

Abstract
We report the clinical evolution of a prostate cancer, metastasizing to lungs and bones, recurring locally, and escaping from anti-androgen therapy. Key event of biological progression of the patient’s tumor was the coincidence of allelic imbalance accumulation and of bone metastases occurrence. The recurrent tumor was established as the transplantable xenograft PAC120 in nude mice, where it grew locally. PAC120 displayed the same immunophenotype of the original tumor (positive for keratin, vimentin, prostatic acid phosphatase, and Leu-7) and expressed human HOXB9, HOXA4, HER-2/neu, and prostate-specific antigen genes, as detected by reverse transcriptase-polymerase chain reaction. It formed lung micrometastases detected by mRNA expression of human genes. Cytogenetic analysis demonstrated numerous alterations reflecting the tumor evolution. PAC120 was still hormone-dependent; its growth was strongly inhibited by the new gonadotropin-releasing hormone antagonist FE 200486 but weakly by gonadotropin-releasing hormone superagonist D-Trp6-luteinizing-hormone releasing hormone (decapeptyl). Tumor growth inhibition induced by anti-hormone therapy was linked to the hormone deprivation degree, more important and more stable with FE 200486 than with D-Trp6-luteinizing-hormone releasing hormone. Surgical castration of mice led to tumor regressions but did not prevent late recurrences. Transition to hormone-independent tumors was frequently associated with a mucoid differentiation or with a neuroendocrine-like pattern. Independent variations of mRNA expression of HER-2/neu and prostate-specific antigen were observed in hormone-independent tumors whereas HOXB9 gene expression was constant. In conclusion, PAC120 xenograft, a new model of hormone-dependent prostate cancer retained the progression potential of the original tumor, opening the opportunity to study the hormone dependence escape mechanism.
Prostate cancer is an increasing dilemma because of its frequency and of the difficulties encountered in its treatment. 1 Research into prostate cancer biology leading to improved therapeutic approaches would benefit from a greater availability of models. 2 Transplantation of human prostate cancer tissue into athymic nude mice is rarely successful, and even when the implanted tissue survives, it usually fails to be maintained by serial transplantation. Establishment of prostatic cancer cell lines is also not easier. Thus, most biological or preclinical studies have been conducted using a limited number of cell lines: DU-145, 3 , PC-3, 4 and LnCaP. 5 Similarly, a very limited number of xenografts of prostate cancers have been obtained. 6-9 The PPC-I model, originating from a primary tumor, is tumorigenic and metastatic in nude mice; 10 the hormone-dependent prostate cancer xenograft, LUCAP23.1, originated from a metastasis 11 and transplantable prostatic tumors obtained in NMRI nude mice. 12 These difficulties were well illustrated by Klein and colleagues 2 who obtained four prostate cancer xenografts in SCID mice. To search for biological information based on alterations common to prostate cancers, more tumor models are required, keeping in mind that their suitability requires that they primarily share common biological features with the original tumor. This must lead to identifying the cellular and molecular markers of progression.
Hormone dependence of prostate cancers represents the main therapeutic target. Endocrine therapy is usually successful initially, but generally fails after a variable period. 13 Surgical castration interrupts the main source of testosterone required for hormone-dependent cancer growth. Although effective, castration does not ensure protection from recurrence of a hormone-independent (HID) cancer. Castration can be achieved by gonadotropin-releasing hormone (GnRH) analogs. The efficacy of available compounds is variable; although they have clearly led to a decrease of serum testosterone or subsequently of prostate-specific antigen (PSA), they do not have a major effect on survival. New GnRH antagonists represent promising therapeutic tools. Loss of hormone dependence marks an essential step in tumor progression, as it deprives patients of the only effective therapy for metastatic prostate cancers. The mechanisms and the molecular determinants of tumor adaptation to hormone deprivation are far from being understood; inactivation of androgen receptors by mutation has been described but remains an exceptional event. Pathways of androgen receptor activation or androgen target genes through MAP kinase and HER-2/neu signal cascade activation have been described, 14-16 constituting a putative way to escape from hormone deprivation.
We report here the clinical evolution in two steps of a prostate cancer; we transplanted a sample obtained during transurethral resection of a primary adenocarcinoma into nude mice and established this tumor as a transplantable xenograft model and studied its progression. The features of the xenografted tumor were compared with those of the corresponding patient tumor tissue, with regard to morphology and immunohistochemical characterization. Karyotype and allelic imbalances (AIs) allowed reconstructing its progression. The hormone-dependent PAC120 xenografts permitted the in vivo testing of a new GnRH antagonist. In view of the history of the patient, which indicated that loss of hormone dependence was a step of progression, we designed experiments to test the ability of PAC120 to adapt to hormone deprivation.
Materials and Methods
Preparation of Specimens for Engraftment in Mice
Tumor material for xenografting was obtained by transurethral resection of a locally recurrent prostate cancer. The sample was free from necrotic and connective tissues and cut into pieces of 5 × 5 mm. Swiss nu/nu (nude) male mice, 5 weeks old, were used as xenograft recipients. They were bred in the animal facilities of Institut Curie and maintained in specified pathogen-free conditions. Their care and housing were in accordance with institutional guidelines as put forth by the French Ethical Committee (Ministère de l’Agriculture, Paris, France) and under the supervision of authorized investigators. Each tumor fragment was subcutaneously implanted under the wall of the lower abdomen, near the prostate of mice, and maintained by serial transplantation. All these procedures were done aseptically, under anesthesia.
Immunohistochemical Studies
For light microscopic examination, 4-μm-thick formalin-fixed paraffin-embedded sections were stained with hematoxylin and eosin (H&E) safran. Immunohistochemical studies, using a panel of antibodies listed in Table 1 ▶ , were performed on the resected tumor of the patient (first resection), on lung metastases, on the second resection, and on the xenografted prostatic tumor after three passages in nude mice. The tissue sections were pretreated using a microwave antigen retrieval method and immunostaining was performed using the avidin-biotin complex. Staining intensity was assessed semiquantitatively: 0 (negative), + (weak), and ++ (strong). The pattern distribution of staining was focal (F), diffuse (D), or occasional to rare cells (R). Appropriate positive and negative controls were used throughout. The Ki67 score was defined as the average number of cells with nuclear staining divided by the total number of cells counted. Five hundred tumor cells were counted and this was repeated three times. The mitotic rate was expressed as the average mitotic count present in 10 high-power fields.
Table 1.
