thawing. T. cruzi lysates were centrifuged at 30,000 × for 30 minutes; supernatants were then aliquoted and stored at −70°C until use.
Histopathological Evaluation
Groups of IL-4+/+ or IL-4−/− mice were sacrificed at different time points after infection. Heart, liver, spleen, and striated muscle were removed and fixed in buffered 10% formalin. Sections were analyzed by optical microscopy after paraffin embedding followed by standard hematoxylin/eosin staining. Inflammatory cells infiltrating heart tissue were counted using digital morphometric evaluation. Images were digitalized using a JVC TK-1280 color video camera adapted to an Axioskop 2 microscope (Carl Zeiss, Göttingen, Germany). The images were analyzed using the AxioVision II program (Carl Zeiss), with which the inflammatory cells were manually counted and integrated by area. The number of parasite nests was also determined by counting in the acquired images. One 100 fields per section were counted in five to ten sections per heart.
Immunohistochemistry
Sections of 5 μm from frozen heart fragments were fixed with cold acetone and incubated with one of the following antibodies: rat anti-mouse CD4 or CD8, (PharMingen, San Diego, CA); rat anti-mouse Fc receptor (2.4G2 cell culture supernatant); peroxidase-conjugated goat anti-mouse IgM (Sigma, St. Louis, MO); T. cruzi-hyperimmunized rat antiserum. The sections pre-incubated with rat antibodies were incubated with horseradish-peroxidase conjugated to anti-rat IgG (Sigma). Reaction was developed using the peroxidase substrate diaminobenzidine (Vector, Burlingame, CA), followed by counterstaining with hematoxylin. The percentage of CD4+ or CD8+ cells was determined by counting the number of positive cells over the total number of inflammatory cells determined as described above.
Reconstitution and Infection of BALB/c nu/nu Mice
BALB/c nu/nu mice received adoptive transfer of thymocytes obtained from newborn wild-type mice or IL-4−/− mice (one thymus per mouse). After 15 days, reconstituted and non-reconstituted control nude mice were infected with 100 Colombian strain trypomastigotes. Mice were sacrificed 30 days later for histopathological evaluation of the hearts, as described above.
In Vitro Stimulation and Measurement of Proliferative Response
Spleen cell suspensions were prepared in RPMI medium (Life Technologies, GIBCO-BRL, Gaithersburg, MD) supplemented with 10% fetal calf serum (Hyclone, Logan, Utah), l-glutamine (2 mmol/L), vitamins, sodium pyruvate (1 mmol/L), Hepes (10 mmol/L), 5 × 10−5 mol of 2-mercaptoethanol, and gentamicin (50 μg/ml) (Sigma). For cytokine determination, spleen cells were cultured in 24-well plates and stimulated with 1 μg/ml of concanavalin A (Con A) (Sigma) or Colombian strain T. cruzi antigen (50 μg/ml). Cell-free supernatants were collected after 72 hours and stored at −20°C for cytokine analysis. To evaluate the proliferative response, splenocytes were plated in 96-well plates at 4 × 105/well in 200 μl and triplicate wells were stimulated with Con A or T. cruzi antigen for 120 hours, as described in the figure legends. After pulsing with 1 μCi of methyl[-3H]thymidine (Amersham, Little Chalfont, England) for 14–18 hours, proliferation was assessed by measurement of [methyl-3H]thymidine uptake in a β-plate counter (Packard, Meriden, CT).
Cytokine and Anti-T. cruzi Antibody Quantification
Supernatants of splenocyte cultures were tested for IFN-γ, IL-2, and IL-4 contents by ELISA, using antibody pairs from PharMingen and following the manufacturer’s instructions. Reaction was developed using the 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate (Kinkergaard & Perry Laboratories, Gaithersburg, MD) and read at 450 nm.
Anti-T. cruzi isotype production was evaluated by ELISA using PF-strain epimastigote lysate as antigen and biotinylated isotype-specific anti-mouse IgM, IgG1, IgG2a, IgG2b, and IgG3 antibodies (PharMingen), followed by streptovidin-peroxidase conjugate (Sigma). Reaction was developed using TMB substrate as described above.
Statistical Analyses
Data were analyzed using Student’s t-test, Wilcoxon’s rank sum test, or Fisher’s exact probability test, as indicated in the text. Differences were considered significant when P < 0.05.
