

Abstract
We explored the role of CD40-CD40L (CD154) in the severe malaria elicited by Plasmodium berghei anka infection in mice. Mortality was >90% by day 8 after infection in +/+ mice, but markedly decreased in CD40−/− or in CD40L−/− mice, as well as in +/+ mice treated with anti-CD40L monoclonal antibody. Parasitemia was similar in the different conditions. Breakdown of the blood-brain barrier was evident in infected +/+, but not in CD40−/− mice. Thrombocytopenia was less severe in CD40−/− mice than in the +/+ controls. Sequestration of macrophages in brain venules and alveolar capillaries was reduced in CD40−/− or in CD40L−/− mice, whereas sequestration of parasitized red blood cells or polymorphonuclear leukocytes in alveolar capillaries was CD40-CD40L-independent. CD40 mRNA was increased in the brain and lung of infected mice whereas CD40L was increased in the lung. Tumor necrosis factor plasma levels were similarly increased in infected +/+ or CD40−/− mice. Expression of CD54 and its mRNA levels in the brain were moderately decreased in CD40-deficient mice. Thus the mortality associated with severe malaria requires CD40-CD40L interaction that contributes to the breakdown of the blood-brain barrier, macrophage sequestration, and platelet consumption.
Infection of mice by Plasmodium berghei anka (PbA) results, in susceptible strains, to a lethal syndrome, commonly named cerebral malaria, in which mice die 7 to 9 days after infection in a state of coma associated with neurological manifestations, 1,2 Prominent in this syndrome are a breakdown of the blood-brain barrier, microhemorrhages, and sequestration of macrophages and platelets in the cortical venules. 3-6 In addition, there is a sequestration of macrophages, polymorphonuclear leukocytes (PMNs), parasitized red blood cells (pRBCs), and platelets in other organs, notably the lung. 5-7 Hence, because this syndrome is not limited to the brain, it is also referred as severe malaria (SM). Various studies using antibodies, recombinant cytokines, or knock-out mice have shown that the secretion of tumor necrosis factor (TNF) is an important effector of the mortality of SM. 8-10 Further studies with TNF receptor (TNFR)-deficient mice have shown that mortality is dependent on the TNFR2 and not the TNFR1, 11 whereas the reverse is true in the majority of immunological and/or infectious diseases. 12
TNF production is induced by an immune response, elicited by the presence of the parasite in the blood. 13 Indeed, depletion of CD4 T lymphocyte prevents the acute mortality of PbA infection in mice, apparently by decreasing TNF production. 1,13 TNF might be responsible for some of the manifestations of SM, such as the hypoglycemia and the sequestration of cells in the microcirculation. TNF is known to increase the expression of the adhesion molecules CD54 (ICAM-1) and CD106 (VCAM) on endothelia, 14-16 which might contribute to increasing the adhesion of leukocytes and other cells and thereby disturbing the microcirculation in the brain and other organs. This pathogenic hypothesis is supported by the increased expression of CD54 and CD106 in the brain microcirculation during SM and the delayed mortality seen in mice treated with anti-CD11a mAb (LFA-1, a β2 integrin determinant) 6,17 or in CD54-deficient mice. 18
CD40 is a cell receptor belonging to the TNF receptor superfamily that can modulate cell proliferation, differentiation, and death. 19 Studies based on mice genetically deficient in CD40 or its ligand CD40L (CD154) as well as the use of anti-CD40L mAb have demonstrated that this system plays an important role in both humoral and cell-mediated immunity. 20,21 Presence of CD40 has been reported on B lymphocytes, platelets, mast cells, endothelial cells, and dendritic cells, whereas the source of CD40L includes T lymphocytes, macrophages, and platelets. 21,22 Response to infectious agents; rejection of allograft; autoimmune diseases, such as encephalitis, arthritis, atherosclerosis and pulmonary fibrosis, are attenuated in mice with a perturbation of the CD40-CD40L signaling. 21 Understanding of the role of CD40-CD40L in cell-mediated immunity is presently incomplete because CD40-CD40L seems to be critical for the resistance to some intracellular parasites such as Leishmania, 23 but not others such as mycobacteria. 24 CD40-CD40L signaling has been reported to induce the expression of adhesion molecules CD54, CD62E, and CD106 on endothelial cells from the umbilical vein, 25 that might be relevant to the pathogenesis of SM, as discussed above.
In this report, we explored the role of CD40-CD40L in the course of PbA-induced SM. Mortality was completely abrogated in CD40−/−, CD40L−/−, as well as in mice treated with the anti-CD40L mAb, indicating an essential role of this system in the pathogenesis of SM.
Materials and Methods
Mice
CD40L−/− and CD54−/− mice, 6 isolated on the C57BL/6 background, were obtained from the Jackson Laboratory (Bar Harbor, ME). CD40−/− were obtained from R. Geha, Boston, MA. 26 Mice were bred in our animal facilities as well as the C57BL/6J (B6) also obtained from the Jackson Laboratory, which were used as wild-type (+/+) controls.
PbA and Treatment
PbA has been passed in rodents 1 and mice were infected by an intravenous injection of 5 × 10 4 parasitized red blood cells (pRBCs).
Anti-CD40L mAb was derived from the hybridoma MR1 of hamster origin. 27 mAbs or nonimmune hamster IgG, as a control, were purified by protein A-Sepharose. Mice were injected with the anti-CD40L mAb or nonimmune hamster IgG (250 μg IgG, ip) on day 6 after infection.
