

Abstract
The nuclear factor (NF)-κB family of eukaryotic transcription factors plays an important role in the regulation of immune response, embryo and cell lineage development, cell apoptosis, cell-cycle progression, inflammation, and oncogenesis. A wide range of stimuli, including cytokines, mitogens, environmental particles, toxic metals, and viral or bacterial products, activate NF-κB, mostly through IκB kinase (IKK)-dependent phosphorylation and subsequent degradation of its inhibitor, the IκB family of proteins. Activated NF-κB translocates into the nucleus where it modulates the expression of a variety of genes, including those encoding cytokines, growth factors, acute phase response proteins, cell adhesion molecules, other transcription factors, and several cell apoptosis regulators. During the past few years, tremendous progress has been achieved in our understanding on how intracellular signaling pathways are transmitted in either a linear or a network manner leading to the activation of NF-κB and subsequent cell growth control. However, a detailed molecular mechanism of NF-κB regulating cell growth has yet to be determined. Elucidation of the relationships between NF-κB activation and cell growth will be important in developing new strategies for the treatment of various human diseases, such as chronic autoimmune disorder and cancer.
After more than a decade of intensive study, a complex body of knowledge has been accumulated, revealing the molecular mechanisms of signal-induced activation of nuclear factor (NF)-κB, a pivotal transcription factor governing the expression of early response genes involved in cell-to-cell interaction, intercellular communication, cell recruitment or transmigration, amplification or spreading of primary pathogenic signals, and initiation or acceleration of tumorigenesis. 1-3 Presently, five mammalian NF-κB family members have been identified and cloned. 4-6 These include NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA(p65), RelB, and c-Rel. All of these NF-κB family members share a highly conserved Rel homology domain responsible for DNA binding, dimerization, and interaction with IκB, the intracellular inhibitor for NF-κB. 7 The C-terminal regions of RelA, RelB and c-Rel contain a transactivating domain that is important for NF-κB-mediated gene transactivation. The C-termini of the precursor molecules for p50 and p52, p105 and p100, contain multiple copies of the so-called ankyrin repeat, which is found in IκB family members, including IκB-α, IκB-β, IκB-ε, Bcl3, and Drosophila cactus.
A wide range of signals, which typically include cytokines, mitogens, environmental and occupational particles, toxic metals, intracellular stresses, viral or bacterial products, and UV light, induce expression of early response genes through the NF-κB family of transcription factors. 2,4,8-10 In resting cells, NF-κB is sequestered in the cytoplasm in an inactive form through its association with one of several inhibitory molecules, including IκB-α, IκB-β, IκB-ε, p105, and p100. Activation of the NF-κB-signaling cascade results in a complete degradation of IκB or partial degradation of the carboxyl termini of p105 and p100 precursors, allowing the translocation of NF-κB to the nucleus, where it induces transcription (Figure 1) ▶ . Activated NF-κB binds to specific DNA sequences in target genes, designated as κB-elements, and regulates transcription of genes mediating inflammation, carcinogenesis, and pro- or anti-apoptotic reactions.
Figure 1.
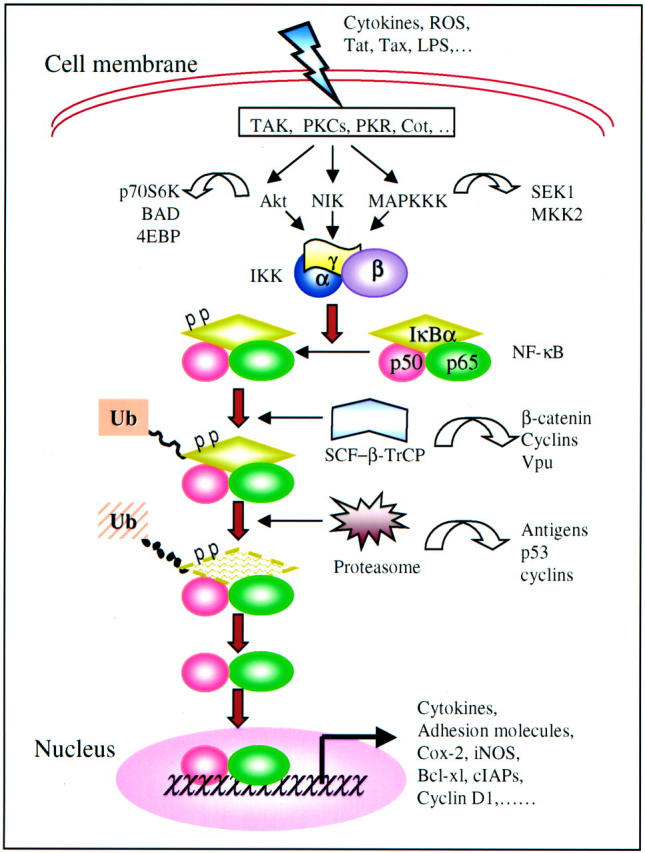
Signaling pathways of NF-κB activation. Extracellular inducers, including cytokines, reactive oxygen species (ROS), and viral and bacterial products, activate IKK through upstream kinases directly or indirectly. Activated IKK phosphorylates N-terminal S32 and S36 residues of IκB-α that is associated with NF-κB p50 and p65 heterodimer. The SCF-β-TrCP complex recognizes phosphorylated IκB-α and modifies IκB-α with polyubiquitin chains. This is followed by proteasome-mediated degradation of IκB-α. After degradation of IκB-α, the activated NF-κB translocates into the nucleus where it binds to the κB-sites of gene promoters or enhancers to up-regulate target gene expression. Line arrows and filled arrows denote the NF-κB signaling pathways; open arrows denote the connections with the by-standing signaling pathways.
Three IκB proteins, IκB-α, IκB-β, and IκB-ε, have been identified, among which IκB-α is the most abundant inhibitory protein for NF-κB. 4 All IκB proteins contain two conserved serine (S) residues within their N-terminal domain. Phosphorylation of these conserved S residues in response to inducers, leads to the immediate polyubiquitination of IκB proteins by the SCF-β-TrCP complex, a step that has been shown recently to be inhibited by the nonpathogenic Salmonella bacteria in gut epithelial cells. 11 This modification subsequently targets IκB proteins for rapid degradation by the 26S proteasome. 12 A high-molecular weight complex that phosphorylates IκB-α or IκB-β has been characterized recently and named IκB kinase (IKK) complex. This complex contains two catalytic subunits, IKK-α and IKK-β, and a structural component named NEMO/IKKγ/IKKAP. 13,14 An earlier report by Cohen and colleagues 15 suggested the presence of a scaffold protein named IKK complex-associated protein (IKAP) in the IKK complex, which could not be confirmed in other studies. A later study by the group, who originally identified IKAP, indicated that the observed association of IKAP with IKK was because of a nonstrict elusion condition of chromatographic extracts during the purification of IKK. 16 Recently, two groups independently reported the identification of a novel protein, CIKS/Act1, associated with the IKK complex and suggested that CIKS/Act1 functions as an anchoring proteins in the assembly of the IKK complex and in providing a possible connection between IKK and c-Jun-N-terminal kinase signaling. 17,18 IKK-α and IKK-β share 50% sequence homology. Both proteins contain an amino terminal kinase domain, a carboxyl terminal region with a leucine zipper, and a helix-loop-helix domain. In vitro or in vivo studies indicate that although both IKK-α and IKK-β are capable of phosphorylating IκB-α on ser32 and ser36, IKK-β is more potent in IκB-α phosphorylation induced by proinflammatory stimuli. However, a distinct IKK complex, named IKKi/ε that does not contain IKK-α, -β, or -γ, was recently identified in T cells. 3,19 IKKi/ε shares 27% homology with IKK-α and IKK-β and possibly mediates NF-κB-activating kinase signaling and PMA/PKCε-induced S36 phosphorylation of IκB-α and NF-κB activation. 3,19
Although the signaling pathways leading to the activation of NF-κB have been well defined, a number of questions remain to be answered. For example, it is unclear exactly how many protein subunits comprise a naïve and activated IKK complex, respectively; how the various signaling pathways converge on this kinase complex; what upstream kinases contribute to the phosphorylation and activation of IKK. It is also unclear whether other substrates, in addition to IκB or NF-κB family members, can be phosphorylated by IKK. Regarding the functional aspects of NF-κB transcription factor, we know that although NF-κB is important and involved in the regulation of cell apoptosis, cell-cycle transition, and carcinogenic transformation, 20 the detailed interconnections among NF-κB activation, cell cycle, and apoptosis are still undefined. Considering the fact that aberrant activation of NF-κB is associated with a wide range of human diseases, elucidation of molecular mechanisms determining NF-κB activation and expression of its various functions may lead to the development of novel preventive and therapeutic strategies for many diseases including chronic inflammation and cancer.
