

Abstract
To determine the regulatory role of plasminogen in hepatic repair following a chronic liver injury, we injected carbon tetrachloride (CCl4) biweekly into mice lacking plasminogen (Plg0) and plasminogen-sufficient littermates (Plg+). On gross examination, we found that Plg0 livers became enlarged and pale with foci of red nodules as early as 4 weeks after CCl4 injection, while Plg+ livers appeared minimally affected by 6 weeks. Microscopically, Plg0 livers had a pronounced pericentral linking, with accumulation of centrilobular eosinophilic material in injured areas, which resulted in a significant increase in liver mass and total protein. Immunohistochemistry revealed that fibrin accumulated progressively in injured regions. However, the combined genetic loss of plasminogen and fibrinogen did not correct the abnormal phenotype. Mason’s trichrome staining revealed intense signal in centrilobular regions and electron microscopy showed a marked increase in fibrillary material demonstrating an excessive accumulation of extracellular matrix in Plg0 mice. The zone-specific increase in matrix components was associated with an increase in the number of activated hepatic stellate cells within injured sites of Plg0 livers. Taken together, these data suggest that the progressive accumulation of fibrin-unrelated matrix substrates in Plg0 livers after a chronic injury results from the combined effects of impaired proteolysis and increased matrix production.
The extracellular matrix (ECM) of the liver provides physical support for parenchymal and non-parenchymal cells, modulates cell differentiation and migration, and facilitates nutrient exchange between blood and hepatocytes. 1,2 ECM is a complex scaffolding of macromolecules including three main components: collagens, glycoproteins, and proteoglycans. Components of the ECM change drastically after a liver injury with low-density, basement-membrane ECM organizing into a high-density, interstitial ECM. These changes must be reversed by tissue and serine proteases for the liver to restore a normal lobular architecture. For example, matrix metalloproteinase (MMP)-1, MMP-2, and MMP-9 are tissue proteases with important proteolytic properties for the hepatic matrix. 3,4 Recently, we demonstrated that plasminogen plays a central regulatory role in hepatic matrix remodeling in vivo. 5
Plasminogen is a liver-derived serine protease with a well-established role in vascular hemostasis, cell migration, and tissue remodeling through fibrin degradation. 6 The genetic inactivation of the plasminogen gene in mice allows for normal development, growth to adulthood, and reproduction. 6 However, plasminogen deficiency results in the multi-system accumulation of fibrin, which creates a physical impediment to epithelial cell migration and leads to microvascular thrombosis, delayed tissue repair, and a reduced life expectancy. 6-11 Plasminogen is also central to the control of liver repair after an acute injury, but does so in a fashion independent of fibrin degradation. Without plasminogen, restoration of normal hepatic architecture is impaired following an acute toxic injury with persistence of necrotic hepatocytes within injured zones and poor reorganization of hepatic matrix in the immediate reparative process. 5 Because of the defective matrix remodeling in these livers, we reasoned that plasminogen-deficient mice could provide a valuable in vivo model to study the role of impaired proteolysis in fibrogenesis. To further explore the regulatory role of plasminogen in remodeling of matrix components in a model of hepatic fibrogenesis, we induced chronic liver injury in plasminogen-deficient mice. Our working hypothesis was that absence of plasminogen leads to the accumulation of matrix components and abnormal lobular reorganization following a chronic liver injury.
Materials and Methods
Reagents and Instruments
Carbon tetrachloride (CCl4) was obtained from Aldrich (Milwaukee, WI). The sedatives ketamine and xylazine were obtained from Phoenix Pharmaceuticals (St. Joseph, MO) and acepromazine maleate was obtained from Fort Dodge Laboratories (Fort Dodge, IA). The Bio-Rad protein assay was purchased from Bio-Rad (Hercules, CA). Cell proliferation was determined using the cell proliferaton kit (Amersham Pharmacia, Piscataway, NJ). The Vectastatin ABC-AP and -HRP detection systems (Vector Laboratories, Burlingame, CA) and Fast Red TR/Naphthol AS-MX (Sigma, St. Louis, MO) were used for fibrin immunohistochemistry. Staining of hepatic stellate cells was performed using anti-human smooth muscle actin/anti-human smooth muscle actin-alpha (α-SMA) antibody containing horseradish peroxidase and anti-human desmin antibody (Dako, Denmark) and M.O.M. immunodetection kit solution (Vector Laboratories). Gels containing 8% Tris-glycine acrylamide, 10% gelatin gels, and PVDF membranes for Western blotting were obtained from Novex (San Diego, CA). A Storm 860 processor (Molecular Dynamics, Inc., Sunnyvale, CA) was used to detect specific signals from chemifluorescence.