Immunophenotype of the Different Prostate Cancer Samples
| Marker | Source | Dilution | Primary tumor* 1st resection | Lung metastasis | Primary tumor 2nd resection | PAC120 xenograft from the 2nd resection |
|---|---|---|---|---|---|---|
| Keratin (KL1) | Immunotech | 1 /100 | ++ (D†) | ++ (D) | ++ (D) | ++ (D) |
| EMA | DAKO | 1 /40 | ++ (D) | ++ (D) | ++ (D) | ++ (D) |
| Vimentin | DAKO | 1 /20 | 0 | 0 | 0 | 0 |
| PSA | Eurodiagnostics | 1 /100 | 0 | 0 | 0 | 0 |
| PAP | DAKO | 1 /200 | ++ (F†) | + (F) | ++ (F) | ++ (F) |
| Leu-7 | Becton | 1 /10 | ++ (F) | + (F) | ++ (F) | + (R)† |
| Chromogranin A | Biosoft | 1 /50 | 0 | nd | 0 | 0 |
| Synaptophysin | Boehringer | 1 /10 | 0 | nd | 0 | 0 |
| NSE | DAKO | 1 /200 | 0 | nd | + (F) | + (R) |
| Ki67 | Immunotech | 1 /50 | Ratio = 5% | nd | Ratio = 40% | Ratio = 20% |
| bcl2 | DAKO | 1 /50 | 0 | 0 | 0 | 0 |
| CD82 | Gift‡ | 1 /500 | nd | nd | 0 | nd |
*High-grade part of the tumor.
†Staining diffuse (D), focal (F), or on rare (R) cells. Staining intensity was assessed semiquantitatively; 0 (negative), + (weak), ++ (strong).
‡Gift of Dr. H. Conjeaud, INSERM U283, Hopital Cochin, Paris.
nd, not done; EMA, epithelial membrane antigen; PSA, prostate-specific antigen; PAP, prostatic acid phosphatase; NSE, neuron-specific enolase.
Karyotype and Chromosome Painting
Tumors removed from the mice were mechanically disrupted and seeded in Dulbecco’s modified Eagle’s medium (4.5 g of glucose/L; Gibco) with 15% fetal calf serum. Metaphases were harvested after a 2.5-hour colchicine block, 72 hours after the initiation of the cultures. Chromosome spreads were obtained according to previously described techniques. 17 Karyotypes were established after R-banding, and chromosome painting was used for all chromosomes to allow greater precision. On the whole lot, more than 100 metaphases were studied, of which 10 were karyotyped. Fluorescence in situ hybridization was performed as described. 18 Biotinylated paints (Oncor Appligene, Illkirch, France) were used as probes. The biotinylated probes were detected by goat anti-biotin antibody (Vector Laboratories, Burlingame, CA) and fluoresceinated anti-goat rabbit antibody (Biosys, Compiègne, France). Chromosomes were counterstained with 0.1 mg/ml of propidium iodide.
Allelic Imbalances
DNA was extracted from clinical specimens. Formalin or Bouin-fixed, paraffin-embedded tumor samples with corresponding normal tissues surrounding the tumoral tissue were used. Normal and neoplastic tissues were identified on H&E-stained tissue sections. Manual microdissection of selected areas on serial sections was done. Ten slides of 10-μm sections were microdissected and DNA purified as previously described. 19 The markers used and their chromosomal locations are listed in Table 2 ▶ . Microsatellite markers were detected by polymerase chain reaction (PCR) amplification with standard methods, except that samples were subjected to 35 cycles of denaturation for 40 seconds at 94°C, and 30 seconds of annealing. The final extension step at 72°C was lengthened to 10 minutes. The magnesium concentration and annealing temperature were optimized for each primer set. Products were diluted 1:3 in denaturing loading buffer and then heat-denatured; 1.5 μl of each sample was loaded on 6% acrylamide gel containing 7.5 mol/L urea. DNA was then transferred to nylon membrane filters. Probes (CA repeat oligonucleotide or specific primer) were labeled with 32P-dCTP. The membrane filters were hybridized overnight at 42°C with the labeled probe, washed, and autoradiographed at −80°C for an appropriate period. For detection of AI, each tumor DNA sample was run in adjacent tracks, together with corresponding normal-tissue DNA. Normal DNA that was polymorphic at a given locus was considered informative. The signal intensity of the fragments was determined by means of densitometry and blind inspection by three independent observers. AI was considered to be present when the relative intensity of the two alleles in tumor DNA differed from the relative intensity in normal DNA. The reduction in signal intensity that was observed for one allele with respect to normal tissue DNA was 50 to 100%. Each analysis was performed at least twice (independent PCR amplification, gel separation, and quantification).
Table 2.
Allelic Imbalances on Six Chromosomal Regions in DNA from Tumor Samples
| Chromosome location Polymorphic loci studied | 8p21-p22 D8S133 D8S261 | 16q24 D16S507 | 10q22-q23 D10S541 D10S1765 | 13q12-q13 D13S153 D13S171 D13S2491 | 17q21 D17S800 D17S855 | 18q21 D18S39 D18S42 |
|---|---|---|---|---|---|---|
| DNA origin | ||||||
| At the diagnosis | AI† | AI | no AI | no AI | no AI | no AI |
| Lung metastasis* | ||||||
| At the recurrence | AI | AI | AI | AI | AI | AI |
| Primary tumor‡ | ||||||
| PAC120 xenograft | AI | AI | AI | AI | AI | AI |
*Obtained from corresponding microdissected histological preparations, as described in Materials and Methods.
†AI, allelic imbalance.
‡Second resection.
Detection of PSA by Northern Blot Analysis
Denatured RNA in 50% formamide/2.2 mol/L formaldehyde and 1× MOPS (3-(N-morpholino) propanesulfonic acid buffer) were size-fractionated on a gel containing 1.2% agarose, 2.2 mol/L formaldehyde, 1× MOPS. RNA was then transferred overnight to Hybond-N membrane (Amersham Pharmacia Biotech, Uppsala, Sweden) by the capillary method. The membrane was UV-crosslinked (0.12 J/cm2) and baked 2 hours at 80°C. RNA were prehybridized for 30 minutes at 68°C with the QuikHyb hybridization solution (Stratagene, La Jolla, CA) and hybridized overnight in this buffer at the same temperature. After appropriate washing, the membrane was exposed to Kodak AR X-ray film at −70°C using intensifying screens for 2 to 7 days. The 32P-labeled probe used was generated by the random primer method from the exon 3 of the hKLK3 gene encoding PSA.
Detection of Micrometastasis by Reverse Transcriptase (RT)-PCR
Brain, lung, and liver of mice bearing PAC120 tumors were harvested 6 to 8 weeks after grafting and immediately frozen for subsequent RNA extraction. RNA was prepared using a commercially available kit (Trizol, Life Technologies). RT-PCR was performed using human HOXB9, 20 PSA primers, 21 HER-2/neu primers {AGAGTGAATGTGTGTGGAGTTATGGTGT(S) and ATTCAGAGTCAATCATCCAACACATTT(AS)} and HOXA4 primers {CATTCTCCGGTTCTGAAACCAGATC(S) and TGTACCCCTGGATGAAGAAGATCC(AS)}. The quality of all RNA was confirmed by ethidium bromide staining for ribosomal RNA and by RT-PCR using murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers {GACCCCTTCATTGACCTCAACTACA(S) and CATGTGGGCCATGAGGTCCACCAC(AS)} as a control.