Roles of NF-κB Activation in Cell Apoptosis
Programmed cell death, or apoptosis, is an essential mechanism for any multicellular organism to eliminate cells that are in excess or potentially dangerous. 21 Most apoptotic cells are characterized by unique morphological features, such as membrane blebbing, cell shrinking, cytosolic and nuclear condensation, and breakdown of chromosomal DNA. Depending on the use of different initiating caspases, signal-induced apoptosis can be roughly divided into receptor-mediated extrinsic apoptosis and mitochondrial-mediated intrinsic apoptosis (Figure 2) ▶ . 22,23 The extrinsic apoptotic pathway is triggered as a consequence of ligand binding to death receptors, including tumor necrosis factor (TNF)-R, Fas, OX40, CD40, and 4–1BB, which contain conserved protein-protein-binding domains termed death domains. These receptors recruit procaspases, mainly caspase-8, via adapter molecules. The intrinsic apoptotic pathway is mediated by mitochondria through release of apoptosis-promoting factors, including cytochrome c, apoptosis-inducing factor, and Diablo/Smac. 24-27 Cytochrome c forms a complex with a cytosolic protein named Apaf-1, a flavoprotein with homology to plant ascorbate reductases and bacterial NADH oxidases, to activate caspase-9. 27 Whereas apoptosis-inducing factor released from the intermembranous space of damaged mitochondria induces apoptosis in a caspase-independent manner, Smac/Diablo released from mitochondria promotes apoptosis by binding to and antagonizing XIAP, cIAP1, and cIAP2, allowing the activation of caspases. 25,26 Both activated caspase-8 from the extrinsic apoptotic pathway and activated caspase-9 from the intrinsic apoptotic pathway cleave and activate effector caspases, mainly caspase-3 to execute an apoptotic process. These apoptotic pathways, however, were compromised in many cases because of the activation of caspase-independent signaling cascades that function to block the apoptotic responses. A good example is TNF-R-mediated activation of NF-κB that induces expression of anti-apoptotic proteins, including caspase inhibitors, such as cIAP1, cIAP2, and XIAP, and mitochondria membrane stabilizers, such as Bcl-xl and Bfl-1. 28-30
Figure 2.

Possible targeting point of anti-apoptotic signals from NF-κB. Intrinsic (open arrows) and extrinsic (filled arrows) apoptosis pathways are depicted. The effector caspases, such as caspase-3 and caspase-7, are activated by upstream initiator caspases, caspase-8 and caspase-9. The initiator caspases themselves are activated by either ligands binding to the death receptor complex or cytochrome c released from damaged mitochondria. An anti-apoptotic effect of NF-κB is achieved through its up-regulation of IAPs that inhibits caspases and Bcl-xl that protects mitochondria from further damaging.→, activation; ⊣, inhibition.
Tumor suppressor protein, p53, has been considered to be one of the major contributors of cell apoptosis in response to a variety of stress inducers. As a transcription factor, p53 is able to up-regulate the expression of genes involved in either reactive oxygen species production or reactive oxygen species metabolism, including quinone oxidoreductase (Pig 3), proline oxidase (Pig 6) homologues, glutathione transferase (Pig 12), and glutathione peroxidase (GPx). 31 Moreover, p53 also activates the expression of several genes that directly control or regulate the process of apoptosis. These genes include Bax, Fas, Fas ligand (FasL), IGF-BP3, PAG608, 32 ei24 (Pig 8), 33 and Noxa. 34
With the identification of role of NF-κB in transcriptional regulation of several pro-apoptotic genes, such as fas and fasl, controversy raged as reports demonstrating that NF-κB protected cells from apoptosis in some types of cells were matched by a similar number of reports demonstrating that it did not in other types of cells. 35,36 Based primarily on earlier studies, NF-κB was initially considered a pro-apoptotic factor because of its rapid activation in cells in response to apoptotic signals and its involvement in the expression of some apoptotic genes, including TNF-α, c-myc, and fasl. 37,38 More recent work, however, has altered this view and revealed an anti-apoptotic effect of NF-κB in response to a variety of apoptotic stimuli.
The direct evidence for the anti-apoptotic effects of NF-κB is provided by gene knockout studies in which the genes encoding either members of NF-κB family proteins or upstream kinases were disrupted. RelA (p65)-deficient mice die during embryonic development through apoptosis of hepatocytes. 39 IKK-β gene knockout mice and IKK-β/IKK-α double-knockout mice die as embryos and show massive liver cell apoptosis, 40-42 a phenotype similar to the response of NF-κB p65 gene knockout mice. In addition, knockout of the IKK-α gene results in perinatal lethality of mice with an increased thickness of the skin because of the deficiency of keratinocyte differentiation. 43 Male mice with an inactivated X-linked gene encoding IKK-γ/NEMO, an essential modulator of the IKK complex for NF-κB activation, die at mid-gestation because of a massive apoptosis of cortical and medulla lymphocytes in the thymus, in addition to degeneration of the liver. 44,45 Female mice deficient in the IKK-γ/NEMO gene manifest a unique dermatopathy because of the apoptosis of keratinocytes and consequent abnormal pigmentation, a characteristic strikingly similar to that of the human X-linked dominant, male-lethal genetic disease—incontinential pigmenti or Bloch-Sulzberger Syndrome. Cross-breeding of relA or IKK-β gene knockout mice with TNF-R1 or TNF-α gene knockout mice revealed partial rescue of embryonic lethality, suggesting that NF-κB deficiency sensitized cells in response to TNF-α-mediated cytotoxicity. 46,47 Similarly, mice deficient in both TNF-R1 and IKK-β showed an attenuated embryonic liver apoptosis. 48
Other compelling evidence linking NF-κB with an anti-apoptotic effect is based on the studies indicating that NF-κB is a priming factor for liver regeneration after partial hepatectomy. 49 This priming effect of NF-κB might be through its transcriptional regulation for survival genes or anti-apoptotic genes whose products can block stress signal-induced cell death, a process critically involved in cell proliferation and transformation. Candidate anti-apoptotic genes targeted by NF-κB include those encoding the cell-cycle regulatory protein cyclin D1, 50-52 the mitochondrial membrane-stabilizing proteins Blf-1 and Bcl-xl, 53,54 the caspase inhibitors cIAP1/cIAP2 and XIAP, and the TNF receptor-associated factors TRAF1 and TRAF2. 55 It should be noted that several reports suggest that NF-κB is also a pro-apoptotic factor in FasL-induced cell death. 56,57 This argument is primarily based on earlier observations that NF-κB can regulate the artificial promoter activity of the fasl gene, a gene encoding an important activator of apoptosis through a CD95/Fas- and Fas-associated death domain (FADD)-mediated caspase-8 activation pathway. 22 However, both promoter truncation studies of the fasl gene and somatic cell mutagenesis studies of IKK-γ indicate that NF-κB is not required for the fasl gene expression. 58,59 Nevertheless, pro-apoptotic or anti-apoptotic effects of NF-κB might depend on the cellular context in combination with a bewildering variety of activators.