Results
IL-4−/− Mice Are More Resistant to Infection by Colombian-Strain T. cruzi
Parasitemia levels in T. cruzi-infected wild-type BALB/c mice were about five times higher than in IL-4 knockout mice (Figure 1A ▶ ; P < 0.02, < 0.01 and < 0.002 on the 15th, 21st, and 25th or 30th days postinfection, respectively; Wilcoxon’s rank sum test; statistical analysis was not meaningful after the 30th day postinfection due to bias arising from the death of many wild-type animals). Moreover, whereas 55% of the animals in the wild-type mouse group died before or on the 40th day of infection, only 15% of infected IL-4−/− mice did not reach the 60th day after infection (Figure 1B ▶ ; P = 0.019 on the 60th day postinfection; Fisher’s exact probability test). Tissue parasitism in spleen, liver and striated muscle was also lower in IL-4−/− mice than in IL-4+/+ mice. Heart tissues from IL-4−/− mice had three- to fourfold less parasite nests than those of wild-type mice (P < 0.05, Wilcoxon’s rank sum test; Figure 1C ▶ ). After the fourth month of infection, no intact parasite, and only a single macrophage containing parasite antigenic material, was detected in those heart tissues by immunohistochemistry.
Figure 1.

Infection and heart inflammation in IL-4−/− and wild-type BALB/c mice. Groups of 20 IL-4−/− (triangles) or wild-type (circles) mice were infected with T. cruzi. A: Parasitemia. B: Percentage of surviving mice. C: Number of parasite nests in the heart. D: Number of inflammatory cells in the heart. Each symbol in A represents the median of the 9 to 20 surviving mice and in C and D represents the mean value of three to five surviving mice. Vertical bars represent the standard deviations of the means.
Myocarditis Is Exacerbated in T. cruzi-Infected IL-4−/− Mice
Inflammatory infiltrates in tissues of all infected mice were composed mainly of mononuclear cells. In the acute phase of the disease (up to 40 days after infection), foci of inflammatory infiltration of similar intensity were found in the liver and in striated muscles of animals from both groups, causing intense myocytolysis, in the presence of parasites. Large spleens, with intense cell proliferation and myeloid metaplasic reaction with megakariocytes were found in mice from both groups. Infected wild-type mouse spleens, however, were significantly larger than those of IL-4−/− mice (mean weights ± SD were 625 ± 63 and 373 ± 60 mg, respectively; P < 0.0001, Student’s t-test).
Interestingly, the intensity of myocarditis was higher, with 3- to 10-fold more mononuclear cells, in IL-4-deficient than in wild-type mice, despite the lower heart parasitism (P < 0.05, Wilcoxon’s rank sum test; Figures 1D and 2, A and B ▶ ▶ ). Inflammation was multifocal and mononuclear cells were frequently found attached to myocardial fibers.
Figure 2.
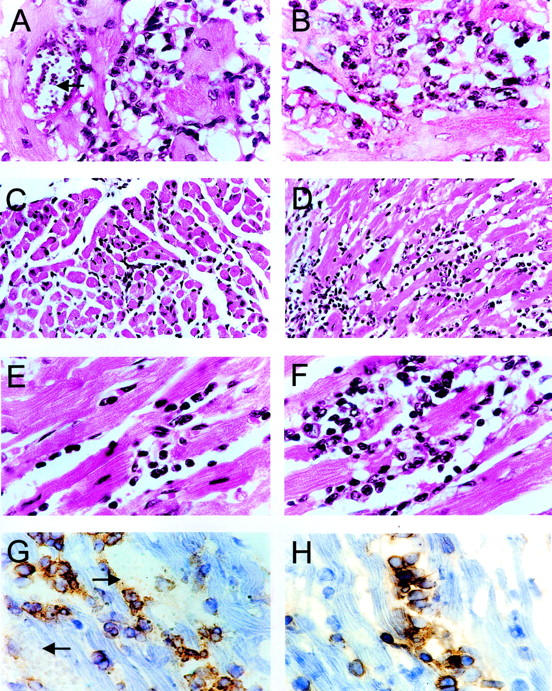
Pathology of hearts from T. cruzi-infected BALB/c mice. A, C and E: Heart sections of wild-type mice. B, D and F: Heart sections of IL-4−/− mice, 1 (A and B), 4 (C and D) and 7 (E and F) months after infection, stained by H&E. G and H: Immunoperoxidase staining (DAB) in heart sections of IL-4−/− mice using an anti-CD8 antibody 1 month after T. cruzi infection (G) or an anti-CD4 antibody 7 months after infection (H). Magnifications: C and D, ×200; A, B, E-H, × 400). Arrows indicate parasite nests.