Blood
Blood (0.015 ml) was isolated from the retro-orbital plexus of ethrane-anesthetized mice using heparinized capillaries and diluted in ethylenediaminetetraacetic acid (1% final), in accord with the Swiss national guidelines. Blood elements were counted in a cell counter (Casy 1, Schärfe system, D-72760 Reutlingen, Fluka, 9470-CH). Parasitemia was counted on blood smears stained with May-Grunwald-Giemsa.
Assessment of Vascular Leak
Mice (three wild-type and three mutant) were injected intravenously with 0.2 ml of 1% Evans blue on day 6, shortly before death of the wild-type mice. One hour later, mice were sacrificed and the staining of brain sections was assessed as an indicator of increased capillary permeability.
Light and Electron Microscopy
Mice were sacrificed by an intraperitoneal injection of nembutal and the aorta was cut. Brain was isolated and immersion-fixed in formaldehyde 5% in ethanol 70%. Frontal sections were prepared for ordinary histology. Venules within the cortex were examined and the endothelial cells and macrophages were counted. Endothelial cells were counted (>50/sections) as well as the number of macrophages and the sequestration was evaluated by the macrophage/endothelial ratio. Megakaryocytopoiesis was evaluated on sections of the spleen, by counting at a magnification of 400-fold, the number of megakaryocytes/microscopic field, as described previously. 28
To examine the lung, the thorax was opened and the lung was fixed by intratracheal instillation of glutaraldehyde (5% in 0.1 mol/L cacodylate buffer) and processed for epon embedding. A section was taken in the left lobe across the hilus. Thin sections were prepared from two blocks/mouse taken from the parenchyma. Thin sections were examined with a Philips 400 electron microscope at 60 kV. Cells within alveolar capillaries were examined and red blood cells (RBCs), parasitized RBCs (pRBCs), platelets, and PMNs were counted. Approximately 100 RBCs were counted per each individual thin section. RBCs were used as a neutral indicator of blood stasis and sequestration was evaluated by the platelets/RBCs and PMN/RBC ratios.
Immunohistochemistry
Frozen tissue sections were immunostained as described elsewhere. Briefly, 5-μm sections were incubated overnight at room temperature with rat mAbs: anti-CD54 and anti-CD106 mAbs, as described previously, 6 or anti-CD40 mAb FGK45. 29 After washing, sections were incubated 1 hour with fluorescein isothiocyanate-labeled goat anti-rat IgG (Southern Biotechnology, Bioreba, Reinach, Switzerland).
Evaluation of TNF Serum Level
TNF-α was evaluated on the citrated plasma, using the mouse enzyme-linked immunosorbent assay Dy 410 (R&D Systems, Inc, Minneapolis, MN), which detects both free and receptor-bound TNF, with a sensitivity of 12 pg/ml.
Evaluation of CD40L, CD40, TNF-α, and CD54 mRNA Levels
TNF-α (TNF), 30 CD40L, 31 CD40, 32 and CD54 33 mRNA levels were evaluated on Northern blots, using 32P-labeled dUTP riboprobes. Quantification of mRNA was performed using the Arcus II system of quantification of scanned gels (Agfa, Mortsel, Belgium) with the Image Quant Analysis (Molecular Dynamics, Sunnyvale, CA) integration. The values were normalized to the 18S RNA and the amount of mRNA was expressed in arbitrary units. 34
Statistical Evaluation
Significance analysis of survival curves was determined by Fisher’s exact test. Groups of values were compared using Student’s t-test or the nonparametric Mann and Whitney test. 35
Results
Morbidity and Mortality
Infection with PbA results, in susceptible mice, in a lethal complication 7 to 9 days after infection. 8 More than 90% of the infected +/+ mice were dead on day 8 after infection, while the CD40−/− (Figure 1A) ▶ or the CD40L−/− (not shown) did survive beyond day 12. A few hours before death, mice became hypothermic and comatose, and because a temperature below 33°C is invariably followed by death, this criteria was used as an end point in the majority of the experiments for ethical reasons. Thus, hypothermia was evident on days 7 to 8 after infection in infected +/+, but absent or markedly attenuated in CD40−/−, CD40L−/− mice, as well as in +/+ mice treated with anti-CD40L mAbs (Figure 1, B and C) ▶ .
Figure 1.