Further support for the anti-apoptotic effect of NF-κB comes from observations in which NF-κB can protect cortical neurons from β-amyloid peptide-induced apoptosis in Alzheimer’s disease. 60,61 Exposure of cortical neurons to β-amyloid peptide increased levels of IκB-α mRNA and protein and a consequent decrease in NF-κB activity. 60 Elevation of NF-κB activity by pretreatment of these cells with an antisense oligonucleotide to IκB-α protected them from β-amyloid peptide-induced apoptosis. Conversely, blockade of NF-κB activity by κB decoy DNA was associated with enhanced β-amyloid peptide-induced mitochondrial dysfunction and cell apoptosis. 61 Moreover, data from the studies of rodent models of stroke or cardiac arrest suggested that NF-κB might possibly prevent ischemic neuronal degeneration. 62 The protective role of NF-κB on neurons was attributed to NF-κB-mediated transcription of genes encoding Bcl-2, Mn-SOD, and proteins regulating cellular calcium homeostasis. 63
The vast majority of studies focused on the regulatory roles of NF-κB on apoptosis suggest that NF-κB is acting on the upstream pathways of apoptosis, either negatively or positively. Conversely, a few recent studies have demonstrated the possible regulation of apoptotic molecules on NF-κB. Of potential interest regarding regulation of NF-κB by apoptotic molecules are the observations of cross-competition between NF-κB and p53, a major pro-apoptotic protein. 64-69 The molecular events identified thus far as mediators of cross-competition between NF-κB and p53 can be roughly classified into two categories: the upstream kinases or other regulatory molecules that relay input signals into NF-κB and p53, and co-factors that affect transcriptional activities of NF-κB and p53. An earlier study conducted by Jung and colleagues 70 indicated that ATM, a major kinase responsible for DNA damage-induced N-terminal phosphorylation of p53, was involved in IκB-α phosphorylation in SV40 large T-transformed fibroblasts in response to ionizing radiation. In nontransformed fibroblasts, however, Ashburner and colleagues 71 demonstrated a lack of involvement of ATM in IκB-α phosphorylation. In an in vitro study, Liu and colleagues 72 reported that DNA-dependent protein kinase (DNA-PK), a kinase phosphorylating p53 in response to DNA damage, was able to phosphorylate the carboxyl terminus of IκB-α protein. On the functional level, the first evidence of mutual functional regulation between NF-κB and p53 was from the observation that p53 could antagonize NF-κB activity by cross-competition for a limiting pool of the co-activator, p300. 65 In contrast, two recent studies indicated that p53 might activate NF-κB through an unknown mechanism 73 or stimulate the activity of NF-κB through induction of its target gene, p21Waf1, which inhibits cyclin E/Cdk2 activity and blocks its ability to compete with NF-κB for co-factors, such as p300 and CBP. 74
Further evidence indicating that apoptotic molecules regulate NF-κB comes from study of caspase cleavage of NF-κB p65 subunit or IκB-α protein. 75-77 The cleavage of p65 by caspase-3 leads to a loss of the carboxyl-terminal transactivation domain. 75 The carboxyl terminal truncated p65 is transcriptionally inactive. The cleavage of IκB-α by caspase-3 has been observed in γ-radiation-induced apoptosis and NF-κB inhibition-induced apoptosis. 77,78 A caspase-3 cleavage site has been identified in the region of amino acids 26 to 32 of human IκB-α and 32 to 37 of chicken IκB-α protein. 76 The cleavage site of caspase-3 on IκB-α with the amino acid sequence D-R-H-D-S resembles the consensus caspase-3 cleavage site, D-X-X-D-G/S/A, where X represents any amino acid residue. Cleavage of IκB-α by caspase-3 creates a N-terminal truncated IκB-α protein that is resistant to degradation by proteasome in response to inducers of NF-κB, but is able to bind to and suppress NF-κB. Therefore, the role of caspase-3 cleavage on IκB-α and p65 is to ensure that the anti-apoptotic gene is suppressed and apoptotic process is not interrupted once the cells are committed to apoptotic elimination.
In contrast, several studies indicate that caspases might also participate in the activation of NF-κB under certain circumstances. One example supporting this notion is the involvement of Dredd, a caspase encoded by Drosophila dredd gene, in the endoprotease cleavage of the Relish protein. 79 Structurally similar to human p100 and p105, two precursor proteins of NF-κB family, Relish contains a N-terminal Rel homology domain and a C-terminal IκB-like region. On lipopolysaccharide stimulation, Relish undergoes a rapid cleavage between the Rel homology domain and IκB-like region. Proteasome inhibitors failed to prevent the cleavage of Relish. In contrast, introducing a dominant-negative mutant of Dredd to inhibit the caspase activity of Dredd significantly blocked the cleavage of Relish, indicating caspase, rather than proteasome, is required for the activation of NF-κB-like protein in Drosophila. In addition, in mammalian cells, caspase-8, caspase-10, and MRIT, three death effector domain-containing proteins, have been shown to be able to activate upstream signals, such as NIK and IKK, leading to the activation of NF-κB. 80 This activity seems to be mainly dependent on the interaction between the prodomain of caspases and IKK. Furthermore, several recent studies suggested that other apoptosis-inducing proteins, such as Nod2, an Apaf-1 family member, and BclI0 and vCLAP, two caspase-recruitment domain-containing proteins, could also activate NF-κB through the interaction with IKK-γ subunit of IKK complex. 81-83 However, it is hard to reconcile these observations with the notions that NF-κB is repressed in the cells undergoing apoptosis. 33,77,78
NF-κB and Cell-Cycle Regulation
It has been known for decades that multiple signals are required to maintain proper cell growth and tissue homeostasis. 84 Most cells within a normal tissue may be forced out of the active cell cycling into a quiescent (G0) state from which they may re-enter cell cycling under some future circumstances. In a mature tissue, cells may be induced to terminal differentiation by relinquishing their proliferative or cell-cycling potential. Control of the orderly progression of dividing cells through the G1, S, G2, and M phases of the cell cycle in eukaryotic cells relies on a series of cell-cycle regulatory proteins, mainly cyclins that exert their function by binding to and activating a number of specific cyclin-dependent kinases (CDKs). The CDK activity is further modulated by kinases and phosphatases that phosphorylate and dephosphorylate CDK, respectively. Moreover, CDKs are subject to regulation by association with one of a number of specific CDK inhibitors or cell-cycle checkpoint proteins, such as p21Waf1, p16INK4a, p27Kip1, 85 and growth arrest and DNA-damage protein 45 (GADD45) (Figure 3) ▶ . 86
Figure 3.

Involvement of NF-κB in cell-cycle regulation. NF-κB may facilitate cell-cycle transition from G1 to S phase by antagonizing the activation or function of p53 and up-regulating cyclin D1 gene expression. NF-κB may also promote G2- to M-phase transition by down-regulating the expression of GADD45, a G2/M phase blocker that inhibits CDC2/cyclin B complex.→, activation; ⊣, inhibition.
Overwhelming evidence during recent years demonstrates that a variety of stress inducers, including DNA-damaging agents, activate checkpoint function of cells, leading to a cell-cycle arrest. Several checkpoints exist in the G1/S phase, G2 phase, and M phase of cell cycle. In mammalian cells, the control of the S-phase checkpoint requires the p53 tumor suppressor protein that governs the expression of CDK inhibitor, p21Waf1. 87,88 The activation of the G2/M phase checkpoint is dependent on the phosphorylation-dependent inactivation of CDC25C phosphatase by checkpoint kinases 1 or 2 (Chk1 or Chk2) and the induction of GADD45, an inhibitor for the G2/M phase cyclin B/CDC2 complex. 89,90 An additional checkpoint, the spindle checkpoint, has been identified in a later stage of M phase. 84,85 This checkpoint arrests mitotic progression if the spindle is not properly assembled, or if the chromosomes are not correctly oriented and attached to the spindle. All of the checkpoints are essential for maintaining genomic stability by allowing cells to have enough time to repair damage, thus, protecting the organism from the deleterious consequences of mutation.