After the acute phase of infection (namely around three to four months after infection), hearts of wild-type mice had a healed appearance, with scarce inflammatory foci (Figures 1D and 2C) ▶ ▶ . In contrast, a conspicuous multifocal inflammatory reaction, with or without associated fibrosis, was observed in hearts from IL-4−/− mice (Figure 2D) ▶ .
Myocarditis increased in severity from the fourth month to the seventh month of infection in mice from both groups. Wild-type mice, however, still had a mild disease, with little fibrosis (Figure 2E) ▶ , whereas IL-4−/− mice had severe multifocal myocarditis, with mononuclear cells frequently adhering to cardiac fibers undergoing myocytolysis (Figure 2F) ▶ . In the latter mice fibrosis was very prominent, particularly in the atria, both around intact heart fibers and in areas of active inflammation.
Increased Proportion of CD4+ T Cells in Hearts of IL-4−/− Mice
The mononuclear infiltrate in hearts of mice from both groups was mainly composed by macrophages (Fc receptor+ and surface IgM− cells), B cells (surface IgM+ cells; these constituted no more than 5% of the infiltrate), and T lymphocytes (CD8+ or CD4+ cells; these constituted no more than 25% of the infiltrate), with higher numbers of CD8+ cells in relation to CD4+ cells 30 days after infection (Figure 3) ▶ . At four and seven months after infection, however, CD4+ cells predominated, inverting the CD8+/CD4+ ratio. This was significantly more intense in IL-4−/− mice than in wild-type mice at seven months after infection (Figure 3 ▶ , P < 0.05, Wilcoxon’s rank sum test). The majority of CD8+ cells in the acute phase of infection surrounded parasite nests (Figure 2G) ▶ , whereas CD4+ cells were frequently found in intimate association with damaged fibers (Figure 2H) ▶ .
Figure 3.

CD4+ and CD8+ cells in heart of wild-type or IL-4−/− mice. Numbers of CD4+ (solid bars) and CD8+ (open bars) cells were evaluated in sections of hearts obtained from surviving mice at the indicated times after T. cruzi infection by immunohistochemistry. Each bar represents the mean of results from six sections per heart obtained from three to six mice per group.
Increased Th1 Response in T. cruzi-Infected IL-4−/− Mice
At 30 days after infection, IL-4−/− mouse splenocytes produced two to three times more IFN-γ in response to T. cruzi antigen in vitro than those of wild-type mice (Figure 4A) ▶ . Moreover, non-specific stimulation by concanavalin A (Con A) led splenocytes from IL-4−/− mice to produce IL-2 (not shown) and proliferate at normal levels, whereas splenocytes from wild-type mice had markedly reduced responses (Figure 4C ▶ ; P < 0.0357, Student’s t-test).
Figure 4.

Immune response in T. cruzi-infected wild-type (open bars) and IL-4−/− (closed bars) mice. A–C: Levels of IFN-γ (A) and IL-2 (B) in culture supernatants, and DNA synthesis (C) in T. cruzi antigen- or Con A-stimulated spleen cell cultures. Spleen cell suspensions were prepared from organs collected at the indicated times after infection. D: Levels of T. cruzi-specific IgM, IgG1, IgG2a, IgG2b, and IgG3 antibodies in sera from individual mice, prepared from blood samples collected seven months after T. cruzi infection. Each bar represents the mean of data obtained from three to six mice. Vertical bars represent standard deviations of means.