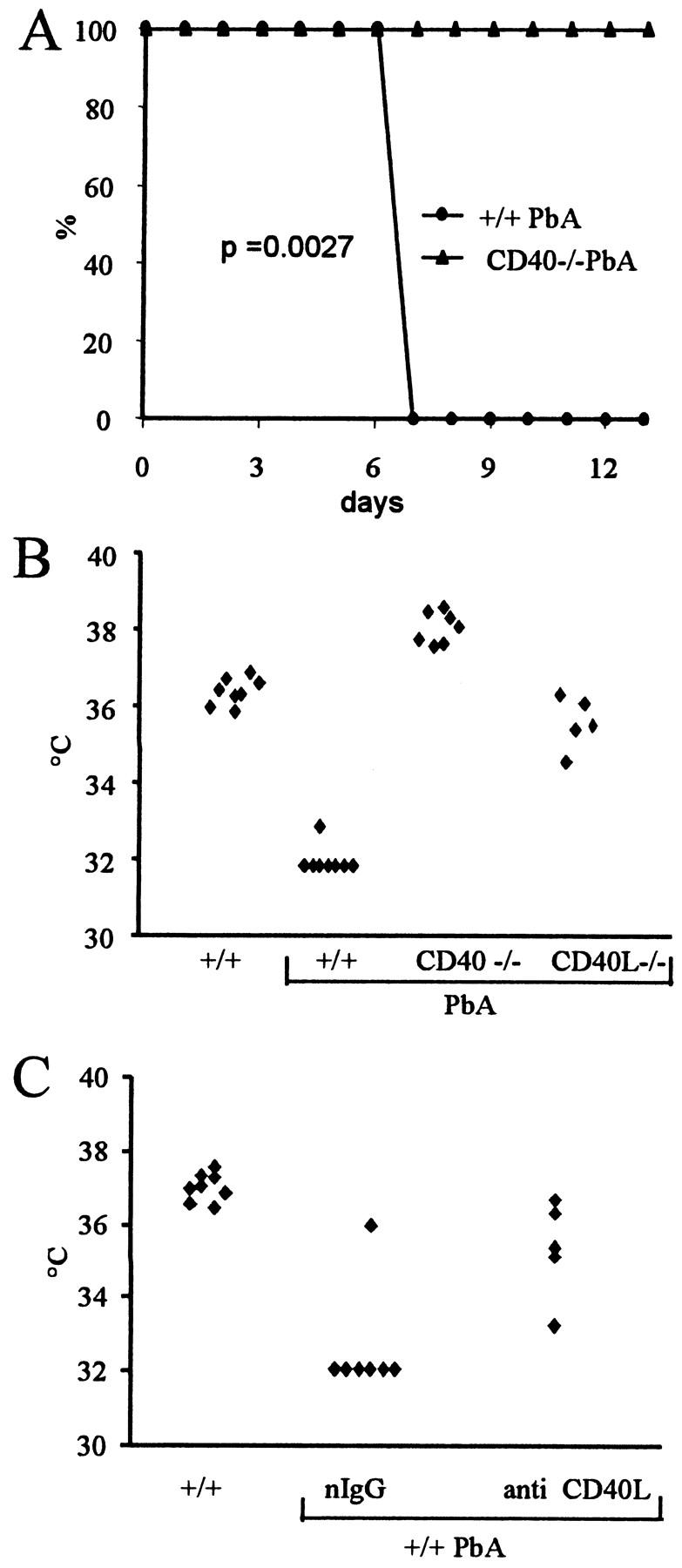
Mortality of PbA-infected mice. A: Survival of PbA-infected mice in days after infection (n = 10). B: Temperature on days 7 to 8 after PbA infection. Infected CD40−/− and CD40L−/− are significantly different from the +/+ (P < 0.05). C: Temperature on day 7 after infection in infected +/+ mice treated with hamster nonimmune IgG (nIgG) or anti-CD40L mAb on day 6. The two groups are significantly different (P < 0.05).
Blood-Brain Barrier
A breakdown of the blood-brain barrier, detected by a leakage of Evans blue, as well as an increase of the vascular permeability in other organs, such as the lung, are evident during PbA-induced SM. 3,6 A marked increase of the leak was evident in infected +/+, but not in CD40−/− mice (Figure 2) ▶ .
Figure 2.

Blood-brain barrier. Noninfected +/+ and infected +/+ and CD40−/− mice were injected with Evans blue 7 days after infection. There is an increase of leak of Evans blue in the brain (top) and lung (bottom) in infected +/+, but not in infected CD40−/− mice. Infected CD40−/− are similar to a noninfected +/+ mouse.
Parasitemia
Parasitemia was similar in +/+, CD40−/− and CD40L−/− mice until day 8 after infection when the +/+ died (Figure 3) ▶ while in the CD40−/− and CD40L−/− mice, parasitemia kept increasing the following days. Mortality was therefore not related to difference in parasitemia.
Figure 3.

Parasitemia in +/+, CD40−/− and CD40L−/− mice. Mean (+SD, n = 10) of the percentage of pRBCs various times after infection. Wild-type (+/+) mice were all dead by day 8 after infection.
In +/+ mice treated with anti-CD40L mAb, no significant change appeared in the percentage of parasitized red blood cells (not shown).
Splenomegaly after PbA Infection
Seven days after infection, the spleen increased approximately threefold in size, and this was evident in +/+ as well as in CD40−/− mice, whereas in CD40L−/− mice, the splenomegaly was less pronounced (Figure 4) ▶ . In contrast, the size of the lymph nodes did not change significantly in +/+ infected mice (not shown).
Figure 4.

Splenomegaly after PbA infection. Weight of the spleen (mean + SD, n = 5) 7 to 8 days after infection.
Platelets
SM is associated with a severe thrombocytopenia because of a reduced platelet life span. 36 On the 7th day after infection, +/+ mice were severely thrombocytopenic, but thrombocytopenia was attenuated significantly, but to a modest extent, in CD40−/− mice and more markedly in CD40L−/− (Figure 5A) ▶ . Thrombocytopenia was also significantly attenuated in +/+ mice treated with the anti-CD40L mAb, compared to those treated with the control IgG (Figure 5B) ▶ .
Figure 5.

Platelet counts during SM. A: Platelet counts 7 days after infection. Wild-type mice (+/+) are significantly thrombocytopenic, compared to noninfected mice (n = 8, P < 0.01), whereas thrombocytopenia is significantly (P < 0.05) attenuated in infected CD40−/− and CD40L−/−, compared to +/+ mice. B: Values are the mean (+SD, n = 7) of the platelet counts 7 days after infection. Infected +/+ mice were treated on the 6th day after infection with nIgG or anti-CD40L mAb.