The relationship between NF-κB and apoptosis has been intensively explored during the last few years, whereas only limited information is available regarding the possible involvement of NF-κB in cell-cycle regulation in cellular response to a variety of stress signals. A critical role for NF-κB in cell-cycle progression was suggested by earlier observations that NF-κB activity was elevated during the G0 to G1 cell-cycle transition in mouse fibroblasts. 91 A series of recent studies has begun to elucidate that in addition to fibroblasts, NF-κB activation was required for cell cycling in other types of cells, such as regenerating liver cells and estrogen receptor-negative breast cancer cells. 91-95 It was also found that the levels of NF-κB activation were linked to signaling that controls cell-cycle progression in HeLa cells and Jurkat T cells. 74,96 Inhibition of NF-κB caused impairment of cell-cycle progression in human glioma cells 97 and a retarded G1/S transition in HeLa cells. 98 The identification of NF-κB binding sites in the promoter region of cyclin D1 gene provided direct evidence for the contributions of NF-κB to the cell cycle. 50-52,95 Cyclin D1, in association with cyclin-dependent kinases, CDK4 and CDK6, promotes G1/S phase transition through CDK-dependent phosphorylation of pRb, thereby releasing the transcription factor E2F, which is required for the activation of S phase-specific genes. 99-101 Two NF-κB binding sites in the human cyclin D1 promoter have been identified. Inhibition of NF-κB by a degradation-resistant IκB-α caused a pronounced reduction of serum-induced cyclin D1 expression accompanied by a decrease of cyclin D1-associated kinase activity and delayed phosphorylation of pRb.
In contrast, several recent reports also indicated that NF-κB activation is necessary to cause cell-cycle arrest and/or induce cells to commit to terminal differentiation. Overexpression of NF-κB p65 or c-Rel arrests G1/S cell-cycle transition in pro-B cells and HeLa cells, respectively. 96,102 In HeLa cells, overexpression of c-Rel arrests cells at the G1/S phase because of the stabilization of p53 protein, which can subsequently activate the expression of p21Waf1, a potent inhibitor of CDK2. 96 The elevated levels of p21Waf1 correlated with the accumulation of the hypophosphorylated form of pRb and a decrease in E2F DNA binding. It is unclear how overexpression of c-Rel resulted in a prolonged half-life of p53 protein. In pro-B cells, although overexpression of c-Rel exhibited no effect on cell-cycle regulation, overexpression of p65 caused G1 arrest and subsequent apoptosis. This G1-arresting effect of p65, however, seems to be dependent on cell developmental stage, because overexpression of p65 did not cause G1 arrest in mature B cells. It remains unsettled whether manipulation of NF-κB signaling using protein overexpression can lead to consequences that are physiologically relevant. If it is, one may speculate that inhibition of NF-κB should cause over-cycling or hyperproliferation. Indeed, data obtained by gene inactivation of IKK-α in the mouse indicated an unexpected excessive proliferation of the skin basal layer because of the absence of epidermal differentiation. 103,104 NF-κB activity could not be found in keratinocytes from IKK-α-null mouse skin. These results point to a unique role for NF-κB in the epidermis, that is, NF-κB forces keratinocytes out of cell cycle and subsequent terminal differentiation. In this regard, the response of the epidermal keratinocytes to NF-κB seem to be opposite from that of other cell types, such as lymphocytes and macrophages, where NF-κB seems to promote cell-cycle transition. 50-52,95
Although most of the studies so far addressed the effects of NF-κB on G1/S phase regulation, the question of whether NF-κB also contributes to G2/M phase transition has not been explored. In a recent study in human bronchial epithelial cell line, BEAS-2B, we found that NF-κB inhibition by stable expression of a kinase mutated form of IKK-β potentiated toxic metal-induced G2/M cell-cycle arrest. 105 First, flow cytometric analysis demonstrated that at 48 hours after arsenite treatment, BEAS-2B cells expressing a kinase-mutated form of IKK-β showed a marked dose-dependent increase of cells arrested in the G2/M phase and a corresponding decrease in the number of cells in G1 phase. Second, a dose-dependent induction of GADD45 protein was observed in cells treated with arsenite. This induction of GADD45 by arsenite seems to be dependent on the activation of c-Jun-N-terminal kinase, because blockage of c-Jun-N-terminal kinase activation by expression of a dominant-negative SEK1 vector decreased the induction of GADD45. On the other hand, inhibition of NF-κB by expressing a kinase-mutated form of IKK-β increased GADD45 induction by arsenite. Third, analysis for the expression of CDC25 family members revealed that arsenite induced de novo CDC25A expression, but markedly reduced the levels of CDC25B and CDC25C proteins, two phosphatases dephosphorylating and activating CDC2/cyclin B complex required for the transition of the cell cycle from G2 to M phase. The effects of Cr(VI) on the regulation of the cell cycle were also determined and revealed to be more complicated. Although Cr(VI) was able to induce GADD45 and suppress both CDC25B and CDC25C, it had no effect on CDC25A. Cell-cycle-profiling studies showed that whereas a lower concentration of Cr(VI) (0.25 μg/ml) promoted cell-cycle transition, higher concentrations of Cr(VI) (1 to 4 μg/ml) arrested cells at S phase. In the case of vanadate-induced cell-cycle regulation, the cell-cycle-arresting effect of vanadate seems to be dependent on the status of NF-κB activation. In normal epithelial cells, vanadate exhibited less effect on cell-cycle transition. However, in the cells where NF-κB activation was specifically inhibited, vanadate showed a marked G2/M phase-arresting effect. However, vanadate was unable to induce the expression of GADD45, an inhibitor of cyclin B/CDC2 complex required for G2/M transition (Chen et al, unpublished observations).
NF-κB and Oncogenesis
The ability of NF-κB to suppress apoptosis and to regulate cell-cycle transition clearly indicates that NF-κB may participate in many aspects of oncogenesis. Indeed, elevation of NF-κB activity is evident in a number of human cancers, including breast cancer, 106 non-small cell lung carcinoma, 107 thyroid cancer, 108 T- or B- lymphocyte leukemia, 109 melanoma, 110 colon cancer, 111 bladder cancer, 112 and several virally induced tumors. 113-115 The earliest evidence for a role for NF-κB in oncogenic transformation has been derived from the fact that v-Rel, a highly oncogenic retroviral homologue of c-Rel, causes carcinogenesis in avian lymphoid cells. 108 Later studies suggested that v-Rel also has the capacity of transforming mammalian cells in vivo. 116 Transgenic mice expressing v-Rel under the control of the T-cell-specific lck promoter develop T-cell lymphomas. Inhibition of NF-κB by overexpression of a degradation-resistant IκB-α delays the development of T-cell lymphomas and prolongs the survival of v-Rel transgenic mice. 116
Chromosomal alterations of NF-κB family genes provided additional evidence for the role of NF-κB in oncogenesis. It has been demonstrated that genes encoding c-Rel, NF-κB2 (p100/p52), p65/RelA, and Bcl-3 proteins are all located within breakpoint regions of the genome that are involved in oncogenic rearrangements or amplifications. Rearrangement of nfkb2 gene by t(10,14) chromosomal translocation causes deletions of sequences encoding the ankyrin repeat motif of p100. Consequently, this carboxyl terminal truncated p100 is constitutively located in the nucleus of cells, which has been originally found in a case of B-cell non-Hodgkin’s lymphoma and observed in a number of lymphoid neoplasms, particularly cutaneous lymphomas. 117-119 Rearrangement and amplification of c-Rel gene has also been found in numerous non-Hodgkin’s lymphomas and cancer cell lines. 108,120 The bcl-3 gene, which encodes an IκB-like protein that regulates transcriptional activity of NF-κB p50 or p52 homodimer, was identified as a [t(14, 19)(q32;q13.1)] chromosomal translocation in many cases of chronic lymphocytic leukemia. 121 Unlike rearrangement of the nfkb2 gene, alterations at the bcl-3 locus do not truncate or change the coding sequence, but rather cause overexpression of bcl-3 mRNA. In malignant Hodgkin and Reed-Sternberg (H/RS) cells from Hodgkin’s lymphoma, mutations in the IκB-α gene have been detected and are suggested to cause a sustained activation of NF-κB. 122
Accumulating evidence reveals that tumorigenesis or oncogenesis is a multistep process and that these steps reflect defections in regulatory circuits that govern normal cell proliferation, differentiation, and death. 123 Although abnormal activation or function of NF-κB has been clearly demonstrated in the initiation or facilitation of oncogenesis, the central question that has to be answered is: how many and what steps are influenced by NF-κB. NF-κB has been shown to antagonize the function of p53 as discussed earlier. 65 Obviously, this antagonism of p53 by NF-κB will result in the evasion of cells from stress-induced cell-cycle arrest and/or programmed cell death and consequently sensitize the cells for genomic instability. Furthermore, NF-κB could promote cell-cycle transition by a direct transcriptional up-regulation of the cyclin D1 gene. 50-52,95 Although it remains to be confirmed, this increased expression of cyclin D1 may possibly provide cells with an uncontrolled or limitless replicative potential. Up-regulation of anti-apoptotic genes, such as cIAP1, cIAP2, XIAP, and bcl-xl, by NF-κB, 4 is an additional mean of cells to escape from or resist signal-induced apoptosis. Other NF-κB-regulated genes include those encoding intercellular adhesion molecule-1, 5 extracellular matrix protein tenascin-C, 124 vascular endothelial growth factor, 124 chemokines, and cyclooxygenase-2. 124 These gene products are directly associated with the tumor cell metastasis and tumor tissue angiogenesis.