Gene-Targeted Mice
Mice with a targeted disruption of the genes coding for plasminogen (Plg0), fibrinogen (Fib0), or both (Plg0/Fib0) were 1 to 4 months old and of a mixed genetic background and, therefore, all studies were conducted using littermate controls. 6,12,13 Polymerase chain reaction-based genotyping was performed as previously described. 5 Animal protocols were approved by the Institutional Animal Care and Use Committee of the Children’s Hospital Research Foundation.
Liver Injury
Gene-targeted and control mice were injected biweekly with 0.25 μl of CCl4 per gram body weight as a 12.5% solution in corn oil intraperitoneally. 14,15 Mice were examined daily and sacrificed after 1, 4, and 6 weeks of injections. A separate group of Plg0 and Plg+ mice were injected with 0.9% NaCl (saline) in a similar fashion and served as controls. At the time of sacrifice, mice were weighed and anesthesized intramuscularly with 0.1 ml of ketamine:xylazine:acepromazine (4:1:1) per 30 g of body weight. Blood samples were obtained by cannulation of the inferior vena cava. The liver was removed and weighed, and the anterior lobe of the liver was fixed in 10% formalin overnight, followed by paraffin embedding. Determination of serum albumin, alanine aminotransferase (ALT), and bilirubin was performed in plasma using an automated enzymatic assay with the Vistros Chemistry Systems 950. 16
Protein and DNA Contents and Cell Proliferation
Frozen liver samples were used for determination of protein (Bio-Rad Assay) and DNA contents. 16,17 The proliferative response after CCl4 injury was measured by the incorporation of bromodeoxyuridine (BrdU) into hepatocytes detected immunohistochemically in liver sections as previously described. 16 For each liver sample, the hepatocyte-labeling index (percentage of hepatocytes incorporating BrdU) was calculated by counting BrdU-labeled and unlabeled hepatocytes in 10 high-power fields (HPF) (100 hepatocytes nuclei per field) by an investigator unaware of the mouse genotype. 16
Fibrinogen and Trichrome Stains
Fibrinogen immunostaining of liver sections was performed using rabbit anti-mouse fibrinogen antiserum, the Vectastatin ABC-AP detection system, and Fast Red TR/Naphthol AS-MX as described previously. 6 The standard Mason’s trichrome staining procedure was used to detect ECM components (eg, collagen) in liver sections of control and experimental animals.
Hepatic Stellate Cell Detection
Paraffin-embedded sections of mouse liver were deparaffinized with xylene and treated with 3% hydrogen peroxide to inhibit endogenous peroxidase. Following antigen retrieval, 16 sections were initially incubated with blocking reagent. For desmin immunostaining, anti-human desmin antibody was applied to the sections followed by biotinylated anti-mouse IgG. Specific signal was obtained with the Vectastatin ABC reagent and diaminobenzidine substrate, and counterstaining with Harris’ hematoxylin. The same protocol was followed for α-SMA immunostaining in liver sections, except that incubation with a secondary antibody was not necessary because horseradish peroxidase was already present on the antibody. The number of desmin or α-SMA-labeled hepatic stellate cells (HSCs) of each liver sample was calculated by counting stained HSCs in one high-power field above and below 10 central veins and portal tracts by an investigator unaware of the mouse genotype.
Gelatin-Based Zymography and Western Blotting
Gelatin-based zymography was performed as previously described. 18 Briefly, liver samples were homogenized and 240 μg of protein lysate from each liver sample was mixed with non-reducing sample buffer and loaded into gelatin-containing polyacrylamide gels. Following electrophoresis, the gels were incubated with 2.5% Triton-X solution twice for a total of 60 minutes and were incubated overnight in 0.1 mol/L glycine solution at 37°C. The gels were then stained with Coomassie blue solution and destained in a solution of 10% acetic acid and 20% methanol to reveal lytic zones. Positive controls consisted of 1 μg of human recombinant MMP-9 (Oncogene Research Products, San Diego, CA). The relative concentration of active MMP-9 in tissue extracts was established by Western analysis as described previously, using antibodies that recognize the active form of the metalloprotease (Oncogene Research Products). Detection of a specific signal was obtained by chemifluorescence (Chemicon International, Temecula CA). To control for loading variability, the relative concentration of albumin was determined using an anti-mouse albumin antibody (Bethyl Laboratories, Montgomery, TX).