Cell Sequestration
PbA-induced SM is associated with the sequestration of various cell types, notably in the brain and lung. In the brain, macrophage sequestration was evident in the cortical venules of infected +/+ mice and, as seen in Figure 6 ▶ , this was markedly reduced in infected CD40−/− or CD40L−/− mice. In mice treated with the anti-CD40L mAb, the modification of sequestration was less conclusive. In some mice, sequestration was decreased, whereas in others, it was modified by an increase of lymphocyte sequestration together with macrophages. Sequestration of PMNs is not known to occur in the cerebral blood vessels of PbA-infected mice and was not observed in the present study (not shown). pRBCs are present in the brain microcirculation and were sometimes observed in contact with the venular endothelium by transmission electron microscopy (not shown), in accord with a recent report. 37 However, when the percentage of pRBCs in the brain venules and capillaries was evaluated by transmission electron microscopy, it was not higher than in the systemic circulation, in contrast to what was observed in alveolar capillaries (see below), therefore arguing against a selective retention of pRBCs in brain microcirculation. Microhemorrhages are common in PbA-infected CBA mice, 5 whereas in contrast, these are rare in mice of C57BL/6 background, and were therefore not evaluated in the present experiments.
Figure 6.

Cell sequestration in brain venules. A and B: Cortical venules of infected +/+ (A) or CD40−/− (B) mice. An important sequestration of leukocytes is evident in A, but not in B (original magnification, ×400). C: Macrophages and endothelial cells in cortical venules were counted on H&E-stained sections by light microscopy. The results are expressed as the mean (+SD, n = 5) of the ratio of macrophages/endothelial cells seen in cortical venules.
In the lung, sequestration in the alveolar capillaries includes macrophages, PMNs, platelets, and pRBCs. 6 As evaluated by semiquantitative electron microscopy, sequestration of macrophages was significantly decreased in CD40−/−, whereas sequestration of PMNs, pRBCs, and platelets was similar to that seen in +/+ mice (Figure 7, A and B ▶ , and Figure 8A ▶ ). Similar results were obtained in three infected CD40L−/− mice studied (not shown). Using blood smears, it has been reported that the percentage of pRBCs is higher in the lung than in the peripheral blood. 7 With the present method, we also observed a higher percentage of pRBCs in the alveolar capillaries than in the large blood vessels of the lung, indicating a selective adhesion of pRBCs in alveolar capillaries (Figure 7C ▶ and Figure 8B ▶ ). This was similar in +/+ and CD40−/− infected mice (Figure 8B) ▶ , as well as in CD40L−/− mice (not shown). In CD54−/−, in contrast to CD40−/−, no difference between the percentage of pRBCs in large blood vessels and alveolar capillaries was observed (Figure 8B) ▶ . These observations indicate that the sequestration of pRBCs in alveolar capillaries requires CD54, but not CD40.
Figure 7.
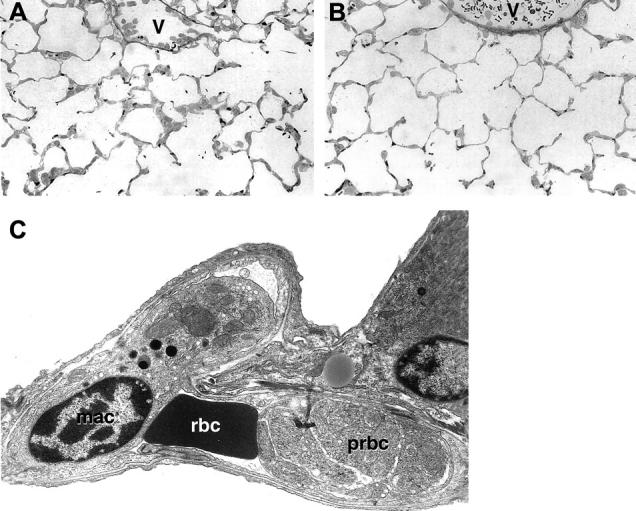
A and B: Alveolar capillaries from an infected +/+ (A) or CD40−/− (B) mice. An increased cellularity of the alveolar septa is evident in A but not in B, which is similar to noninfected +/+ mice (not shown). Macrophage sequestration is also evident in the venules (v) in A but not in B. C: An alveolar capillary from an infected +/+ is seen by transmission electron microscopy. A macrophage (mac), a red blood cell (rbc), and a parasitized red blood cell (prbc) in an advanced stage of maturation are seen inside the capillary. Original magnifications: ×400 (A and B), ×6500 (C).
Figure 8.

Cells in pulmonary blood vessels. A: RBCs, pRBCs, macrophages (mac), PMNs, and platelets (plt) within alveolar capillaries were counted by transmission electron microscopy. Results are the mean (±SD, n = 5), of the ratio with RBCs seen in individual mice. Macrophage and PMN sequestration was significantly (P < 0.05) increased in infected +/+, compared to noninfected +/+ mice, whereas macrophage and PMN sequestration was also significantly less important in infected CD40−/−, compared to +/+ mice. B: pRBCs in large blood vessels and alveolar capillaries were counted in large blood vessels and in the alveolar capillaries observed on thin sections by transmission electron microscopy. The results are the mean (±SD, n = 5) of the percentage of pRBCs.
Evaluation of CD40 and CD40L mRNAs
We examined the mRNA levels in the brain, lung, and spleen of controls or PbA-infected mice (Figure 9) ▶ . The CD40 mRNA levels were significantly increased in the brain and lung of infected mice, compared to noninfected mice (Figure 9) ▶ . By histochemistry, a weak staining of the cortical venules was observed (not shown).
Figure 9.

CD40 and CD40L mRNA levels. RNA was isolated from the brain, lung, and spleen from normal or infected B6 mice 7 days after infection. Blots obtained from two individual mice are shown on top and the mean (+SD, n = 5) values expressed in arbitrary units are shown as bar graphs.