After the acute phase of infection (4 to 7 months after infection), spleen cells from infected and uninfected wild-type mice and from IL-4−/− mice had identical responses to Con A and T. cruzi antigen, in terms of proliferation (Figure 4C ▶ and data not shown). However, splenocytes of IL-4−/− mice still produced higher levels of IFN-γ on in vitro T. cruzi-antigen stimulation than wild-type mouse splenocytes (Figure 4A ▶ ; P < 0.0309, Student’s t-test). This was associated with increased IgG3 (P < 0.0207, Student’s t-test) and reduced IgG1 (P < 0.0021, Student’s t-test) anti-T. cruzi antibody levels (Figure 4D) ▶ . The production of IL-2 by splenocytes obtained from both groups of mice on stimulation with T. cruzi antigen was similar in all time points analyzed (Figure 4B) ▶ .
T. cruzi Infection Causes Intense Myocarditis in nu/nu BALB/c Mice Reconstituted with IL-4−/− Thymocytes
T. cruzi-infected BALB/c nu/nu mice had intense parasitemia and succumbed to the acute infection. Their hearts, 30 days after infection, had a remarkable high parasite load without any inflammation (Figure 5A) ▶ . In contrast, hearts from infected nu/nu mice, previously reconstituted with thymocytes from both wild-type and IL-4−/− mice, had inflammation (Figure 5B) ▶ . The inflammation, which was diffuse and composed by mononuclear cells, similar to that observed in infected wild-type or IL-4−/− mice, was more intense in nu/nu mice reconstituted with IL-4−/− than with wild-type mouse thymocytes (two-fold more inflammatory cells). In addition, the number of parasite nests was lower in IL-4-reconstituted nu/nu mice (Figure 5) ▶ .
Figure 5.

Parasitism and inflammation intensities in hearts of T. cruzi-infected nude mice. Mice were reconstituted with wild-type (Wild type) or IL-4−/− (IL-4−/−) BALB/c splenocytes or were not reconstituted (None). Each bar represents the mean number of parasites nests per cm 2 (A) or the mean number of inflammatory cells per mm 2 (B) in heart sections of 2–4 mice. Vertical bars represent the standard deviations of the means.
Discussion
The results described herein, obtained when BALB/c mice were infected with 100 myotropic, Colombian-strain T. cruzi, revealed an enhancing effect of IL-4 on the infection: IL-4−/− mice had reduced tissue parasitism and mortality when compared to control mice. Infection of mice with low numbers of parasites, mimicking the natural transmission, 21 reproduces well the three phases of T. cruzi infection in human beings, constituting an adequate model of the human disease. 4,22 These results apparently conflict with a recent report that IL-4−/− and control BALB/c mice, when infected with high numbers (5000 parasites) of reticulotropic, Y strain T. cruzi trypomastigotes, did not differ in terms of parasitemia and mortality. 23 In that experiment, in which the intensity of myocarditis was not evaluated, IL-4−/− mouse splenocytes also produced increased levels of IFN-γ in response to T. cruzi antigens. The infective inoculum used, however, by being higher than that which occurs in nature, 21 may not reproduce closely what happens in natural infection. Moreover, T. cruzi strains differ markedly in their biological behavior, 24 a fact that has to be taken into account when interpreting conflicting observations in experimentally or naturally infected mammals.
As described in the literature, 25 wild-type mice were immunosuppressed in the acute phase of T. cruzi infection, as demonstrated by their reduced lymphoproliferative response to Con A in vitro. T. cruzi-infected IL-4−/− mice, with reduced parasitism, on the other hand, had more intense in vitro lymphoproliferative response to Con A (Figure 4C) ▶ and less marked splenomegaly than wild-type mice. Whether these two latter phenomena could be ascribed to the reduction in parasitism or could result from a putative effect of IL-4 on the non-specific immunosuppression or on the polyclonal B-cell activation of T. cruzi-infected mammals is open to speculation.
The reduced acute-phase parasitemia and mortality of T. cruzi-infected IL-4−/− mice, in relation to infected normal mice, can be ascribed to an enhanced parasite-specific Th1 immune response. This enhanced response was demonstrated in this paper by the detection of increased amounts of IFN-γ in supernatants of antigen-stimulated splenocytes and increased levels of IFN-γ -dependent IgG3 anti-T. cruzi antibodies in vivo. As expected, while splenocytes from wild-type mice produced IL-4 on in vitro stimulation with Con A and T. cruzi antigen in all time points analyzed, no IL-4 was produced in vitro by IL-4−/− splenocytes (not shown) and IL-4−/− mice had reduced levels of IL-4-dependent IgG1 anti-T. cruzi antibodies. In fact, IFN-γ has been shown to control parasitism in vivo and in vitro. 26-31
On the other hand, the development of heart inflammatory foci in T. cruzi infection was intensified in IL-4−/− mice. This, to our knowledge, has not been previously reported. That it was not due to a putative abnormality in the target organ, and indeed depended on T-cell activity was demonstrated by the passive transfer of the severe myocarditis-susceptibility trait to athymic nude mice by IL-4−/− thymocytes. In addition, the development of inflammation in thymocyte-reconstituted athymic mice, as also reported for the first time herein, is probably the most conclusive demonstration that the heart disease in T. cruzi-infected mammals is immune mediated.