Growth of Xenografts, Orchidectomy, and Hormone Deprivation
Tumor growth was assessed by measuring two perpendicular diameters with a caliper. The volume of each tumor was measured every 3 days. Tumor volume (V) was calculated as previously described: 22 V = a 2 X b/2 where a is the width of the tumor in millimeters and b is the length of the tumor in millimeters. Median of tumor volumes was calculated by group of mice. The relative tumor volume (RTV) was calculated as follows: RTV = Vx/V1, where Vx is the mean tumor volume in mm 3 at any given time and V1 is the mean tumor volume in mm 3 at the beginning of treatment. Tumor growth inhibition was calculated as the ratio between the RTV in the treated group and that of the tumors in the control group at a given time X 100. Tumor growth rate was calculated as the median time necessary to multiply by 5 the initial size of tumors. The statistical significance of the differences between the tumor volumes reached in each group was calculated using Student‘s t-test. Mice were sacrificed when the tumor volumes reached 2000 mm3.
Bilateral orchidectomy was performed under anesthesia. Castrated male mice were used as recipient for HID variants of PAC120. Anti-androgen compounds were administered subcutaneously in the flank of nude mice. D-Trp6-luteinizing-hormone releasing hormone (LHRH) (decapeptyl 3.75; Ipsen/Biotech, Paris, France) was administered at a dose of 0.15 mg/kg, in the excipient provided, once every 2 weeks. FE 200486, was obtained from the Ferring Research Institute Inc., San Diego, CA. FE 200486 was given every 2 weeks at a dose of 2 mg/kg solubilized in 5% mannitol, in a 0.1-ml volume. Testosterone supplementation of castrated male nude mice was done using testosterone pellets, 12.5 mg/pellet, 60-day release (Innovative Research of America, FL), grafted subcutaneously in the flank, a week after castration and 24 hours before tumor graft.
Testosterone Assay
Mice were treated and bled after different delays after start of treatment, as indicated in the text. Twenty mice were used as controls; 5 to 10 mice were included in groups of treatment. The serum was discarded and serum testosterone levels measured by radioimmunoassay (Linco Res., MO), according to the manufacturer’s protocol. Data are expressed in median of pg/ml.
Clinical Findings
A 51-year-old man was admitted to our hospital with dyspnea. Pulmonary radiography showed multiple lung metastases and extensive investigation led to the discovery of a prostatic neoplasm. The plasma PSA level was 16 ng/ml. A transurethral resection was performed and the patient was treated with GnRH agonist and anti-androgen. The PSA normalized (below 4 ng/ml, for 15 months), but lung metastases remained unchanged. Different combinations of chemotherapy were given to the patient without any valuable benefit. Two years after diagnosis, the plasma PSA increased to 10 ng/ml, whereas recurrent hematuria with dysuria required a palliative transurethral resection of a voluminous prostate cancer; tumor samples were grafted into male nude mice. The patient received chemotherapy, associated with anti-androgen therapy, without any effect. A few months later the patient developed bone metastases in the hip and sacrum. The patient died 36 months after diagnosis. The natural history of the disease is reported in Figure 1 ▶ .
Figure 1.

Global scheme of clinical, biological, and experimental progression of the prostate cancer.
Results
Histological Examination and Markers of the Clinical Samples and of Xenografts
Tumor tissue samples from the patient were examined at two stages of the disease: at diagnosis, samples of the lung metastases and of prostate tissue, and at the second transurethral resection. In the initial biopsies, histology of the primary prostatic tumor showed a high-grade adenocarcinoma with a Gleason score of 9 (5 + 4) (Figure 2, A and B) ▶ . The neoplastic cells exhibited enlarged nuclei with a prominent nucleolus. Fifteen mitoses were counted per 10 high-power field. These same features were found in the lung metastases. In the first resection, a focus of prostate cancer with Gleason pattern 2 was also present in the transition zone (Figure 2C) ▶ , displaying a high PSA staining (Figure 2D) ▶ . The histology of the sample obtained from the patient at the second resection was very similar to the high-grade tumor.
Figure 2.
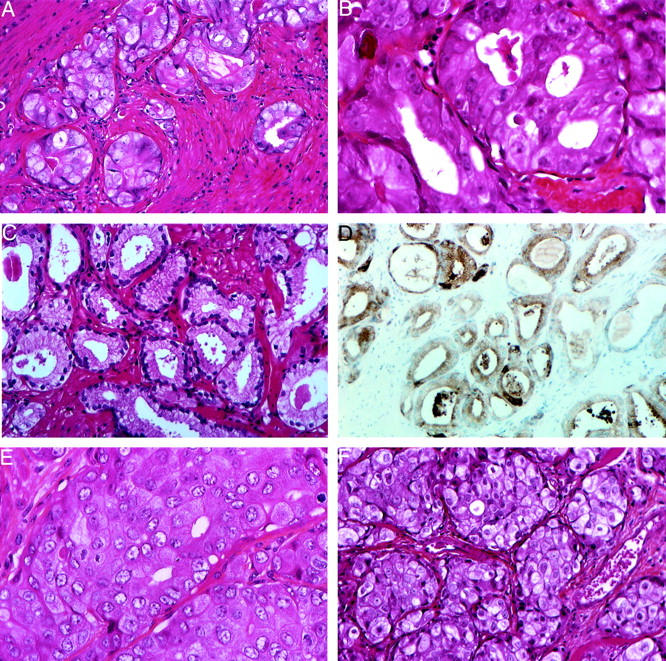
Histology and immunohistochemistry of the prostate cancer from the patient and after xenografting. A and B: Second resection of a human primary prostatic adenocarcinoma, composed of glands appearing irregular, fused, sometimes with a cribriform pattern [HES: original magnifications: ×200 (A) and ×400 (B)]. B: At higher magnification nuclei appeared enlarged with a prominent nucleolus. In C and D (HES; original magnification, ×250), a focus of Gleason pattern 2 adenocarcinoma in the transition zone was prostate cancer, similar to that shown in A and B; tumoral glands are loosely packed, round, of medium size with variability in shape with definite separation from each other by stroma, the cells have pale cytoplasm and are frankly nucleolated; immunostaining with anti-PSA antibody was positive (D). E: Histology of the PAC120 xenograft shows a high-grade adenocarcinoma with gland lumens in some areas. Nucleoli are prominent (HES; original magnification, ×400).
Macroscopically, PAC120 xenograft showed a dense purple tumor, highly vascularized. It consisted in Gleason pattern 5 (Figure 2E) ▶ ; glandular differentiation was rare, and mitotic index was high. Two weeks after surgical castration, mitotic activity was absent; few necrotic areas were detected, without evidence of apoptotic cells (Figure 2F) ▶ . Immunohistochemistry performed on the clinical tumor samples and the PAC120 xenograft (Table 1) ▶ showed a very close immunophenotype, with a high expression of cytokeratin (KL1), epithelial membrane antigen, and more focally CD57 antigen (Leu7). Scattered tumor cells were positive for neurone-specific enolase in the second resection and in PAC120. There was no staining for vimentin, chromogranin A, and synaptophysin. PSA was found positive only in the focus of Gleason pattern 2, found in the biopsy of the initial tumor (Figure 2D) ▶ . The other samples were negative for PSA and positive for prostatic acid phosphatase (not shown). PSA mRNA transcript was highly expressed as a strong signal in the 1.6-kb range was obtained when total RNA was hybridized with a hKLK3 DNA probe by the Northern blotting (data not shown). PSA was also detected in PAC120 protein extract by Western blotting using a polyclonal anti-PSA antibody (data not shown) and by RT-PCR as a 240-bp transcript. PSA was detected at a level of 0.55 ng/ml in the serum of nude mice, bearing PAC120 tumors of 2200 mm 3 of median size.