CD40L mRNA levels were significantly increased in the lung, whereas in the brain or spleen they were not markedly modified (Figure 9) ▶ . However, taking into account the approximately threefold increase of the spleen size, it is likely that the amount of CD40L mRNA per spleen is increased. These observations suggest an increase of CD40L production in infected mice.
Expression of TNF, CD54, CD106, and CD31
TNF mRNA levels were significantly elevated in the brain, lung, and spleen of infected +/+ mice. Surprisingly, in the lung and spleen, the mRNA levels were significantly higher in CD40−/− or in CD40L−/− than in +/+ mice RNA (Figure 10) ▶ . In the brain in contrast, TNF mRNA levels were increased in CD40−/− and abrogated in CD40L−/− (Figure 10) ▶ . In mice treated with the anti-CD40L mAb, the TNF mRNA levels in the brain, lung, and spleen showed similar trends to those seen in CD40L−/− (not shown).
Figure 10.

TNF and CD54 mRNAs levels. RNA was isolated from brain, lung, and spleen of normal or infected mice on the 7th day after infection. Blots obtained with two individual mice are shown on top and the mean (+SD, n = 5) of the values observed are shown as a bar graph. Left: TNF mRNA. Right: CD54 mRNA. Values of infected +/+ were compared with noninfected controls and infected CD40−/− and CD40L−/− were compared with infected +/+ mice.
TNF was also assayed in the plasma by enzyme-linked immunosorbent assay. TNF plasma levels were of <10, 248 (42), and 538 (82) pg/ml for noninfected, infected +/+, and CD40−/− mice, respectively (mean + SD, n = 6).
Expression of CD54 was evaluated both at the mRNA level and by histochemistry. In the brain, the infection is known to increase the CD54 mRNA levels as well as the expression of CD54 in venules. CD54 mRNA levels were slightly and similarly decreased in both CD40−/− and CD40L−/− infected mice, compared to +/+ (Figure 10) ▶ . By histochemistry of brain section, CD54 was clearly increased in venules of infected +/+ compared to controls. Expression of CD54 was also increased in infected CD40−/− mice, without obvious difference with infected +/+ mice (not shown).
In the lung, CD54 mRNA levels were slightly but not significantly increased by infection in +/+ mice. Surprisingly, in infected CD40−/− or CD40L−/− mice the levels were significantly different from the infected +/+ but in opposite direction, markedly reduced in CD40−/−, and significantly increased in CD40L−/− mice (Figure 10) ▶ . In the spleen, the modifications were similar to those seen in the lung (Figure 10) ▶ . Expression of CD106 in brain venules was increased in both infected +/+ and CD40−/− mice, as seen by histochemistry, without detectable difference between them (not shown).
CD31 is present in a variety of cells, including endothelial cells and platelets. No important modifications in the distribution of PECAM were evident in +/+ mice, infected or not or in infected CD40−/− mice (not shown).
Discussion
The reason why the patients or the mice die during the acute phase of malarial infection is still controversial in reason of the multiplicity of the pathogenic mechanisms that might be involved. Here we report that CD40-CD40L needs to be added to the numerous cascades already known to contribute to the development of the lethal complication.
PbA infection resulted in an approximately threefold increase in the spleen size in 7 days, 38 which was evident in +/+ and in CD40−/−, but less pronounced in CD40L−/− mice. The splenomegaly might contribute to the severity of the disease because the mortality is prevented by splenectomy. 39 In humans, lymphoproliferative diseases have been reported to be associated with an increase of the amount of CD40L in plasma. 40,41 We were unable to measure the CD40L plasma levels in infected mice, but the evaluation of the mRNA levels in tissue showed an increased level in the lung, and an increased level per spleen if the spleen size (Figure 4) ▶ is taken into account. Expression of the cell receptor CD40, judged from its mRNA was increased in the brain and lung, observations that argue for an increase of CD40L-CD40 signaling during PbA infection.
TNF is believed to play a central role in many of the alterations observed during SM, including the mortality. Because CD40/CD40L deficiency is reported to disrupt cell-mediated immunity, it was obviously of importance to evaluate whether the absence of CD40-CD40L signaling decreases TNF production. In PbA-infected mice, we observed similar TNF plasma levels in +/+, CD40−/−, and CD40L−/− mice. Accordingly, TNF mRNA levels in the spleen and lung of infected CD40−/− or CD40L−/− mice were not decreased and were even significantly higher in CD40L−/− than in +/+ mice. These observations indicate that absence of CD40 or CD40L did not markedly affect systemic TNF production and that the blockade of the events leading to SM resulting from the abrogation of CD40-CD40L signaling must be exerted downstream of TNF production. This interpretation is also in accord with the fact that the anti-CD40L mAb did prevent the occurrence of SM, even when given on day 6 after infection, ie, immediately before the effector phase of the disease. Surprisingly in the brain, TNF mRNA levels were modified in opposite direction in CD40−/− or CD40L−/− mice and almost abrogated in the latter. This observation suggests that CD40L might exert a specific influence on the production of TNF in the brain, independent of CD40. This discrepancy between the cerebral TNF mRNA levels in the brain seen in CD40 and CD40L also indicates firstly, that TNF mRNA levels in the brain are not correlated with morbidity, (ie, body temperature) because CD40- and CD40L-deficient mice were not sick, but with high or low cerebral TNF mRNA levels, respectively, and secondly, that TNF mRNA levels in the brain are not correlated with macrophage sequestration, because this was similarly reduced in CD40−/− and CD40L−/− mice (Figure 10) ▶ .