The key role that NF-κB plays on multiple steps of oncogenesis makes this factor a central and favorable target for therapeutic intervention of cancer, especially, certain types of leukemia or lymphomas. 125 Indeed, experimental data suggest that inhibition of NF-κB by antisense oligonucleotides to relA, degradation resistant IκB-α, and aspirin or nonsteroidal anti-inflammatory drugs, could enhance the efficacy of cancer chemotherapies and radiation. 126,127 Studies by Wang and co-workers 55,128 showed that inhibition of NF-κB by infecting the cells with an adenovirus carrying a modified form of IκB-α (superrepressor IκB-α) leads to dramatically enhanced apoptosis of HT1080 fibrosarcoma cells in response to ionizing radiation or daunorubicin treatment. Consistent with these reports, working with pancreatic cancer cell lines exposed to VP16 or doxorubicin, Arlt and colleagues 129 demonstrated recently that NF-κB inhibition by pharmacological proteasome inhibitors or transfection of the cells with a N-terminal-truncated IκB-α variant efficiently reduces chemoresistance of these cells. Using similar or different approaches to inhibit NF-κB, this effect has also been noted in a variety of other cell types including non-small cell lung cancers, 130 head and neck squamous carcinomas, 131 human myeloblastic leukemia cells, 132 colorectal cancer, 133 and bladder cancer cells. 112 Despite these encouraging observations, however, care has to be taken when using different approaches to inhibit NF-κB that might be attributable to the process of oncogenesis. Indeed, different approaches for the inhibition of NF-κB does not necessarily lead to the same extents of inactivation of NF-κB because of the existence of functionally and stoichiometrically different NF-κB complexes that respond to different activation signals. 5 Also, the inhibitory effect of NF-κB inhibitors can vary considerably between different cell types because of unique simultaneous or asynchronous events triggered by these inhibitors in any given cell type. 134
Summary
The detailed molecular mechanisms by which the NF-κB transcription factor contributes to cell growth control, such as cell apoptosis, cell-cycle transition, and oncogenesis remain to be further determined. One of the major challenges in understanding mechanisms of cell growth regulation by NF-κB in response to environmental stress is to elucidate how signal transduction pathways are activated and how signaling cross-talk and specificity are achieved when several signaling pathways are activated simultaneously by stress inducers that elicit different cellular responses. For instance, why does activation of the NF-κB, an anti-apoptotic transcription factor, coincide with obvious apoptotic features in cells undergoing stress responses? Because many stress inducers and their mediators are highly reactive but nonspecific, an activation of only one specific signaling pathway is hard to achieve in the cells in response to a particular inducer. Even in a single signaling pathway, because of their highly reactive and nonspecific characteristics, certain stress inducers and their mediators can in principle induce conflicting signals by affecting signaling molecules at different levels. A good example is the effects of oxidative stress on NF-κB signaling pathway. It has been frequently observed in certain types of cells that oxidative stress amplified or potentiated NF-κB activation, whereas at the same time oxidation of IKK or NF-κB proteins inhibited NF-κB function. Translating the knowledge gained by studying the connections among NF-κB activation, cell apoptosis, cell-cycle regulation, and oncogenesis may aid in identifying novel preventive and therapeutic measures for diseases, such as chronic inflammation and cancer.
Acknowledgments
We thank our colleague Dr Murali Rao for helpful suggestions and critique of the manuscript; and to the many friends whose primary valuable work in the related fields we could not directly acknowledge because of space constrains. Dr. Fei Chen thanks the Health Effects Laboratory Division of National Institute for Occupational Safety and Health for support through a cooperative agreement from the Association of Teachers of Preventive Medicine and the Centers for Disease Control and Prevention of the United States.
Footnotes
Address reprint requests to Dr. Fei Chen, PPRB of NIOSH, 1095 Willowdale Rd., Morgantown, WV 26505. E-mail: lfd3@cdc.gov.
Supported by a Career Development Award in Genetics under a cooperative agreement from the Centers for Disease Control and Prevention through the Association of Teachers of Preventive Medicine (to F. C.).
References
- 1.Zandi E, Karin M: Bridging the gap: composition, regulation, and physiological function of the IκB kinase complex. Mol Cell Biol 1999, 19:4547-4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karin M, Ben-Neriah Y: Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 2000, 18:621-663 [DOI] [PubMed] [Google Scholar]
- 3.Israel A: The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol 2000, 10:129-133 [DOI] [PubMed] [Google Scholar]
- 4.Chen F, Castranova V, Shi X, Demers LM: New insights into the role of nuclear factor-κB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem 1999, 45:7-17 [PubMed] [Google Scholar]
- 5.Baldwin AS, Jr: The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 1996, 14:649-683 [DOI] [PubMed] [Google Scholar]
- 6.Ghosh S, May MJ, Kopp EB: NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998, 16:225-260 [DOI] [PubMed] [Google Scholar]
- 7.Siebenlist U, Franzoso G, Brown K: Structure, regulation and function of NF-κB. Annu Rev Cell Biol 1994, 10:405-455 [DOI] [PubMed] [Google Scholar]
- 8.Pahl HL: Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18:6853-6866 [DOI] [PubMed] [Google Scholar]
- 9.Gilmore TD: The Rel/NF-κB signal transduction pathway: introduction. Oncogene 1999, 18:6842-6844 [DOI] [PubMed] [Google Scholar]
- 10.Sun SC, Ballard DW: Persistent activation of NF-κB by the tax transforming protein of HTLV-1: hijacking cellular IκB kinases. Oncogene 1999, 18:6948-6958 [DOI] [PubMed] [Google Scholar]
- 11.Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, Rao AS, Madara JL: Prokaryotic regulation of epithelial responses by inhibition of IκBα ubiquitination. Science 2000, 289:1560-1563 [DOI] [PubMed] [Google Scholar]
- 12.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T: The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 1994, 78:773-785 [DOI] [PubMed] [Google Scholar]
- 13.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV: IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science 1997, 278:866-869 [DOI] [PubMed] [Google Scholar]
- 14.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A: IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 1997, 278:860-866 [DOI] [PubMed] [Google Scholar]
- 15.Cohen L, Henzel WJ, Baeuerle PA: IKAP is a scaffold protein of the IκB kinase complex. Nature 1998, 395:292-296 [DOI] [PubMed] [Google Scholar]
- 16.Krappmann D, Hatada EN, Tegethoff S, Li J, Klippel A, Giese K, Baeuerle PA, Scheidereit C: The IκB kinase (IKK) complex is tripartite and contains IKKγ but not IKAP as a regular component. J Biol Chem 2000, 275:29779-29787 [DOI] [PubMed] [Google Scholar]