Statistical Analysis
Values are shown as means ± standard deviation (S.D.) Statistical significance between two genotypes was assessed by the unpaired t-test. If more than two genotypes were compared, statistical analysis was performed using the General Linear Model and PDIFF option in SAS version 8.01 for Windows. One-way analyses of variance were performed using the number of hepatic stellate cells, liver mass, serum ALT, and percentage of positive-BrDU cells as dependent variables and genotype as the independent variable. The PDIFF option was used to generate pair-wise comparisons among genotypes. A p-value of 0.05 or less was considered significant.
Results
Abnormal Remodeling in Plg0 Livers
To examine the impact of plasminogen deficiency on matrix remodeling following a chronic injury, we injected CCl4 biweekly into both Plg+ and Plg0 mice for 1, 4, or 6 weeks. Livers of Plg+ mice had a normal appearance in the first 4 weeks of injections and exhibited a mild lacy appearance after 6 weeks. In contrast, livers of Plg0 mice exhibited a diffusely lacy and pale appearance at 1 and 4 weeks, becoming enlarged with a grossly pale micronodular surface with scattered red foci at 6 weeks (Figure 1) ▶ . Saline-treated mice of both genotypes exhibited a normal hepatic appearance throughout the experiment. Microscopically, Plg+ livers developed pericentral necrosis 48 hours after the first dose of CCl4, and this lesion resolved after the first week of CCl4 injections. Plg+ livers subsequently appeared normal through 4 weeks of CCl4; at 6 weeks, minimal accumulation of eosinophilic material was noted in centrilobular area. A more pronounced accumulation of this material was already present in the centrilobular areas of Plg0 livers at 1 week. After 6 weeks of biweekly CCl4 injections, Plg0 livers displayed a pattern of pericentral bridging that, together with persistent necrotic cells, produced the micronodular/pale appearance, with remaining normal periportal hepatocytes comprising the red foci (Figure 2) ▶ . Taken together these data show that livers of Plg+ mice are able to restore and maintain normal lobular organization until 6 weeks of biweekly CCl4 injections. In contrast, livers of Plg0 mice already display a persistent centrilobular injury following two injections of CCl4 (first week of injections) in a similar fashion to the outcome previously reported following an acute CCl4 insult. 5 Thereafter, this disordered lobular structure progresses in severity over time.
Figure 1.

Plg0 livers develop an early diseased appearance following CCl4. Plg+ livers display a lacy appearance following 6 weeks of CCl4 injections. In contrast, Plg0 livers are lacy and pale at 1 and 4 weeks and become enlarged with a pale micronodular surface and red foci at 6 weeks. Control livers are from Plg+ and Plg0 mice injected with saline biweekly for 6 weeks.
Figure 2.

Accumulation of eosinophilic material in Plg0 livers. Plg+ livers maintain a normal histological appearance until 4 weeks of CCl4 injections when they develop a mild accumulation of eosinophilic material (arrows) in the centrilobular area. Plg0 livers have a more prominent accumulation of this material in the centrilobular area by 1 week that progresses to pericentral linking by 4 weeks. Normal histology is observed in control livers from Plg+ and Plg0 mice injected with saline biweekly for 6 weeks. Magnification, × 200.
Despite the striking histological difference between the genotypes, ongoing hepatocellular injury was mild as demonstrated by an increase in the plasma ALT of only 28 ± 46% in Plg+ mice and 66 ± 77% in Plg0 mice at 6 weeks of injections above pretreatment values (baseline levels for ALT in saline-treated mice, mean ± SD: 86.8 ± 34.6 IU/L). Consistent with these findings, we did not find a significant difference between the proliferative response of the genotypes with BrdU labeling ranging from 0.5 to 0.7% for Plg+ livers and 0.9 to 1.7% for Plg0 livers when measured at 1, 4, and 6 weeks. These data suggest that the low-grade proliferative response that follows chronic injury provides sufficient cellular mass to maintain adequate metabolic and excretory functions. This view is also supported by maintenance of normal plasma albumin and bilirubin levels throughout the study period (data not shown).