One of the effects of TNF is to increase the expression of adhesion molecules on endothelial cells, an activity shared with IL-1. 25,42 Published reports, based on response of endothelial cells in vitro, suggest that CD40 signaling might also contribute to increasing the expression of adhesion molecules CD54 or CD106. 25 In the present model, the role of CD40 and CD40L in the expression of CD54 was not identical and also organ-specific, ie, in the lung and spleen, CD54 expression was CD40-dependent but CD40L-independent. Because in these organs, the CD54 mRNA levels were significantly higher in CD40L−/− than in +/+ mice, this suggests that CD40L can also inhibit the expression of CD54. In the brain, in contrast to lung and spleen, expression of CD54 was decreased to a modest but similar extent in both CD40−/− and CD40L−/− mice, an observation compatible with the possibility that expression of CD54 in the brain contributes to the mortality of SM. 11 The increased expression of CD54 in the brain of infected CD40L−/−, whose cerebral TNF mRNA was almost abrogated, indicates that circulating, but not cerebral, TNF is sufficient to up-regulate CD54 in cerebral venules.
Both CD40 and CD40L deficiency markedly decreased the sequestration of macrophages in brain venules and alveolar capillaries. Because the modest down-regulation of CD54 does not seem to be a sufficient explanation, these observations raise the possibility that macrophages bearing CD40L might bind to the endothelial CD40, although we could not detect a significant expression of CD40 in cerebral venules by histochemistry, in accord to what was observed in a study of multiple sclerosis. 43 Nevertheless, the decrease of macrophage sequestration in the brain and lung of CD40−/− or CD40L−/− does correlate well with the decreased mortality. Sequestration of pRBCs as a possible pathogenic mechanism of PbA infection has been primarily overlooked so far, because its occurrence in the brain microcirculation has not been clearly demonstrated. pRBCs are indeed present in the brain microcirculation, 37 but in our experiments performed with parasitemia <10%, we did not detect a significantly higher parasitemia in the brain microcirculation than in large blood vessels, arguing against a specific retention of pRBCs in the brain. In contrast, parasitemia was significantly higher in the alveolar capillaries than in the major blood vessels of the lung (Figure 8A) ▶ , arguing for a specific retention of pRBCs in the alveolar capillaries. In addition, pRBCs arrested in the alveolar capillaries are the most mature pRBCs (Figure 7C) ▶ , in accord to what has been reported for other rodent parasites. 6,7 It has been suggested that pRBCs might contribute to the severity of SM by interfering with the microcirculation and/or by the release of toxic molecules damaging the downstream circulation. 44 pRBC sequestration was significantly increased in alveolar capillaries of CD40−/− or CD40L−/− mice (Figure 8B) ▶ , indicating that it does not require CD40-CD40L signaling, whereas it was not evident in CD54−/− mice, as we reported previously. 6 Because pRBCs sequestration was obvious in CD40−/− or CD40L−/− mice that were otherwise not sick, these results argue that pRBC sequestration is not sufficient to induce alone the manifestations of SM. Sequestration of PMNs has also been reported to contribute to the mortality of SM, 5 but the present results indicate that PMN sequestration, which was evident in infected CD40−/− mice and is therefore CD40-independent, is also not sufficient to elicit alone the complications of SM.
SM, as are other systemic inflammatory disorders such as endotoxemia or acute respiratory distress syndrome, is associated with a thrombocytopenia, because of a drastic reduction of platelet life span. 45 Platelet loss might occur by platelet adhesion in the microcirculation, as it has been reported 46 and/or by the fragmentation of platelets in plasma and the generation of microparticles. 47 Both processes might influence the course of the disease, by the release, local or systemic, of platelet factors. In our experiments, blockade of CD40L by either genetic disruption or mAb did significantly attenuate thrombocytopenia, whereas the effect of CD40 deficiency was also significant but less pronounced. These results indicate that CD40L, whose production increases during infection, as discussed above, is an important effector of the PbA-induced thrombocytopenia. A significant contribution of CD40-CD40L in malaria-induced thrombocytopenia is in accord with a recent observation indicating that platelets bear CD40 and CD40L and that CD40L-mediated platelet activation can result in vascular damage. 22 Because injection of TNF can induce a thrombocytopenia 48 and because the thrombocytopenia of SM can be attenuated by anti-TNF (unpublished observation) as well as with anti-CD40L antibody, these observations indicate that both TNF and CD40L contribute to the platelet loss of SM. 48
Interestingly CD40, as well as CD54 deficiency, 6 did decrease both platelet loss and breakdown of the blood-brain barrier. Pathogenesis of vascular leak in the brain, 49 as well as its possible role in the mortality are poorly understood. This vascular leak is evident in cortex and white matter and interests therefore most likely capillaries, unlike macrophage sequestration that is limited to venules. In the retina, leak seems to start in venules, and extends subsequently to capillaries. 16 Because in other territories TNF can induce a vascular leak, 50,51 it is possible that TNF contributes to the vascular leak of SM. However, the vascular leak was absent in CD40−/− mice despite elevated TNF plasma levels or elevated cerebral TNF mRNA levels, indicating that this effect of TNF might require the presence of CD40.
Thus, CD40-CD40L plays a crucial role in the mortality induced by PbA infection and the present results indicate that CD40-CD40L is necessary for the breakdown of the blood-brain barrier, macrophage sequestration, and platelet consumption associated with SM.
Footnotes
Address reprint requests to Pierre F. Piguet, MD, Department of Pathology, 1 rue M. Servet, CMU, 1211 Geneva, Switzerland. pierre. piguet@medecine.unige.ch.