Tumor Progression as Seen by Cumulative Genomic Anomalies
AIs were found in the DNA extracted from the clinical samples from the biopsies of lung metastases performed at the diagnosis and from samples obtained at the second resection and in PAC120. The markers used are listed in Table 2 ▶ . They displayed two different profiles of genetic alterations: only two AI of 8p21-22 and 16q24 were detected in the lung metastases, four additional AI (17q21, 10q22-23, 13q12-q13, and 18q21) were found in the recurrent tumor. The six AIs were present in PAC120 xenografts.
Karyotype of PAC120 comprised 55 to 69 chromosomes of which approximately half were rearranged. Most rearranged chromosomes were in duplicate. Although no karyotypes were identical, most rearrangements were observed in all karyotypes, indicating a relative stability.
The formula was: 55-69, −X, der(X)t(X;19)(q24;?)x2, del(1)(p33), hsr(1){p35;hsr(1;17)}, der(1)t(hsr1;3;8;17)(1qter->1p35::hsr1;17::8?->8?::3q13->3qter), der(2)t(2;14)(q32;q23), +3, der(3)t(3;17)(q13;q23(x2, der(4)t(4;20)(p11;q12), i(4)(q10), −5, add(5)(q12)x2, der(5)t(5;17)(p12;q22(x2, del(6)(p22), +der(7)t(4;7)(q22;q21), del(8)(q11q22), der(8)t(X;8)(q23;p11)x2, +der(8)t(3;8)(q13;q23), del(9)(p12)x2, +i(9)(p10), der(10)t(8;10)(?;15(x2, der(10)t(10;15)(q23;q24), der(11)t(1;11)(q12;p15), −12,+der(12)t(12;19;21)(12q24->12p11::19? ->19?::21q11->21qter)x2, −13, −15, +der(15)t(10;15)(q23;q25(x2, +del(16)(q?p?), add der(17;18)(q10;p10)(q21), der(17)t(5;15;17)(17pter->17q22::15? ->15?::5q23->5qter)x2, −19, del(20)(p11), −21, −21, −21, del(22)q11), der(22)t(Y;22)(q11;p12)+0-6 mar.
All of the rearrangements were confirmed with the use of chromosome painting. Assuming a monosomic-type karyotype evolution, the following scheme can be reconstructed: at early stages, many chromosomes were rearranged and/or lost, leading to an hypodiploid karyotype. Endoreplication occurred, generating duplicate derivative chromosomes such as the der(3)t(3;17) and del(9), among others (Figure 3A) ▶ . Chromosome rearrangements continued to occur after endoreduplication, leading to either a single copy of new derivatives, such as the del(8)q11q22), or complex single copy chromosomes deriving from a previously formed and duplicated derivative such as the der(1)t,(1;3;8;17) probably derived from the der(1)hsr(1;17) (Figure 3B) ▶ .
Figure 3.

Cytogenetic features of PAC120 xenograft. A: Reconstitution of the karyotype evolution; a first group of alterations observed in two copies occurred before endoreduplication; another group of alterations observed in only one copy, in addition to the three copies of the parental homologous chromosome, indicating that the alterations occurred after endoreduplication; for five rearrangements, this classification was not possible. B: Reconstitution of chromosomes 1 and 11 after painting.
Micrometastasis in Mice Bearing the PAC120 Xenograft
Search for micrometastasis was done by the RT-PCR assay using human-specific oligomers of HOXB9, PSA, and HER-2/neu. Micrometastases were detected in lungs of mice bearing PAC120 xenografts. These three human genes PSA, HER-2/neu, and HOXB9 were expressed in seven distinct PAC120 xenografts (Figure 4) ▶ , although at variable levels. In lung extracts, PSA was detected in four lungs, HER-2/neu in four, and HOXB9 in two. No human gene signal was detected in the lungs of mice 1 and 2. In lung extracts, expression of a murine GAPDH, was used as a control of total deposited RNA. Finally, five lungs of the seven tested displayed a positive signal corresponding to the presence of human RNA; no signals were detected in the RNA extracted from brain or liver of the corresponding mice.
Figure 4.

mRNA transcripts of human PSA, HER-2/neu, HOXA4, and HOXB9 genes and of the murine GAPDH gene. mRNA was extracted either from seven different primary xenografts of PAC120, collected at a size of 1.2 to 2 cm 3 or from corresponding lung.
Androgen Dependence of PAC120 Xenografts, Efficacy of Surgical and Pharmacological Castration, and Hormone Escape
Initial reduction of circulating PSA after treatment with an anti-androgen therapy indicated that the tumor was initially hormone sensitive. PAC120 established in male mice was not transplantable in castrated males or in females. Surgical castration of mice bearing PAC120 tumors led to almost complete regression; however, all tumors locally recurred, 160 to 220 days after castration (Figure 5) ▶ . Hormone-dependent PAC120 tumor-bearing male mice were treated with the new GnRH FE 200486 (antagonist) and the long acting formulation D-Trp6-LHRH (agonist) (decapeptyl 3.75). A single injection of 2 mg/kg of FE 200486 decreased the serum testosterone level of 2669 to 104 pg/ml within 24 hours. Serum testosterone was decreased below 100 pg/ml for 9 days (Figure 6) ▶ and returned to normal level between day 16 and day 23. FE 200486 treatment completely suppressed the growth of PAC120 xenografts, as long as the treatment was administered (Figure 7) ▶ . When the treatment was stopped after the fourth injection (at day 45), tumors regrew 35 to 45 days later. Repeated injections of D-Trp6-LHRH reduced transiently the serum testosterone level (266 pg/ml 24 hours after the first injection) but less efficiently than the antagonist (Figure 6) ▶ . Decapeptyl produced a significant but modest growth inhibition (36% of growth inhibition, optimal at day 42, P < 0.003).
Figure 5.

Relative tumor growth of PAC120 tumors, effects of surgical castration. PAC120 fragments were grafted into 10 male nude mice; 4 to 6 weeks after grafting, tumors appeared; when the tumor size reached 500 mm 3 (on average), the mice were surgically castrated (day 1). Individual growth curves were established. Five mice sacrificed at day 100 displayed complete regression; the five other mice survived to day 220, when they were sacrificed. Mean tumor growth in control mice is shown (filled circles).
Figure 6.

Serum testosterone levels in nude mice, treated with D-Trp6-LHRH (open squares) and FE 200486 (filled diamonds) as a function of time, as medians. FE 200486 was diluted in 5% mannitol was injected as a single dose subcutaneously at 2 mg/kg. GnRH superagonist D-Trp6-LHRH (decapeptyl 3.75) diluted in 5% mannitol was given daily, subcutaneously, at a dose of 0.15 mg/kg.