- 17.Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, Haag M, Stark GR: Act1, an NF-κB-activating protein. Proc Natl Acad Sci USA 2000, 97:10489-10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U: CIKS, a connection to IκB kinase and stress-activated protein kinase. Proc Natl Acad Sci USA 2000, 97:10494-10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, Kaneko Y, Nimura Y, Motoyama N, Ikeda K, Karin M, Nakanishi M: NAK is an IκB kinase-activating kinase. Nature 2000, 404:778-782 [DOI] [PubMed] [Google Scholar]
- 20.Huang S, DeGuzman A, Bucana CD, Fidler IJ: Nuclear factor-κB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin Cancer Res 2000, 6:2573-2581 [PubMed] [Google Scholar]
- 21.Strasser A, O’Connor L, Dixit VM: Apoptosis signaling. Annu Rev Biochem 2000, 69:217-245 [DOI] [PubMed] [Google Scholar]
- 22.Ashkenazi A, Dixit VM: Death receptors: signaling and modulation. Science 1998, 281:1305-1308 [DOI] [PubMed] [Google Scholar]
- 23.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 24.De Laurenzi V, Melino G: Apoptosis. The little devil of death. Nature 2000, 406:135-136 [DOI] [PubMed] [Google Scholar]
- 25.Du C, Fang M, Li Y, Li L, Wang X: Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102:33-42 [DOI] [PubMed] [Google Scholar]
- 26.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL: Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102:43-53 [DOI] [PubMed] [Google Scholar]
- 27.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G: Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397:441-446 [DOI] [PubMed] [Google Scholar]
- 28.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17:3247-3259 [DOI] [PubMed] [Google Scholar]
- 29.Reed JC: Bcl-2 family proteins. Oncogene 1998, 17:3225-3236 [DOI] [PubMed] [Google Scholar]
- 30.Dahl AM, Klein C, Andres PG, London CA, Lodge MP, Mulligan RC, Abbas AK: Expression of bcl-X(L) restores cell survival, but not proliferation off effector differentiation, in CD28-deficient T lymphocytes. J Exp Med 2000, 191:2031-2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B: A model for p53-induced apoptosis. Nature 1997, 389:300-305 [DOI] [PubMed] [Google Scholar]
- 32.Amundson SA, Myers TG, Fornace AJ, Jr: Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene 1998, 17:3287-3299 [DOI] [PubMed] [Google Scholar]
- 33.Gu Z, Flemington C, Chittenden T, Zambetti GP: ei24, a p53 response gene involved in growth suppression and apoptosis. Mol Cell Biol 2000, 20:233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N: Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288:1053-1058 [DOI] [PubMed] [Google Scholar]
- 35.Barkett M, Gilmore TD: Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 1999, 18:6910-6924 [DOI] [PubMed] [Google Scholar]
- 36.Aggarwal BB: Apoptosis and nuclear factor-κB: a tale of association and dissociation. Biochem Pharmacol 2000, 60:1033-1039 [DOI] [PubMed] [Google Scholar]
- 37.Hsu SC, Gavrilin MA, Lee HH, Wu CC, Han SH, Lai MZ: NF-κB-dependent Fas ligand expression. Eur J Immunol 1999, 29:2948-2956 [DOI] [PubMed] [Google Scholar]
- 38.Matsui K, Fine A, Zhu B, Marshak-Rothstein A, Ju ST: Identification of two NF-κB sites in mouse CD95 ligand (Fas ligand) promoter: functional analysis in T cell hybridoma. J Immunol 1998, 161:3469-3473 [PubMed] [Google Scholar]
- 39.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D: Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376:167-170 [DOI] [PubMed] [Google Scholar]
- 40.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M: The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J Exp Med 1999, 189:1839-1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV: Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity 1999, 10:421-429 [DOI] [PubMed] [Google Scholar]
- 42.Li Q, Estepa G, Memet S, Israel A, Verma IM: Complete lack of NF-κB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev 2000, 14:1729-1733 [PMC free article] [PubMed] [Google Scholar]
- 43.Gerondakis S, Grossmann M, Nakamura Y, Pohl T, Grumont R: Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene 1999, 18:6888-6895 [DOI] [PubMed] [Google Scholar]
- 44.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M: Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell 2000, 5:969-979 [DOI] [PubMed] [Google Scholar]
- 45.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M: NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol Cell 2000, 5:981-992 [DOI] [PubMed] [Google Scholar]
- 46.Rosenfeld ME, Prichard L, Shiojiri N, Fausto N: Prevention of hepatic apoptosis and embryonic lethality in RelA/TNFR-1 double knockout mice. Am J Pathol 2000, 156:997-1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y: Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci USA 1999, 96:2994-2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM: Severe liver degeneration in mice lacking the IkB kinase 2 gene. Science 1999, 284:321-325 [DOI] [PubMed] [Google Scholar]
- 49.Fausto N, Laird AD, Webber EM: Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J 1995, 9:1527-1536 [DOI] [PubMed] [Google Scholar]
- 50.Joyce D, Bouzahzah B, Fu M, Albanese C, D’Amico M, Steer J, Klein JU, Lee RJ, Segall JE, Westwick JK, Der CJ, Pestell RG: Integration of Rac-dependent regulation of cyclin D1 transcription through a nuclear factor-κB-dependent pathway. J Biol Chem 1999, 274:25245-25249 [DOI] [PubMed] [Google Scholar]
- 51.Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M: NF-κB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol 1999, 19:2690-2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS, Jr: NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 1999, 19:5785-5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen F, Demers LM, Vallyathan V, Lu Y, Castranova V, Shi X: Involvement of 5′-flanking κB-like sites within bcl-x gene in silica-induced Bcl-x expression. J Biol Chem 1999, 274:35591-35595 [DOI] [PubMed] [Google Scholar]
- 54.Chen C, Edelstein LC, Gelinas C: The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol 2000, 20:2687-2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS, Jr: NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281:1680-1683 [DOI] [PubMed] [Google Scholar]
- 56.Ivanov VN, Lee RK, Podack ER, Malek TR: Regulation of Fas-dependent activation-induced T cell apoptosis by cAMP signaling: a potential role for transcription factor NF-κB. Oncogene 1997, 14:2455-2464 [DOI] [PubMed] [Google Scholar]
- 57.Kasibhatla S, Brunner T, Genestier L, Echeverri F, Mahboubi A, Green DR: DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-κB and AP-1. Mol Cell 1998, 1:543-551 [DOI] [PubMed] [Google Scholar]
- 58.Rivera-Walsh I, Cvijic ME, Xiao G, Sun SC: The NF-κB signaling pathway is not required for Fas ligand gene induction but mediates protection from activation-induced cell death. J Biol Chem 2000, 275:25222-25230 [DOI] [PubMed] [Google Scholar]
- 59.Latinis KM, Norian LA, Eliason SL, Koretzky GA: Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J Biol Chem 1997, 272:31427-31434 [DOI] [PubMed] [Google Scholar]
- 60.Bales KR, Du Y, Dodel RC, Yan GM, Hamilton-Byrd E, Paul SM: The NF-κB/Rel family of proteins mediates Ab-induced neurotoxicity and glial activation. Brain Res Mol Brain Res 1998, 57:63-72 [DOI] [PubMed] [Google Scholar]
- 61.Guo Q, Robinson N, Mattson MP: Secreted b-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-κB and stabilization of calcium homeostasis. J Biol Chem 1998, 273:12341-12351 [DOI] [PubMed] [Google Scholar]