The inverse relationship between tissue parasitism and inflammatory response in hearts of IL-4−/− mice was striking. In fact, only a single macrophage containing T. cruzi antigen could be observed in several sections of heart tissue obtained from four mice four months after infection. These findings, however, cannot be used as a conclusive evidence that CChC is mediated by non-parasite-specific immune responses, 32 since very little parasite antigen, undetectable by immunohistochemical analysis, could theoretically maintain T cells specifically activated and recruiting large numbers of effector macrophages. Evidence for autoimmune phenomena in the pathogenesis of CChC comes from elsewhere. 4-9
At the acute phase of the infection, when parasitism was brought under control, heart-infiltrating lymphocytes were predominantly CD8+. These lymphocytes, rather than CD4+ cells, were frequently found in the vicinity of parasites or parasite antigens. These findings indicate that CD8+ cells may be directly involved in parasite control, either by releasing IFN-γ and/or by lysing infected cells before the full differentiation of amastigotes into trypomastigotes, and are consistent with data showing severe, lethal infection in β2-microglobulin or TAP-1 knockout mice. 33 Later on, during the intermediary and chronic phases of the infection, the percentage of CD4+ cells increased and supplanted the percentage of CD8+ cells, mainly in IL-4−/− mice. Contrasting to what was found for CD8+ cells in the acute phase, these cells were frequently found in close association with myocytes undergoing degenerative changes. These findings are consistent with the hypothesis that CD4+ T cells mediate CChC and with the fact that anti-CD4-antibody treatment, and not anti-CD8, cures experimental CChC in mice. 5,22
As for the reduction of parasitism, the enhanced heart alterations in IL-4−/− mice could be easily ascribed to an intensified Th1 response, since Th1 responses have been shown to be more aggressive than Th2 responses to host tissues in several situations. 13 In fact, an association of severity of myocarditis and increased IFN-γ levels has been described in human beings. 16,17
IL-4, by regulating an IFN-γ-producing response, may play an important role in preventing the development of incapacitating heart disease in T. cruzi-infected animals. In fact, the intense carditis and extensive fibrosis found in hearts of IL-4−/− mice during the chronic phase of T. cruzi infection closely resembles the severe CChC found in human patients. 2 The modulating activity of IL-4-producing T cells may maintain a balance between parasitism and tissue integrity in the indeterminate phase of the infection, during which mild inflammatory foci resolve into focal fibrosis and perhaps a contained Th1 response would keep parasitism under relative control. 3 The progressive destructive process in CChC could therefore result from a failure of a pathogenic Th1 response to be down-regulated by IL-4. This failure could, in its turn, depend on host genetic characteristics, on age-dependent changes of the immune system, 34 superposition of infections by unrelated microorganisms, and/or by T. cruzi re-infection. 35
Acknowledgments
We thank Dr. José O. Mengel, who kindly provided the breeding pairs of BALB/c IL-4−/− mice.
Footnotes
Address reprint requests to Ricardo Ribeiro dos Santos, M.D., Ph.D. Centro de Pesquisas Gonçalo Moniz. Rua Waldemar Falcão, 121 - Brotas - Salvador, BA, Brazil 40295-001. E-mail: rrsantos@e-net.com.br.
Supported by grants from the Brazilian National Research Council (CNPq) and FIOCRUZ.