Figure 7.

Individual growth curves of PAC120 established in male nude mice; effects of FE 200486 on the growth of mice. FE 200486 diluted in 5% mannitol was administered subcutaneously at a dose of 2 mg/kg, every 2 weeks. For five mice, the treatment was stopped after the fourth injection (dotted lines), or continued for five others (solid lines). The average growth control is shown (filled circles).
HID Variants of PAC120 Xenografts, Morphological Changes, and Gene mRNA Expression
PAC120 bearing mice were castrated when the local tumor reached 250 to 500 mm3. After several months, HID tumors started to grow. Six HID variants were independently derived, designated as HID25, -28, -33, -16, -19, and -34. Recurrence latencies were, respectively, of 15, 7, 8, and 12 months for the two latter variants. Time delays before the second passage in precastrated males were of 3, 5, 5, 3, 4, and 6 months, respectively. HID28 tumors were transplanted either into intact or castrated males. After transplantation, latency for tumor appearance of HID28 tumors was 45 and 180 days, respectively, and growth rate was 18 and 35 days. Testosterone delivered by slow-releasing pellets in castrated males increased similarly the growth rate of HID tumors (data not shown). HID tumors were studied at the second passage; the microscopic pattern of HID33 (Figure 8A) ▶ was close to that of the hormone-dependent PAC120 tumor, some glandular structures were still present, the mitotic rate was high. HID19 (Figure 8B) ▶ consisted in large sheets of pleomorphic tumor cells, without glandular differentiation. HID34 tumors were characterized by a focal neuroendocrine-like pattern (Figure 8C) ▶ ; HID28 and HID25 presented mixed forms composed of well-limited mucoid cell areas contiguous with areas morphologically similar to PAC120 (Figure 8, D and E) ▶ , or as fully mucoid tumors (Figure 8F) ▶ .
Figure 8.
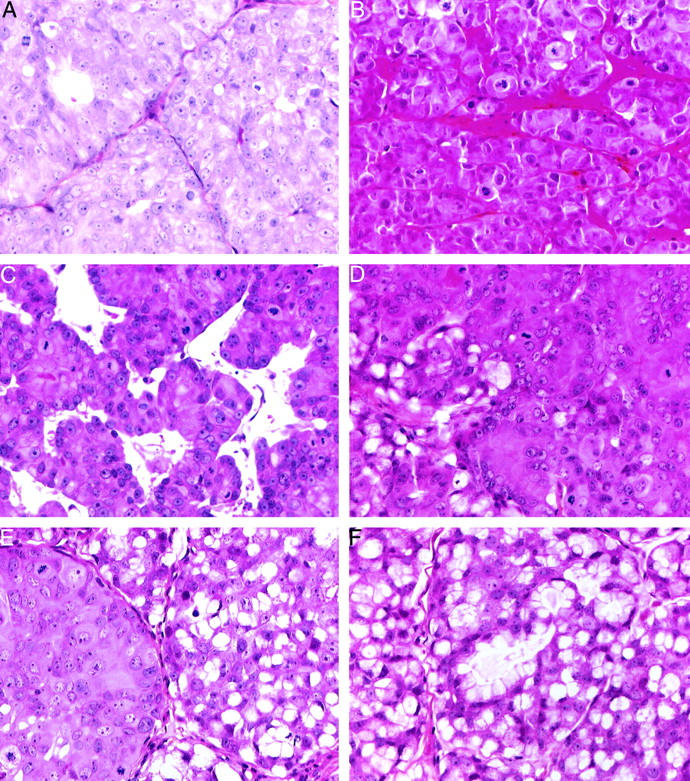
Histological features of hormone-independent recurrences of PAC120, the HID xenografts, at the second transplantation in castrated male (HES; original magnification, ×400). A: HID33, a microscopic pattern close to that of the hormone-dependent PAC120 tumor. B: HID19, large sheets of tumor cells, without glandular differentiation. C: HID-34, focal neuroendocrine-like pattern. D and E: HID-28 and HID-25, mixed forms composed of well-limited mucoid cell areas contiguous with areas morphologically similar to PAC120. F: HID25, a fully mucoid adenocarcinoma.
Expression of mRNA transcripts of PSA tested by RT-PCR was variable whereas that of HER-2/neu and HOXB9 were constant (Figure 9) ▶ . Five of the six distinct HID25 tumors expressed PSA, but at variable levels. HID16 and HID34 expressed the 240-bp transcript of PSA, whereas HID19 and HID33 did not. Levels of PSA in the serum of mice bearing HID variants was 0.26 ng/ml and below detection threshold, respectively.
Figure 9.

mRNA transcripts of human PSA, HER-2/neu, and HOXB9 genes. mRNA was extracted from hormone-independent tumors. Products of RT-PCR of six different HID-25 tumors were deposited into lanes 8 to 13, HID 33, HID-16, HID-19, and HID-34 into lanes 14 to 17, respectively.
Discussion
We report here the case of a patient with prostate cancer, with a classical progressive disease, although atypical for its initial presentation of lung metastases and because of his young age. After transurethral surgery and anti-androgen therapy, the patient succumbed at the end of the third year with recurrence of the primary tumor and a wave of bone metastases. This clinical history comprises the two key events in the evolution of prostate cancers, ie, escape from hormone dependence and metastatic dissemination. We characterized some biological features of this tumor at different steps in its progression, which was further prolonged by xenografting in nude mice. This allowed us to evaluate the pertinence of correlation between biological markers and tumor progression.
Disease progression is linked with genomic instability. 23,24 We observed here two waves of metastatic dissemination: firstly, massive lung involvement that did not progress for 3 years under anti-androgen treatment; then, later, bony metastases that grew rapidly, preceding death by a few months. These two waves of dissemination did not have the same prognostic significance and seemed to be related to different genetic events, the second wave corresponding to an increased genomic instability.
A link between metastatic dissemination and hormone independence is currently observed and has been reproduced experimentally. 2 However, this case report illustrates the complexity of such a relationship. During the first phase of the disease, lung metastases appeared long before escape from hormone deprivation. The second wave of metastases occurred as the primary tumor recurred under anti-androgen treatment. Thus, metastatic dissemination to bones and hormone dependence would be correlated, at the opposite of metastatic dissemination to lungs which was not.
Although escape from hormone deprivation is considered to be an endpoint, which is clinically true, this does not mean that transition to hormone independence is an irreversible event. Moreover, clinically, the disease progressed under anti-androgen therapy, whereas the tumor was not transplantable into castrated animals, demonstrating that it was still hormone-dependent. This could indicate that the hormone deprivation was not sufficient in the patient to inhibit the growth of his tumor. Our experimental study suggests that growth of hormone-independent variants in castrated males results either from an adaptation to hormone deprivation, or to a selection of a pre-existing subpopulation of HID cells, as emphasized by Isaacs and Coffey 25 , but not from a deficiency of androgen-dependent pathways because they are still able to benefit from hormone supply when grafted into intact male mice.