Because of the enlarged appearance of the Plg0 livers and the increase in eosinophilic material noted microscopically, liver mass was determined. Liver mass of Plg+ mice increased by 22 ± 13% in mice treated with CCl4 above saline-treated mice at 6 weeks (p = 0.002). However, a much more pronounced increase was observed in livers of Plg0 mice, which were 77 ± 25% larger than saline-treated Plg0 livers at one week (p = 0.007) and reached a maximum increase of 317 ± 72% at 6 weeks (p < 0.001). This increase was not due to differences in hepatic DNA content (543 ± 94 μg and 500 ± 146 μg at 6 weeks for Plg+ and Plg0, respectively, p = 0.46). Rather, this difference was largely a function of increased total protein in livers of Plg0 mice (Figure 3A) ▶ . Based on immunohistochemical analysis, one component of the accumulated protein was fibrin, which is one established proteolytic target of plasmin (Figure 3B) ▶ . Therefore, we next determined the mechanistic role of fibrin accumulation in the inability of Plg0 livers to restore lobular integrity during chronic injury.
Figure 3.
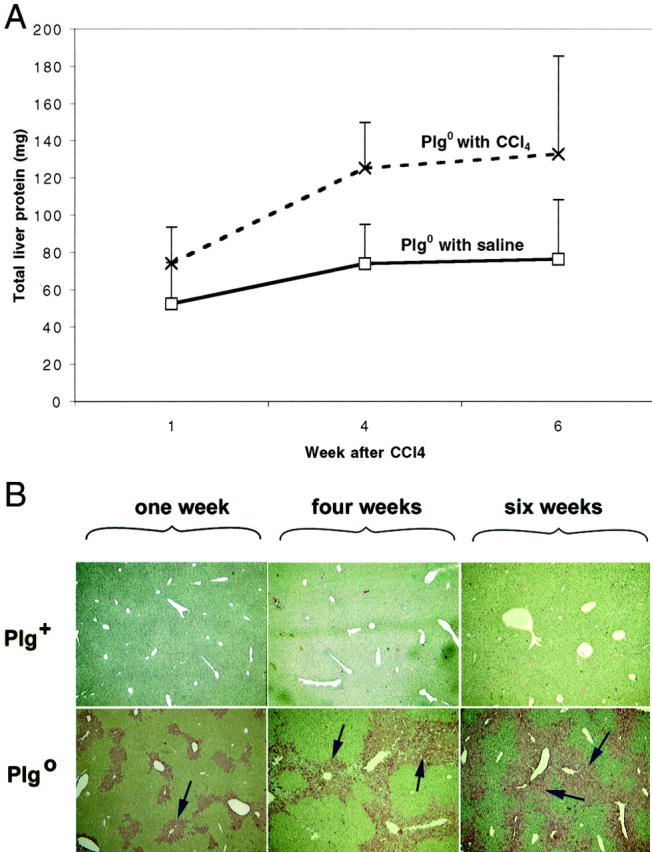
A: Protein content of Plg0 livers increases following CCl4. Plg0 livers display a progressive increase in the protein content following CCl4 administration reaching a level of 40 to 75% above Plg0 saline controls throughout the experiment (p < 0.04). B: Fibrin accumulation in Plg0 livers. Immunohistochemistry shows that Plg+ livers have minimal accumulation of fibrin following CCl4 injections, while Plg0 livers have a progressive accumulation of fibrin (arrows) in the centrilobular regions at 1 to 6 weeks. Magnification, × 200.
Fibrin Deficiency Does Not Prevent Abnormal Matrix Deposition in Plg0 Livers
To directly determine the role of fibrin(ogen) in the abnormal repair observed in Plg0 mice following a chronic injury, we administered CCl4 to mice with a combined deficiency in plasminogen and fibrinogen. We found an early development of gross and microscopic injury in Plg0/Fib0 livers, similar to that displayed in Plg0 mice. Interestingly, livers of Fib0 mice developed a mild lacy appearance by 6 weeks of treatment that was similar to livers of Plg+/Fib+ mice (Figure 4) ▶ . Microscopically, a considerable accumulation of eosinophilic material was present in the centrilobular regions of Plg0/Fib0 at 4 weeks, which progressed to a pattern of pericentral bridging after 6 weeks of CCl4 injections (data not shown), indistinguishable from that displayed by Plg0 livers after the same length of repeated toxic injury (Figure 2) ▶ . These data demonstrate that fibrin accumulation in Plg0 livers is not solely responsible for the defect in repair, and that fibrin deposition is not mechanistically coupled to the abnormal matrix deposition.