This study was supported by grant no. 31-56839.99 from the Swiss National Science Foundation.
References
- 1.Wright DH: The effect of neonatal thymectomy on the survival of golden hamsters infected with Plasmodium berghei. Br J Exp Pathol 1968, 49:379-384 [PMC free article] [PubMed] [Google Scholar]
- 2.White NJ, Ho M: Advances in parasitology. The Pathophysiology of Malaria. 1992:pp 84-173 Academic Press, New York [DOI] [PubMed]
- 3.Thumwood CM, Hunt NH, Clark IA, Cowden WB: Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology 1988, 96:579-589 [DOI] [PubMed] [Google Scholar]
- 4.Clark IA, Macmicking JD, Gray KM, Rockett KA, Cowden WB: Malaria mimicry with tumor necrosis factor. Contrasts between species of murine malaria and plasmodium falciparum. Am J Pathol 1992, 140:325-336 [PMC free article] [PubMed] [Google Scholar]
- 5.Senaldi G, Vesin C, Chang R, Grau G, Piguet PF: An effector role of neutrophils in the pathogenesis of murine severe malaria. Infect Immun 1994, 62:1144-1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Favre N, Willimann K, Ryffel B, Weiss N, Imhof BA, Rudin W, Lucas R, Piguet PF: Role of ICAM-1 in the development of murine cerebral malaria. Microbes Infect 1999, 1:961-968 [DOI] [PubMed] [Google Scholar]
- 7.Coquelin F, Boulard Y, Mora-Silvera E, Richard F, Chabaud AG, Landau I: Final stage maturation of the erythrocytic schizonts of rodent Plasmodium in the lungs. C R Acad Sci (III) 1999, 322:55-62 [DOI] [PubMed] [Google Scholar]
- 8.Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P: Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987, 237:1210-1212 [DOI] [PubMed] [Google Scholar]
- 9.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P: Monoclonal antibody against interferon-gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc Natl Acad Sci USA 1989, 86:5572-5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudin W, Eugster HP, Bordmann G, Bonato J, Muller M, Yamage M, Ryffel B: Resistance to cerebral malaria in tumor necrosis factor-alpha/beta-deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. Am J Pathol 1997, 150:257-266 [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas R, Juillard P, Decoster E, Redard M, Burger D, Donati Y, Giroud C, Monso Hinard C, De Kesel T, Buurman WA, et al: Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur J Immunol 1997, 27:1719-1725 [DOI] [PubMed] [Google Scholar]
- 12.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethman H: Mice lacking the tumor necrosis receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364:798-802 [DOI] [PubMed] [Google Scholar]
- 13.Grau GE, Piguet PF, Engers HD, Louis JA, Vassalli P, Lambert PH: L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J Immunol 1986, 137:2348-2354 [PubMed] [Google Scholar]
- 14.Lucas R, Lou JN, Juillard P, Moore M, Bluethmann H, Grau GE: Respective role of TNF receptors in the development of experimental cerebral malaria. J Neuroimmunol 1997, 72:143-148 [DOI] [PubMed] [Google Scholar]
- 15.Imhof BA, Dunon D: Leukocyte migration and adhesion. Adv Immunol 1995, 58:345-416 [DOI] [PubMed] [Google Scholar]
- 16.Ma N, Hunt NH, Madigan MC, Chan Ling T: Correlation between enhanced vascular permeability, up-regulation of cellular adhesion molecules, and monocyte adhesion to the endothelium in the retina during the development of fatal murine cerebral malaria. Am J Pathol 1996, 149:1745-1762 [PMC free article] [PubMed] [Google Scholar]
- 17.Grau GE, Pointaire P, Piguet PF, Vesin C, Rosen H, Stamenkovic I, Takei F, Vassalli P: Late administration of monoclonal antibody to leukocyte function antigen 1 abrogates incipient murine cerebral malaria. Eur J Immunol 1991, 21:2265-2267 [DOI] [PubMed] [Google Scholar]
- 18.Falanga PB, Butcher EC: Late treatment with anti-LFA-1(CD11a) antibody prevents cerebral malaria in a mouse model. Eur J Immunol 1991, 21:2259-2263 [DOI] [PubMed] [Google Scholar]
- 19.Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DCS, Lee YHW, Durkop H, Engelmann H, Scheurich P, et al: Induction of cell death by tumor necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J 1999, 18:3034-3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H: The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1:167-178 [DOI] [PubMed] [Google Scholar]
- 21.Grewal IS, Flawell RA: CD40 and CD154 in cell-mediated immunity. Ann Rev Immunol 1998, 16:111-135 [DOI] [PubMed] [Google Scholar]
- 22.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Mueller-Berghaus G, Kroczek R: CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391:591-594 [DOI] [PubMed] [Google Scholar]
- 23.Kamanaka M, Yu P, Yasui T, Yoshida K, Kawabe T, Hori T, Kishimoto T, Kitutani H: Protective role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity 1996, 4:275-281 [DOI] [PubMed] [Google Scholar]
- 24.Campos-Neto A, Ovendale P, Bement T, Koppi TA, Fanslow WC, Rossi MA, Alderson MR: CD40 ligand is not essential for the development of cell-mediated immunity and resistance to mycobacterium tuberculosis. J Immunol 1998, 160:2037-2041 [PubMed] [Google Scholar]