The validation of this model benefits of the comparison with the original tumor. The histology of the first transurethral resection revealed that the main part of the tumor as the recurrent one was of high grade. The histological grade of the xenograft was also high. Transcripts of PSA, a prostate-specific protease of the kallikrein family, were detected in PAC120; Western blotting confirmed the presence of the encoded protein. PAC120 formed micrometastases in the lungs of nude mice although no macroscopic metastases were detected.
The proliferation index was high in PAC120. A correlation between the Ki67 index and a high Gleason grade has been reported. 26,27 The high Ki67 positivity of the PAC120 is surprising, given to its relatively slow growth. PAC120 did not express bcl-2, and had a mutated p53 (data not shown). KAI-1/CD82, 28 a marker repressed in metastatic cell lines, was not expressed. In the clinical samples and in PAC120, only dispersed neuroendocrine cells were identified both before and after endocrine therapy. After hormone escape, one HID variant displayed a neuroendocrine-like pattern. Neuroendocrine differentiation was observed in prostate cancer, correlated with tumoral aggressiveness, short survival, and early failure of endocrine therapy. 29 Mucoid differentiation was observed in two of the six HID variants. Mucinous adenocarcinoma is a rare subtype of prostate cancer and is considered as poor responder to hormone therapy. 30 On the other hand, production of neutral mucin assessed by histochemistry is frequent, found in up to 55% of prostate carcinomas. 31
Karyotype of PAC120 was near-triploid, resulting from multiple alterations. Most rearranged chromosomes were in duplicate, indicating that this tumor underwent a tetraploidization and should be considered as hypotetraploid. In karyotypes reported in the literature, chromosomes 1, 3, 5, 8, 10, 17, 19, and Y are recurrently lost or rearranged. 32-34 Deletion of chromosome 10 in the q24 region was first noticed by Atkin and Baker 35 and was associated to PTEN gene mutation. 36,37 Loss of the chromosome 8p arm suggests the presence of another suppressor gene. 38 Losses of heterozygosity were described at specific regions of chromosome arms 5q, 6q, 7q, 8p, 10q, 13q, 16q, 17q, and 18, where genes considered as critical for tumor progression, such as α-catenin, E-cadherin, BRCA1, BRCA2, and PTEN, are located. In the late resection sample, AIs were detected at six loci, including 8p21-22, 10q22-q23, 13q12-q13, 16q24, 17q21, and 18q2. Because five of these six chromosomes were found as two copies, in hypotetraploid cells, it is likely that they derived from the duplication of one parental chromosome. The AIs found in PAC120 exactly reproduced the profile of AIs detected in the tumor sample obtained from the patient at the second resection. Interestingly, the initial lesions from the biopsy of the lung metastases displayed only two of these loci, 8p21-22 and 16q24, whereas these lesions did not progress. Conversely, DNA from the recurrent tumor displayed an accumulation of AIs. These anomalies might be associated with the aggressive progression of the disease.
The initial biopsy revealed co-existence of foci of different grades: a small focus of low-grade, PSA-positive, of the transition zone type, and large infiltrate high-grade adenocarcinoma. Co-existence of foci of different grades in the same tumor has been reported previously. 39 This observation suggests that either an initial focus has evolved to a less differentiated one, or there were initially two distinct tumors. Prostate cancer is a hormone-dependent cancer and this is the therapeutic basis of the use of androgen deprivation. Escape from androgen deprivation is a facet of tumor progression. Mutations of androgen receptors have been incriminated but they are not always associated with loss of function and finally are not recognized as the main mechanism of hormone escape. 40
In our study, the original tumor was hormone-dependent. Disease progression in the patient could be interpreted as a loss of hormone dependence, ie, there was tumor growth despite anti-androgen therapy. Yet, PAC120 was still hormone-dependent, which is surprising because of the inefficiency of the anti-hormone therapy in the patient. This suggests that, in some cases, the dogma that late progression of prostate cancers is because of an irreversible escape from hormone dependence is wrong and that the final stage of this disease could be because of mechanisms involving a metabolic adaptation of tumor cells. Our data showed that hormone supply in the castrated male accelerated growth rate of HID tumors, demonstrating that androgen receptors were still functional, and that the HID tumors were still hormone sensitive.
FE 200486 is a new competitive antagonist of the GnRH receptor that has been shown to produce an immediate and sustained inhibition of the release of gonadotrophins and sex steroids in rats and monkeys. In rats, a single injection of FE 200486 at 2 mg/kg can suppress testosterone down to castration level for more than 40 days. 41 In nude male mice, a single injection of FE 200486 at 2 mg/kg suppressed testosterone level and suppression was maintained for 2 weeks. The level of testosterone in mice treated with repeated administrations of D-Trp6-LHRH decreased, the maximal efficacy being obtained after 24 hours, but it increased rapidly. The remarkable anti-tumoral efficacy of FE 200486 as compared to that of decapeptyl provides insights into the activity mechanism of such compounds and the possible processes of escape: the hormone has to be rapidly and highly suppressed, and efficiently maintained at a constant low level. A direct inhibitory effect of FE 200486 on tumor growth, as shown by Lamharzi and colleagues 42 with another GnRH antagonist, was ruled out by its inability to inhibit the growth of HID xenografts, although PAC120 and its HID variants expressed the GnRH receptor gene, as detected by RT-PCR (data not shown). Thus, FE 200486 reproduced the effects of surgical castration.
Surgical castration did not cure the mice, despite initial regressions of the xenografted PAC120 tumors. Recurrent growth of PAC120 under testosterone deprivation indicates a change in the biology of this tumor. These changes were evidenced by alterations of mRNA expression of PSA (decreased) and HER-2/neu (increased); PSA mRNA expression was found in all hormone-dependent tumors but not in all HID. HER-2/neu mRNA expression was found in all tumors but was lower in PAC120 tumors, showing that mRNA expression of PSA and HER-2/neu was not correlated, as observed in other studies. 43 Expression of HOXB9 was constant in all tumors.
Candidates for molecular determinants supporting growth of HID tumors, as reviewed by Djakiew, 44 include fibroblast growth factor, 45 epidermal growth factor, and epidermal growth factor receptor mutations, 46 fibroblast growth factor, 45 HER-2/neu tyrosine kinase, 14,43 IGF-BP or IGF growth factor 47,48 and PDEF, (prostate-derived Ets factor), a novel prostate epithelium-specific Ets transcription factor, which interacts with the androgen receptor and activates PSA genes in an androgen-dependent or -independent manner. 49 All together, these different studies implicate the role of PSA-regulation, HER-2/neu, MAP-kinase, and insulin growth factor and insulin growth factor binding protein proteins, in complex signal cascades; 50,51 this is conforted by the cDNA and tissue array techniques of Bubendorf and colleagues, 48 which detected high expression of IGFBP2 and HSP27.