Figure 4.

Fibrin deficiency does not prevent abnormal matrix deposition. Plg0/Fib0 livers develop an early pale and lacy appearance at 1 week of treatment with CCl4 that progresses to a pale micronodular appearance with scattered small red foci by 6 weeks. Fib0 livers demonstrate a mild, lacy appearance at 6 weeks which is similar to Plg+Fib+ livers.
To determine the nature of the material accumulating in bridging centrilobular areas of Plg0 livers, we examined liver specimens after 4 weeks of CCl4 administration using trichrome staining and electron microscopy. Centrilobular areas of Plg0 livers were intensely stained with Mason’s trichrome (Figure 5) ▶ . At the ultrastructural level, Plg+ livers had small amounts of collagen-like fibrillary material surrounding the central veins, whereas Plg0 livers had a marked increase in the amount of fibrillary material in the sinusoids and subendothelial spaces (Figure 5) ▶ . Taken together, these data identified a prominent accumulation of hepatic ECM in mice lacking plasminogen. Given that plasminogen is relatively poor at degrading collagen but a reasonably effective activator of pro-MMPs in vitro, collagen accumulation in hepatic tissues may result from a failure of plasmin-mediated pro-MMP activation.
Figure 5.
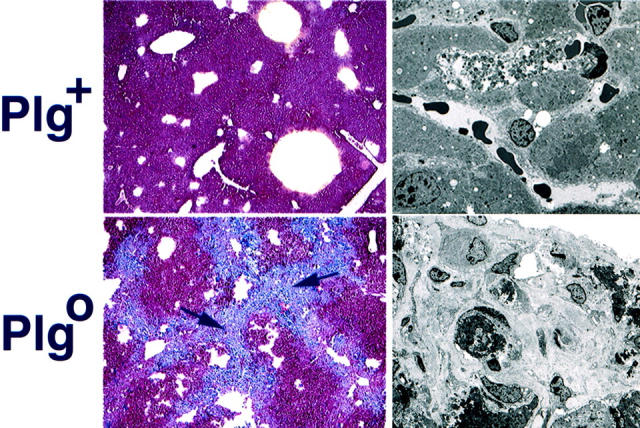
Accumulation of ECM substrates in Plg0 livers. Following 4 weeks of biweekly CCl4 injections, Plg+ livers have no appreciable trichrome staining in the centrilobular region (left). In contrast, Plg0 livers have intense staining at the centrilobular regions and in areas of pericentral linking (arrows; magnification, × 200). Electron microscopy (right) shows a small amount of fibrillary material surrounding the central vein of Plg+ livers. In contrast, Plg0 livers demonstrate a large increase in collagen-like fibrillary material in the areas of defective repair.
MMP-9 Activity Is Not Decreased in Plg0 Livers after Chronic Liver Injury
A potential relationship between plasmin and pro-MMP-9 activation has been proposed in extra-hepatic sites. 8 Therefore, we determined the impact of plasminogen deficiency on hepatic MMP-9 activity in livers collected from Plg+ and Plg0 mice following chronic injection of CCl4 or saline. Gelatin-based zymography assays with liver extracts showed that saline-treated livers contained only small amounts of gelatinase activity. The amount of gelatinase activity in the liver increased similarly in mice of both genotypes after 1 and 4 weeks of CCl4 administration. However, after 6 weeks, the gelatinase activity present in liver extracts from Plg0 mice appeared to be greater than in liver extracts from Plg+ mice (Figure 6A) ▶ . Using an antibody that specifically detects only activated MMP-9, Western blot analysis confirmed that activated MMP-9 was present in the protein lysates of both genotypes treated with CCl4 (Figure 6B) ▶ . These data suggest that the accumulation of collagen-like fibrillary material in Plg0 livers is not due to diminished activation of MMP-9 in the absence of plasmin, but, rather, to the consequences of the failure of plasmin in some other as yet unidentified proteolytic pathways.
Figure 6.