- 62.Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP: Lack of the p50 subunit of nuclear factor-κB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci 1999, 19:8856-8865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mattson MP, Camandola S: NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 2001, 107:247-254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang JP, Hori M, Takahashi N, Kawabe T, Kato H, Okamoto T: NF-κB subunit p65 binds to 53BP2 and inhibits cell death induced by 53BP2. Oncogene 1999, 18:5177-5186 [DOI] [PubMed] [Google Scholar]
- 65.Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T: CREB-binding protein is a nuclear integrator of nuclear factor-κB and p53 signaling. J Biol Chem 1999, 274:1879-1882 [DOI] [PubMed] [Google Scholar]
- 66.Shao J, Fujiwara T, Kadowaki Y, Fukazawa T, Waku T, Itoshima T, Yamatsuji T, Nishizaki M, Roth JA, Tanaka N: Overexpression of the wild-type p53 gene inhibits NF-κB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19:726-736 [DOI] [PubMed] [Google Scholar]
- 67.Ariumi Y, Kaida A, Lin JY, Hirota M, Masui O, Yamaoka S, Taya Y, Shimotohno K: HTLV-1 tax oncoprotein represses the p53-mediated trans-activation function through coactivator CBP sequestration. Oncogene 2000, 19:1491-1499 [DOI] [PubMed] [Google Scholar]
- 68.Pise-Masison CA, Mahieux R, Jiang H, Ashcroft M, Radonovich M, Duvall J, Guillerm C, Brady JN: Inactivation of p53 by human T-cell lymphotropic virus type 1 Tax requires activation of the NF-κB pathway and is dependent on p53 phosphorylation. Mol Cell Biol 2000, 20:3377-3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, Takahashi T, Obata Y, Yoshioka K, Yamamoto K: p300/CBP-dependent and -independent transcriptional interference between NF-κB RelA and p53. Biochem Biophys Res Commun 2000, 272:375-379 [DOI] [PubMed] [Google Scholar]
- 70.Jung M, Kondratyev A, Lee SA, Dimtchev A, Dritschilo A: ATM gene product phosphorylates IκB-α. Cancer Res 1997, 57:24-27 [PubMed] [Google Scholar]
- 71.Ashburner BP, Shackelford RE, Baldwin AS, Jr, Paules RS: Lack of involvement of ataxia telangiectasia mutated (ATM) in regulation of nuclear factor-kB (NF-kB) in human diploid fibroblasts. Cancer Res 1999, 59:5456-5460 [PubMed] [Google Scholar]
- 72.Liu L, Kwak YT, Bex F, Garcia-Martinez LF, Li XH, Meek K, Lane WS, Gaynor RB: DNA-dependent protein kinase phosphorylation of IκBα and IκBβ regulates NF-κB DNA binding properties. Mol Cell Biol 1998, 18:4221-4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ryan KM, Ernst MK, Rice NR, Vousden KH: Role of NF-κB in p53-mediated programmed cell death. Nature 2000, 404:892-897 [DOI] [PubMed] [Google Scholar]
- 74.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ: Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator. Science 1997, 275:523-527 [DOI] [PubMed] [Google Scholar]
- 75.Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW: Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-κB loop. Nat Cell Biol 1999, 1:227-233 [DOI] [PubMed] [Google Scholar]
- 76.Barkett M, Xue D, Horvitz HR, Gilmore TD: Phosphorylation of IκBα inhibits its cleavage by caspase CPP32 in vitro. J Biol Chem 1997, 272:29419-29422 [DOI] [PubMed] [Google Scholar]
- 77.Reuther JY, Baldwin AS, Jr: Apoptosis promotes a caspase-induced amino-terminal truncation of IκBα that functions as a stable inhibitor of NF-κB. J Biol Chem 1999, 274:20664-20670 [DOI] [PubMed] [Google Scholar]
- 78.Jung M, Zhang Y, Dimtchev A, Dritschilo A: Impaired regulation of nuclear factor-κB results in apoptosis induced by γ radiation. Radiat Res 1998, 149:596-601 [PubMed] [Google Scholar]
- 79.Silverman N, Zhou R, Stoven S, Pandey N, Hultmark D, Maniatis T: A drosophila IκB kinase complex required for relish cleavage and antibacterial immunity. Genes Dev 2000, 14:2461-2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L: Activation of the NF-κB pathway by caspase 8 and its homologs. Oncogene 2000, 19:4451-4460 [DOI] [PubMed] [Google Scholar]
- 81.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G: Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem 2000, 276:4812-4818 [DOI] [PubMed] [Google Scholar]
- 82.Ruland J, Duncan GS, Elia A, del Barco Barrantes I, Nguyen L, Plyte S, Millar DG, Bouchard D, Wakeham A, Ohashi PS, Mak TW: Bcl10 is a positive regulator of antigen receptor-induced activation of NF-κB and neural tube closure. Cell 2001, 104:33-42 [DOI] [PubMed] [Google Scholar]
- 83.Poyet JL, Srinivasula SM, Alnemri ES: vCLAP, a caspase recruitment domain-containing protein of equine herpesvirus-2, persistently activates the IκB kinases through oligomerization of IKKγ. J Biol Chem 2001, 276:3183-3187 [DOI] [PubMed] [Google Scholar]
- 84.Nurse P: A long twentieth century of the cell cycle and beyond. Cell 2000, 100:71-78 [DOI] [PubMed] [Google Scholar]
- 85.Shackelford RE, Kaufmann WK, Paules RS: Cell cycle control, checkpoint mechanisms, and genotoxic stress. Environ Health Perspect 1999, 107(Suppl 1):S5-S24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sheikh MS, Hollander MC, Fornance AJ, Jr: Role of Gadd45 in apoptosis. Biochem Pharmacol 2000, 59:43-45 [DOI] [PubMed] [Google Scholar]
- 87.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE: P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer 2000, 83:817-825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jimenez GS, Khan SH, Stommel JM, Wahl GM: p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 1999, 18:7656-7665 [DOI] [PubMed] [Google Scholar]
- 89.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O’Connor PM, Piwnica-Worms H: The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 2000, 275:5600-5605 [DOI] [PubMed] [Google Scholar]
- 90.Hutchins JR, Hughes M, Clarke PR: Substrate specificity determinants of the checkpoint protein kinase Chk1. FEBS Lett 2000, 466:91-95 [DOI] [PubMed] [Google Scholar]
- 91.Baldwin AS, Jr, Azizkhan JC, Jensen DE, Beg AA, Coodly LR: Induction of NF-κB DNA-binding activity during the G0-to-G1 transition in mouse fibroblasts. Mol Cell Biol 1991, 11:4943-4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cressman DE, Greenbaum LE, Haber BA, Taub R: Rapid activation of post-hepatectomy factor/nuclear factor κB in hepatocytes, a primary response in the regenerating liver. J Biol Chem 1994, 269:30429-30435 [PubMed] [Google Scholar]
- 93.Duckett CS, Perkins ND, Leung K, Agranoff AB, Nabel GJ: Cytokine induction of nuclear factor κB in cycling and growth-arrested cells. Evidence for cell cycle-independent activation. J Biol Chem 1995, 270:18836-18840 [DOI] [PubMed] [Google Scholar]
- 94.Yamada Y, Kirillova I, Peschon JJ, Fausto N: Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA 1997, 94:1441-1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Biswas DK, Cruz AP, Gansberger E, Pardee AB: Epidermal growth factor-induced nuclear factor κB activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci USA 2000, 97:8542-8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bash J, Zong WX, Gelinas C: c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol Cell Biol 1997, 17:6526-6536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Otsuka G, Nagaya T, Saito K, Mizuno M, Yoshida J, Seo H: Inhibition of nuclear factor-κB activation confers sensitivity to tumor necrosis factor-a by impairment of cell cycle progression in human glioma cells. Cancer Res 1999, 59:4446-4452 [PubMed] [Google Scholar]
- 98.Kaltschmidt B, Kaltschmidt C, Hehner SP, Droge W, Schmitz ML: Repression of NF-κB impairs HeLa cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene 1999, 18:3213-3225 [DOI] [PubMed] [Google Scholar]