References
- 1.Ribeiro-dos-Santos R, Rassi A, Köberle F: Chagas’ Disease. Schonfeld H Karger S eds. Antiparasitic Chemotherapy, Ciba Foundation Symposium. 1981, :pp 115-134 Elsevier-Experta Medica, Paris-London-New-York [Google Scholar]
- 2.Köberle F: Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol 1968, 6:63-116 [DOI] [PubMed] [Google Scholar]
- 3.Andrade ZA: Mechanisms of myocardial damage in Trypanosoma cruzi infection. Ciba Found Symp 1983, 99:214-233 [DOI] [PubMed] [Google Scholar]
- 4.Ribeiro-dos-Santos R, Pirmez C, Savino W: Role of autoreactive immunological mechanisms in chagasic carditis. Res Immunol 1991, 142:134-137 [DOI] [PubMed] [Google Scholar]
- 5.Ribeiro-dos-Santos R, Rossi MA, Laus L, Silva JS, Savino W, Mengel JO: Anti-CD4 abrogates rejection and reestablishes long-term tolerance to syngeneic newborn hearts grafted in mice chronically infected with Trypanosoma cruzi. J Exp Med 1992, 175:29-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribeiro dos Santos R, Mengel JO, Postol E, Soares RAO, Ferreira-Fernandez E, Soares MBP, Pontes de Carvalho LC: A heart-specific CD4+ T-cell line obtained from a chronic chagasic mouse induces carditis in heart-immunized mice and rejection of normal heart transplants in the absence of Trypanosoma cruzi. Parasite Immunol 2001, 23:93-101 [DOI] [PubMed] [Google Scholar]
- 7.Cunha-Neto E, Duranti M, Gruber A, Zingales B, De Messias I, Stolf N, Bellotti G, Patarroyo ME, Pilleggi F, Kalil J: Autoimmunity in Chagas’ disease cardiomyopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen. Proc Natl Acad Sci USA 1995, 92:3541-3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J: Autoimmunity in Chagas’ disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas’ cardiomyopathy patient. J Clin Invest 1996, 98:1709-1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizzo LV, Cunha-Neto E, Teixera ARL: Autoimmunity in Chagas’ disease: specific inhibition of reactivity of CD4+ T-cells against myosin in mice chronically infected with T. cruzi. Infect Immun 1989, 57:2640-2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarleton RL, Zhang L, Downs MO: “Autoimmune rejection” of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc Natl Acad Sci USA 1997, 94:3932-3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Tarleton RL: Parasite persistence correlates with disease severity and localization in chronic Chagas’ disease. J Infect Dis 1999, 180:480-486 [DOI] [PubMed] [Google Scholar]
- 12.Seder RA, Paul WE: Lymphocyte responses and cytokines. Cell 1994, 76:241-251 [DOI] [PubMed] [Google Scholar]
- 13.Abbas AK, Murphy KM, Sher A: Functional diversity of helper T lymphocytes. Nature 1996, 383:787-793 [DOI] [PubMed] [Google Scholar]
- 14.Hoft DF, Lynch RG, Kirchhoff LV: Kinetic analysis of antigen-specific immune responses in resistant and susceptible mice during infection with Trypanosoma cruzi. J Immunol 1993, 151:7038-7047 [PubMed] [Google Scholar]
- 15.Zhang L, Tarleton RL: Characterization of cytokine production in murine Trypanosoma cruzi infection by in situ immunocytochemistry: lack of association between susceptibility and type 2 cytokine production. Eur J Immunol 1996, 26:102-109 [DOI] [PubMed] [Google Scholar]
- 16.Bahia-Oliveira LM, Gomes JA, Rocha MO, Moreira MC, Lemos EM, Luz ZM, Pereira ME, Coffman RL, Dias JC, Cancado JR, Gazzinelli G, Correa-Oliveira R: IFN-gamma in human Chagas’ disease: protection or pathology? Braz J Med Biol Res 1998, 31:127-131 [DOI] [PubMed] [Google Scholar]
- 17.Correa-Oliveira R, Gomes J, Lemos EM, Cardoso GM, Reis DD, Adad S, Crema E, Martins-Filho OA, Costa MO, Gazzinelli G, Bahia-Oliveira LM: The role of the immune response on the development of severe clinical forms of human Chagas disease. Mem Inst Oswaldo Cruz 1999, 94:253-255 [DOI] [PubMed] [Google Scholar]