In conclusion, we have extensively characterized PAC120, a hormone-dependent human prostatic tumor, as a prolongation of a clinically growing tumor. As a prostate cancer, it was peculiar because of the relatively young age of the patient, the ability of the tumor to be transplanted into nude mice, and its persistent hormone dependence even though it was derived after clinical escape to androgen therapy. It is thus representative of aggressive metastatic prostate cancers. We have used this model to test therapeutic approaches. This model allowed us also to derive several HID variants that could lead to further study mechanisms of adaptation to hormone deprivation of prostate cancers.
Acknowledgments
We thank Dr. Théodore, Institut Gustave Roussy, Villejuif, France; Pr. Zerbib, Hôpital Cochin, Paris, France; Dr. Saltiel, Hôpital Gilles de Corbeil, Corbeil, France; Dr. Natali, Service de Pneumologie, Hôpital d’Instruction des Armées, Versailles, France; Dr. Bellamy, Center Chirurgical du Val-d’Or, Saint-Cloud, France; Dr. Felgères, Dr. Kemeny, and Dr. Vadrot, Laboratoire d’Anatomie Cytologie Pathologiques, Evry, France, for their help in collecting tumor specimens; Dr. H. Conjeaud, INSERM U283, Hopital Cochin for kindly providing us with the antibody antiCD82; D. Flagiello and A Beurdeley-Thomas for their help in HOX and PSA expression evaluation; to V. Bordier and C. Alberti for their excellent technical assistance in animal experimentation; and Dr. S. Agrawal for reviewing the English used in this report.
Footnotes
Address reprint requests to Marie-France Poupon, M.D., Ph.D., Institut Curie, Section de Recherche, UMR-147-CNRS, 26 rue d’Ulm, 75231 Paris, France. E-mail: mfpoupon@curie.fr.
Supported by the CNRS and the Institut Curie, Section de Recherche; and three fellowships from the Association pour la Recherche sur le Cancer, (to F. P.-B.) and the Luxembourg Government R/D BFR 95/035 (to R. B.-G.).
G. D. P. and M. E. L. contributed equally to this work.
References
- 1.Coffey DS: Prostate cancer. An overview of an increasing dilemma. Cancer 1993, 71:880-886 [DOI] [PubMed] [Google Scholar]
- 2.Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, Lamb DJ, Marcelli M, Belldegrun A, Witte ON, Sawyers CL: Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med 1997, 3:402-408 [DOI] [PubMed] [Google Scholar]
- 3.Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF: Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer 1978, 21:274-281 [DOI] [PubMed] [Google Scholar]
- 4.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW: Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 1979, 17:16-23 [PubMed] [Google Scholar]
- 5.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP: LNCaP model of human prostatic carcinoma. Cancer Res 1983, 43:1809-1818 [PubMed] [Google Scholar]
- 6.Hoehn W, Schroeder F, Riemann, Joebsis A, Hermanek P: Human prostatic adenocarcinoma: some characteristics of a serially transplantable line in nude mice (PC-82). Prostate 1980, 1:95-104 [DOI] [PubMed] [Google Scholar]
- 7.Ito Y, Nakasato Y: A new serially transplantable human prostatic cancer (HONDA) in nude mice. J Urol 1984, 132:384-387 [DOI] [PubMed] [Google Scholar]
- 8.Hoehn W, Wagner M, Riemann J, Hermanek P, Williams E, Walther R, Schrueffer R: Prostatic adenocarcinoma PC-EW, a new human tumor line transplantable in nude mice. Prostate 1985, 5:445-452 [DOI] [PubMed] [Google Scholar]
- 9.Pretlow T, Wolman S, Micale M, Pelley R, Kursh E, Resnick M, Bodner D, Jacobberger J, Delmoro C, Giaconia J, Pretlow T: Xenografts of primary human prostatic carcinoma. J Natl Cancer Inst 1993, 85:394-398 [DOI] [PubMed] [Google Scholar]
- 10.Brothman AR, Peehl DM, Patel AM, MacDonald GR, McNeal JE, Ladaga LE, Schellhammer PF: Cytogenetic evaluation of 20 cultured primary prostatic tumors. Cancer Genet Cytogenet 1991, 55:79-84 [DOI] [PubMed] [Google Scholar]
- 11.Royai R, Lange PH, Vessella R: Preclinical models of prostate cancer. Semin Oncol 1996, 23:35-40 [PubMed] [Google Scholar]
- 12.van Weerden WM, de Ridder CM, Verdaasdonk CL, Romijn JC, van der Kwast TH, Schroder FH, van Steenbrugge GJ: Development of seven new human prostate tumor xenograft models and their histopathological characterization. Am J Pathol 1996, 149:1055-1062 [PMC free article] [PubMed] [Google Scholar]
- 13.Denis LJ, Griffiths K: Endocrine treatment in prostate cancer. Semin Surg Oncol 2000, 18:52-74 [DOI] [PubMed] [Google Scholar]
- 14.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C: From HER-2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA 1999, 96:5458-5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, Said J, Reiter RE, Sawyers CL: Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer Res 1999, 59:5030-5036 [PubMed] [Google Scholar]
- 16.Signoretti S, Montironi R, Manola J, Altimari A, Tam C, Bubley G, Balk S, Thomas G, Kaplan I, Hlatky L, Hahnfeldt P, Kantoff P, Loda M: Her-2-neu expression and progression toward androgen independence in human prostate cancer. J Natl Cancer Inst 2000, 92:1918-1925 [DOI] [PubMed] [Google Scholar]
- 17.Couturier J, Dutrillaux B: Conservation of replication chronology of homologous chromosome bands between four species of the genus Cebus and man. Cytogenet Cell Genet 1981, 29:233-240 [DOI] [PubMed] [Google Scholar]
- 18.Muleris M, Zafrani B, Validire P, Girodet J, Salmon RJ, Dutrillaux B: Cytogenetic study of 30 colorectal adenomas. Cancer Genet Cytogenet 1994, 74:104-108 [DOI] [PubMed] [Google Scholar]
- 19.Farabegoli F, Ceccarelli C, Santini D, Trere D, Baldini N, Taffurelli M, Derenzini M: Chromosome 1 aneusomy with 1p36 under-representation is related to histologic grade, DNA aneuploidy, high c-erb B-2 and loss of bcl-2 expression in ductal breast carcinoma. Int J Cancer 1996, 69:381-385 [DOI] [PubMed] [Google Scholar]
- 20.Belotti D, Clausse N, Flagiello D, Alami Y, Daukandt M, Deroanne C, Malfoy B, Boncinelli E, Faiella A, Castronovo V: Expression and modulation of homeobox genes from cluster B in endothelial cells. Lab Invest 1998, 78:1291-1299 [PubMed] [Google Scholar]
- 21.Clements JA, Rohde P, Allen V, Hyland VJ, Samaratunga ML, Tilley WD, Lavin MF, Gardiner RA: Molecular detection of prostate cells in ejaculate and urethral washings in men with suspected prostate cancer. J Urol 1999, 161:1337-1343 [PubMed] [Google Scholar]