- 99.Nevins JR: E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258:424-429 [DOI] [PubMed] [Google Scholar]
- 100.Sherr CJ, Roberts JM: Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995, 9:1149-1163 [DOI] [PubMed] [Google Scholar]
- 101.Weinberg RA: The retinoblastoma protein and cell cycle control. Cell 1995, 81:323-330 [DOI] [PubMed] [Google Scholar]
- 102.Sheehy AM, Schlissel MS: Overexpression of RelA causes G1 arrest and apoptosis in a pro-B cell line. J Biol Chem 1999, 274:8708-8716 [DOI] [PubMed] [Google Scholar]
- 103.Takeda K, Takeuchi O, Tsujimura T, Itami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S: Limb and skin abnormalities in mice lacking IKKα. Science 1999, 284:313-316 [DOI] [PubMed] [Google Scholar]
- 104.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M: Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science 1999, 284:316-320 [DOI] [PubMed] [Google Scholar]
- 105.Chen F, Lu Y, Zhang Z, Vallyathan V, Ding M, Castranova V, Shi X: Opposite effect of NF-κB and c-Jun-N-terminal kinase on p53-independent GADD45 induction by arsenite. J Biol Chem 2001, 276:11414-11419 [DOI] [PubMed] [Google Scholar]
- 106.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE: Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest 1997, 100:2952-2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mukhopadhyay T, Roth JA, Maxwell SA: Altered expression of the p50 subunit of the NF-κB transcription factor complex in non-small cell lung carcinoma. Oncogene 1995, 11:999-1003 [PubMed] [Google Scholar]
- 108.Gilmore TD, Koedood M, Piffat KA, White DW: Rel/NF-κB/IκB proteins and cancer. Oncogene 1996, 13:1367-1378 [PubMed] [Google Scholar]
- 109.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, Royer HD, Scheidereit C, Dorken B: High-level nuclear NF-κB and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood 1996, 87:4340-4347 [PubMed] [Google Scholar]
- 110.Devalaraja MN, Wang DZ, Ballard DW, Richmond A: Elevated constitutive IκB kinase activity and IκBα phosphorylation in Hs294T melanoma cells lead to increased basal MGSA/GRO-α transcription. Cancer Res 1999, 59:1372-1377 [PubMed] [Google Scholar]
- 111.Dejardin E, Deregowski V, Chapelier M, Jacobs N, Gielen J, Merville MP, Bours V: Regulation of NF-κB activity by IκB-related proteins in adenocarcinoma cells. Oncogene 1999, 18:2567-2577 [DOI] [PubMed] [Google Scholar]
- 112.Sumitomo M, Tachibana M, Ozu C, Asakura H, Murai M, Hayakawa M, Nakamura H, Takayanagi A, Shimizu N: Induction of apoptosis of cytokine-producing bladder cancer cells by adenovirus-mediated IκBα overexpression. Hum Gene Ther 1999, 10:37-47 [DOI] [PubMed] [Google Scholar]
- 113.Miwa M, Kushida S, Maeda N, Fang J, Kawamura T, Kameyama T, Uchida K: Pathogenesis and prevention of HTLV-1-associated diseases. Leukemia 1997, 11(Suppl 3):S65-S66 [PubMed] [Google Scholar]
- 114.Berger C, Brousset P, McQuain C, Knecht H: Deletion variants within the NF-κB activation domain of the LMP1 oncogene in acquired immunodeficiency syndrome-related large cell lymphomas, in prelymphomas and atypical lymphoproliferations. Leuk Lymphoma 1997, 26:239-250 [DOI] [PubMed] [Google Scholar]
- 115.Nasti G, Vaccher E, Errante D, Tirelli U: Malignant tumors and AIDS. Biomed Pharmacother 1997, 51:243-251 [DOI] [PubMed] [Google Scholar]
- 116.Carrasco D, Rizzo CA, Dorfman K, Bravo R: The v-rel oncogene promotes malignant T-cell leukemia/lymphoma in transgenic mice. EMBO J 1996, 15:3640-3650 [PMC free article] [PubMed] [Google Scholar]
- 117.Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RS, Dalla-Favera R: B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell 1991, 67:1075-1087 [DOI] [PubMed] [Google Scholar]
- 118.Fracchiolla NS, Lombardi L, Salina M, Migliazza A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A: Structural alterations of the NF-κB transcription factor lyt-10 in lymphoid malignancies. Oncogene 1993, 8:2839-2845 [PubMed] [Google Scholar]
- 119.Liptay S, Seriu T, Bartram CR, Schmid RM: Germline configuration of nfkb2, c-rel and bcl3 in childhood acute lymphoblastic leukemia (ALL). Leukemia 1997, 11:1364-1366 [DOI] [PubMed] [Google Scholar]
- 120.Houldsworth J, Mathew S, Rao PH, Dyomina K, Louie DC, Parsa N, Offit K, Chaganti RS: REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood 1996, 87:25-29 [PubMed] [Google Scholar]
- 121.Michaux L, Dierlamm J, Wlodarska I, Bours V, Van den Berghe H, Hagemeijer A: t(14;19)/BCL3 rearrangements in lymphoproliferative disorders: a review of 23 cases. Cancer Genet Cytogenet 1997, 94:36-43 [DOI] [PubMed] [Google Scholar]
- 122.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT: Mutations in the IκBα gene in Hodgkin’s disease suggest a tumour suppressor role for IκBα. Oncogene 1999, 18:3063-3070 [DOI] [PubMed] [Google Scholar]
- 123.Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000, 100:57-70 [DOI] [PubMed] [Google Scholar]
- 124.Mettouchi A, Cabon F, Montreau N, Dejong V, Vernier P, Gherzi R, Mercier G, Binetruy B: The c-Jun-induced transformation process involves complex regulation of tenascin-C expression. Mol Cell Biol 1997, 17:3202-3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schwartz SA, Hernandez A, Mark Evers B: The role of NF-κB/IκB proteins in cancer: implications for novel treatment strategies. Surg Oncol 1999, 8:143-153 [DOI] [PubMed] [Google Scholar]
- 126.Mayo MW, Baldwin AS: The transcription factor NF-κB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta 2000, 1470:M55-M62 [DOI] [PubMed] [Google Scholar]
- 127.Baldwin AS: Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest 2001, 107:241-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang CY, Cusack JC, Liu R, Baldwin AS: Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nat Med 1999, 5:412-417 [DOI] [PubMed] [Google Scholar]
- 129.Arlt A, Vorndamm J, Breitenbroich M, Folsch UR, Kalthoff H, Schmidt WE, Schafer H: Inhibition of the NF-κB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 2001, 20:859-868 [DOI] [PubMed] [Google Scholar]
- 130.Jones DR, Broad RM, Madrid LV, Baldwin AS, Mayo MW: Inhibition of NF-κB sensitizes non-small cell lung cancer cells to chemotherapy-induced apoptosis. Ann Thorac Surg 2000, 70:930-937 [DOI] [PubMed] [Google Scholar]
- 131.Kato T, Duffey DC, Ondrey FG, Dong G, Chen Z, Cook JA, Mitchell JB, Van Waes C: Cisplatin and radiation sensitivity in human head and neck squamous carcinomas are independently modulated by glutathione and transcription factor NF-κB. Head Neck 2000, 22:748-759 [DOI] [PubMed] [Google Scholar]
- 132.Romano MF, Lamberti A, Bisogni R, Tassone P, Pagnini D, Storti G, Del Vecchio L, Turco MC, Venuta S: Enhancement of cytosine arabinoside-induced apoptosis in human myeloblastic leukemia cells by NF-κB/Rel-specific decoy oligodeoxynucleotides. Gene Ther 2000, 7:1234-1237 [DOI] [PubMed] [Google Scholar]
- 133.Cusack JC, Liu R, Baldwin AS: Inducible chemoresistance to 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxycamptothe cin (CPT-11) in colorectal cancer cells and a xenograft model is overcome by inhibition of nuclear factor-κB activation. Cancer Res 2000, 60:2323-2330 [PubMed] [Google Scholar]
- 134.Bours V, Bentires-Alj M, Hellin AC, Viatour P, Robe P, Delhalle S, Benoit V, Merville MP: Nuclear factor-κB, cancer, and apoptosis. Biochem Pharmacol 2000, 60:1085-1089 [DOI] [PubMed] [Google Scholar]