- 18.Federici EE, Abelmann WB, Neva FA: Chronic and progressive myocarditis in C3H mice infected with Trypanosoma cruzi. Am J Trop Med Hyg 1964, 13:272-280 [DOI] [PubMed] [Google Scholar]
- 19.Brener Z: Biology of Trypanosoma cruzi. Annu Rev Microbiol 1973, 27:347-382 [DOI] [PubMed] [Google Scholar]
- 20.Lemos MV, Menezes H: The effect of an immune RNA (RNAi) against Trypanosoma cruzi infection in mice. Tropenmed Parasitol 1978, 29:119-126 [PubMed] [Google Scholar]
- 21.Brener Z, Andrade ZA, Barral-Netto M (editors): Trypanosoma cruzi e Doença de Chagas, ed 2. Guanabara Koogan, Rio de Janeiro, RJ, 2000,
- 22.Pirmez C, Ribeiro-dos-Santos R: Autoreactivity in chronic experimental Trypanosoma cruzi infection. Ciência e Cultura 1994, 46:418-423 [Google Scholar]
- 23.Abrahamsohn IA, da Silva AP, Coffman RL: Effects of interleukin-4 deprivation and treatment on resistance to Trypanosoma cruzi. Infect Immun 2000, 68:1975-1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrade SG: Influence of Trypanosoma cruzi strain on the pathogenesis of chronic myocardiopathy in mice. Mem Inst Oswaldo Cruz 1990, 85:17-27 [DOI] [PubMed] [Google Scholar]
- 25.Hayes MM, Kierszenbaum F: Experimental Chagas’ disease: kinetics of lymphocyte responses and immunological control of the transition from acute to chronic Trypanosoma cruzi infection. Infect Immun 1981, 31:1117-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed SG: In vivo administration of recombinant IFN-gamma induces macrophage activation and prevents acute disease, immune suppression, and death in experimental Trypanosoma cruzi infections. J Immunol 1988, 140:4342-4347 [PubMed] [Google Scholar]
- 27.Torrico F, Heremans H, Rivera MT, Van Marck E, Billiau A, Carlier Y: Endogenous IFN-γ is required for resistance to acute Trypanosoma cruzi infection in mice. J Immunol 1991, 146:3626-3632 [PubMed] [Google Scholar]
- 28.McCabe RE, Meagher SG, Mullins BT: Endogenous interferon-gamma, macrophage activation, and murine host defense against acute infection with Trypanosoma cruzi. J Infect Dis 1991, 163:912-915 [DOI] [PubMed] [Google Scholar]
- 29.Golden JM, Tarleton RL: Trypanosoma cruzi: cytokine effects on macrophage trypanocidal activity. Exp Parasitol 1991, 72:391-402 [DOI] [PubMed] [Google Scholar]
- 30.Gazzinelli RT, Oswald IP, Hieny S, James SL, Sher A: The microbicidal activity of IFN-γ treated macrophages against Trypanosoma cruzi involves an L-arginine-dependent, nitrogen oxide-mediated mechanism inhibitable by IL-10 and TGF-β. Eur J Immunol 1992, 22:2501-2506 [DOI] [PubMed] [Google Scholar]
- 31.Holscher C, Mohrs M, Dai WJ, Kohler G, Ryffel B, Schaub GA, Mossmann H, Brombacher F: Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect Immun 1998, 66:1208-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palomino SAP, Aiello VD, Higuchi ML: Systematic mapping of hearts from chronic chagasic patients: the association between the occurrence of histopathological lesions and Trypanosoma cruzi antigens. Ann Trop Med Parasitol 2000, 94:571-579 [DOI] [PubMed] [Google Scholar]
- 33.Harty JT, Tvinnereim AR, White DW: CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 2000, 18:275-308 [DOI] [PubMed] [Google Scholar]
- 34.Cardillo F, Falcão RP, Rossi MA, Mengel J: An age-related gamma delta T cell suppressor activity correlates with the outcome of autoimmunity in experimental Trypanosoma cruzi infection. Eur J Immunol 1993, 23:2597-2605 [DOI] [PubMed] [Google Scholar]
- 35.Dias JCP: Epidemiology of Chagas’ disease. Wendel S Brener Z Camargo ME Rassi A eds. Chagas’ Disease (American Trypanosomiasis): Its Impact on Transfusion and Clinical Medicine. 1992, : ISTB, São Paulo [Google Scholar]