- 25.Yellin MJ, Brett J, Baum D, Matsushima A, Szabolcs M, Stern D, Chess L: Functional interactions of T cells with endothelial cells: the role of CD40L-CD40-mediated signals. J Exp Med 1995, 182:1857-1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castigli E, Alt FW, Davidson L, Bottaro A, Mizoguchi E, Bhan A, Geha RS: CD40-deficient mice generated by recombination-activating gene-2-deficient blastocyst complementation. Proc Natl Acad Sci USA 1994, 91:12135-12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A: A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA 1992, 89:6550-6554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piguet PF, Vesin C, Laperousaz CD, Rochat A: Role of plasminogen activators and of urokinase receptor (uPAR, CD87) in platelet kinetics in mice. Hematol J 2000, 1:199-205 [DOI] [PubMed] [Google Scholar]
- 29.Rolink A, Melchers F, Andersson J: The Scid but not the Rag-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity 1996, 5:319-330 [DOI] [PubMed] [Google Scholar]
- 30.Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P: Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med 1989, 170:655-663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendoza RB, Cantwell MJ, Kipps TJ: Immunostimulatory effects of a plasmid expressing CD40 ligand (CD154) on gene immunization. J Immunol 1997, 159:5777-5781 [PubMed] [Google Scholar]
- 32.Grimaldi JC, Torres R, Kozak CA, Chang R, Clark EA, Howard M, Cockayne DA: Genomic structure and chromosomal mapping of the murine CD40 gene. J Immunol 1992, 149:3921-3926 [PubMed] [Google Scholar]
- 33.Horley KJ, Carpeinto C, Baker B, Takei F: Molecular cloning of murine intercellular adhesion molecule (ICAM-1). EMBO J 1989, 8:2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barazzone C, Horowitz S, Donati Y, Vesin C, Rochat A, Rodriguez I, Piguet PF: Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol 1998, 19:573-581 [DOI] [PubMed] [Google Scholar]
- 35.Mann HB, Whitney DR: On a test of whether one or two random variables is stochastically larger than the other. Ann Math Statist 1947, 18:50-60 [Google Scholar]
- 36.Grau GE, Piguet PF, Gretener D, Vesin C, Lambert PH: Immunopathology of thrombocytopenia in experimental malaria. Immunology 1988, 65:501-506 [PMC free article] [PubMed] [Google Scholar]
- 37.Hearn J, Rayment N, Landon DN, Katz DR, de Souza JB: Immunopathology of cerebral malaria: morphological evidence of parasite sequestration in murine brain vasculature. Infect Immun 2000, 68:5364-5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piguet PF, Laperrousaz CD, Vesin C, Donati Y: Incidence of apoptosis in the lymphoid organs of normal or malaria infected mice is decreased in CD18 and urokinase-receptor (UPAR, CD87) deficient mice. Dev Immunol 2000, 8:1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hermsen CC, Mommers E, van de Wiel T, Sauerwein RW, Eling WM: Convulsions due to increased permeability of the blood-brain barrier in experimental cerebral malaria can be prevented by splenectomy or anti-T cell treatment. J Infect Dis 1998, 178:1225-1227 [DOI] [PubMed] [Google Scholar]
- 40.Kato K, Santana-Sahagun E, Rassenti LZ, Weisman MH, Tamura N, Kobayashi S, Hashimoto H, Kipps TJ: The soluble CD40 ligand sCD154 in systemic lupus erythematosus. J Clin Invest 1999, 104:947-955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Younes A, Snell V, Consoli U, Clodi K, Zhao S, Palmer JL, Thomas EK, Armitage RJ, Andreeff M: Elevated levels of biologically active soluble CD40 ligand in the serum of patients with chronic lymphocytic leukaemia. Br J Haematol 1998, 100:135-141 [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA, Thompson RC: Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunol Today 1991, 12:404-410 [DOI] [PubMed] [Google Scholar]
- 43.Howard LM, Amy AJ, Vanderlugt CL, Dal Canto MC, Laman JD, Noelle RJ, Miller SD: Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest 1999, 103:281-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schofield L, Hackett F: Signal transduction in host cells by a glycosylphatidylinositol toxin of malaria parasites. J Exp Med 1993, 177:145-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heffner JE, Repine JE, Crystal RGE (Eds): Platelets. The Lung: Scientific Foundations. New York, Raven Press, 1991, pp 617–627
- 46.Piguet PF, Laperousaz CD, Vesin C, Tacchini-Cottier F, Senaldi G, Grau GE: Delayed mortality and attenuated thrombocytopenia associated with severe malaria in urokinase and urokinase-receptor (UPAR, CD87) deficient mice. Infect Immun 2000, 68:3822-3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes M, Hayward CP, Warkentin TE, Horsewood P, Chorneyko LA, Kelton JG: Morphological analysis of microparticles generation in heparin-induced thrombocytopenia. Blood 2000, 96:188-194 [PubMed] [Google Scholar]
- 48.Tacchini-Cottier F, Vesin C, Redard M, Buurman W, Piguet PF: Role of TNF receptors I and II in TNF-induced platelets consumption in mice. J Immunol 1998, 160:6182-6186 [PubMed] [Google Scholar]
- 49.Stewart PA, Hayakawa K, Farrell CL: Quantitation of blood-brain barrier ultrastructure. Microsc Res Tech 1994, 27:516-527 [DOI] [PubMed] [Google Scholar]
- 50.Brett J, Gerlach H, Nawroth P, Steinberg S, Godman G, Stern D: Tumor necrosis factor cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G-proteins. J Exp Med 1989, 169:1977-1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eunhee SY, Ulich TR: Endotoxin, interleukin-1, and tumor necrosis factor cause neutrophil-dependant microvascular leakage in postcapillary venules. Am J Pathol 1992, 140:659-663 [PMC free article] [PubMed] [Google Scholar]