- 22.Poupon MF, Arvelo F, Goguel AF, Bourgeois Y, Jacrot M, Hanania N, Arriagada R, Le Chevalier T: Response of small-cell lung cancer xenografts to chemotherapy: multidrug resistance and direct clinical correlates. J Natl Cancer Inst 1993, 85:2023-2029 [DOI] [PubMed] [Google Scholar]
- 23.Muleris M, Dutrillaux B: The accumulation and occurrence of clonal and unstable rearrangements are independent in colorectal cancer cells. Cancer Genet Cytogenet 1996, 92:11-13 [DOI] [PubMed] [Google Scholar]
- 24.Isaacs JT: The biology of hormone refractory prostate cancer. Why does it develop? Urol Clin N Am 1999, 26:263-273 [DOI] [PubMed] [Google Scholar]
- 25.Isaacs JT, Coffey DS: Adaptation versus selection as the mechanism responsible for the relapse of prostatic cancer to androgen ablation therapy as studied in the Dunning R-3327-H adenocarcinoma. Cancer Res 1981, 41:5070-5075 [PubMed] [Google Scholar]
- 26.Gallee MP, van Steenbrugge GJ, ten Kate FJ, Schroeder FH, van der Kwast TH: Determination of the proliferative fraction of a transplantable, hormono-dependent, human prostatic carcinoma (PC-82) by monoclonal antibody Ki-67: potential application for hormone therapy monitoring. J Natl Cancer Inst 1987, 79:1333-1340 [PubMed] [Google Scholar]
- 27.Feneley MR, Young MP, Chinyama C, Kirby RS, Parkinson MC: Ki-67 expression in early prostate cancer and associated pathological lesions. J Clin Pathol 1996, 49:741-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong JT, Suzuki H, Pin SS, Bova GS, Schalken JA, Isaacs WB, Barrett JC, Isaacs JT: Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Res 1996, 56:4387-4390 [PubMed] [Google Scholar]
- 29.McWilliam LJ, Manson C, George NJ: Neuroendocrine differentiation and prognosis in prostatic adenocarcinoma. Br J Urol 1997, 80:287-290 [DOI] [PubMed] [Google Scholar]
- 30.Epstein JI, Lieberman PH: Mucinous adenocarcinoma of the prostate gland. Am J Surg Pathol 1985, 9:299-308 [DOI] [PubMed] [Google Scholar]
- 31.Sentinelli S: Mucins in prostatic intra-epithelial neoplasia and prostatic carcinoma. Histopathology 1993, 22:271-274 [DOI] [PubMed] [Google Scholar]
- 32.Sandberg A: Chromosomal abnormalities and related events in prostate cancer. Hum Pathol 1992, 23:368-380 [DOI] [PubMed] [Google Scholar]
- 33.Webb HD, Hawkins AL, Griffin CA: Cytogenetic abnormalities are frequent in uncultured prostate cancer cells. Cancer Genet Cytogenet 1996, 88:126-132 [DOI] [PubMed] [Google Scholar]
- 34.Zitzelsberger H, Szucs S, Robens E, Weier HU, Hofler H, Bauchinger M: Combined cytogenetic and molecular genetic analyses of fifty-nine untreated human prostate carcinomas. Cancer Genet Cytogenet 1996, 90:37-44 [DOI] [PubMed] [Google Scholar]
- 35.Atkin NB, Baker MC: Chromosome 10 deletion in carcinoma of the prostate. N Engl J Med 1985, 312:315. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner S, Giovanella B, Ittmann M, Tycko B, Hibshoosh H, Wigler M, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275:1943-1947 [DOI] [PubMed] [Google Scholar]
- 37.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 38.Bova GS, Carter BS, Bussemakers MJ, Emi M, Fujiwara Y, Kyprianou N, Jacobs SC, Robinson JC, Epstein JI, Walsh PC, Issacs WB: Homozygous deletion and frequent allelic loss of chromosome 8p22 loci in human prostate cancer. Cancer Res 1993, 53:3869-3873 [PubMed] [Google Scholar]
- 39.Grignon DJ, Sakr WA: Zonal origin of prostatic adenocarcinoma: are there biologic differences between transition zone and peripheral zone adenocarcinomas of the prostate gland? J Cell Biochem 1994, 19:267-269 [PubMed] [Google Scholar]
- 40.Wallen MJ, Linja M, Kaartinen K, Schleutker J, Visakorpi T: Androgen receptor gene mutations in hormone-refractory prostate cancer. J Pathol 1999, 189:559-563 [DOI] [PubMed] [Google Scholar]
- 41.Broqua P, Aebi A, Jiang GC, Stalewski J, Semple G, Akinsanya K, Haigh R, Rivière P, Rivier J, Aubert ML, Junien JL: Pharmacological profile of FE 200486, a potent: water soluble and long acting GnRH antagonist. Gynecol Endocrinol 1999, 13:910862263 [Google Scholar]
- 42.Lamharzi N, Schally AV, Koppan M: Luteinizing hormone-releasing hormone (LH-RH) antagonist Cetrorelix inhibits growth of DU-145 human androgen-independent prostate carcinoma in nude mice and suppresses the levels and mRNA expression of IGF-II in tumors. Regul Pept 1998, 77:185-192 [DOI] [PubMed] [Google Scholar]
- 43.Craft N, Shostak Y, Carey M, Sawyers CL: A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 1999, 5:280-285 [DOI] [PubMed] [Google Scholar]
- 44.Djakiew D: Dysregulated expression of growth factors and their receptors in the development of prostate cancer. Prostate 2000, 42:150-160 [DOI] [PubMed] [Google Scholar]
- 45.Dorkin TJ, Robinson MC, Marsh C, Neal DE, Leung HY: aFGF immunoreactivity in prostate cancer and its co-localization with bFGF and FGF8. J Pathol 1999, 189:564-569 [DOI] [PubMed] [Google Scholar]
- 46.Olapade-Olaopa EO, Moscatello DK, MacKay EH, Horsburgh T, Sandhu DP, Terry TR, Wong AJ, Habib FK: Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer 2000, 82:186-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamharzi N, Schally AV, Koppan M, Groot K: Growth hormone-releasing hormone antagonist MZ-5–156 inhibits growth of DU-145 human androgen-independent prostate carcinoma in nude mice and suppresses the levels and mRNA expression of insulin-like growth factor II in tumors. Proc Natl Acad Sci USA 1998, 95:8864-8868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, Mahlamaki E, Schraml P, Moch H, Willi N, Elkahloum AG, Pretlow TG, Gasser TC, Mihatsch MJ, Sauter G, Kallioniemi OP: Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst 1999, 91:1758-1764 [DOI] [PubMed] [Google Scholar]
- 49.Oettgen P, Finger E, Sun Z, Akbarali Y, Thamrongsak U, Boltax J, Grall F, Dube A, Weiss A, Brown L, Quinn G, Kas K, Endress G, Kunsch C, Libermann TA: PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem 2000, 275:1216-1225 [DOI] [PubMed] [Google Scholar]
- 50.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H: Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 1994, 54:5474-5478 [PubMed] [Google Scholar]
- 51.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Hittmair A, Zhang J, Thurnher M, Bartsch G, Klocker H: Regulation of prostatic growth and function by peptide growth factors. Prostate 1996, 28:392-405 [DOI] [PubMed] [Google Scholar]