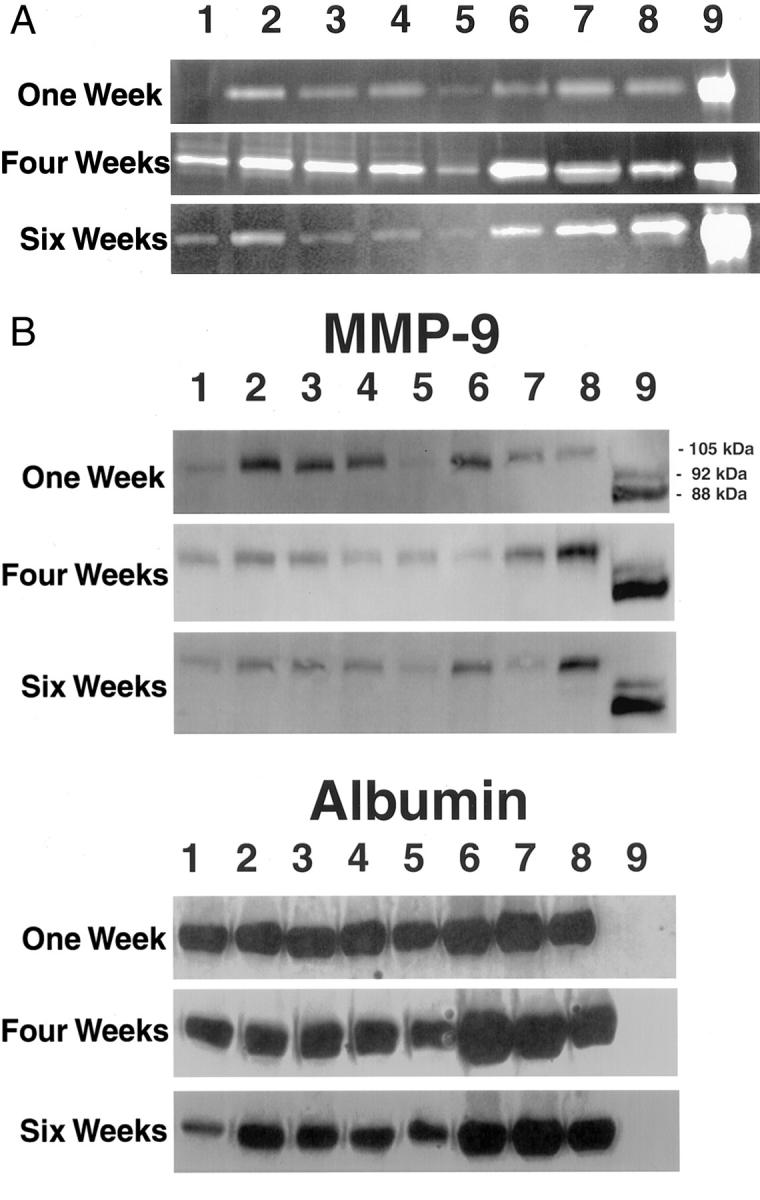
A: MMP-9 activation in Plg+ and Plg0 livers. Gelatin zymography suggests that there is only small amount of MMP-9 in liver extracts prepared from Plg+ (lane 1) and Plg0 (lane 5) mice treated with saline. MMP-9 activity appears to be increased in liver extracts from Plg+ (lanes 2–4) and Plg0 (lanes 6–8) mice treated with CCl4. No significant genotype-dependent difference in MMP-9 activity is observed at either 1 or 4 weeks, but high activity appears to be present in liver extracts of Plg0 mice at 6 weeks. B: Activated MMP-9 is present in Plg+ and Plg0 following chronic toxic injury. Using an antibody that recognizes activated murine MMP-9 (105 kd), a similar level of specific signal is present in both genotypes throughout the experiment. To control for loading variability, Western blotting for albumin is also shown. Lane 1 contains Plg+ liver homogenate treated with saline, while lanes 2–4 contain Plg+ homogenates treated with CCl4. Lane 5 contains Plg0 liver homogenate treated with saline, while lanes 6–8 contain Plgo liver homogenates treated with CCl4. Lane 9 contains human MMP-9 (proenzyme 92 kd, active enzyme 88 kd).
Increased Activation of Hepatic Stellate Cells in Plg0 Livers
In addition to impaired proteolysis, centrilobular matrix accumulation may also result from increased ECM production. Following an injury, production of fibrillary and non-fibrillary matrix components derives primarily from activation of HSCs. 2,3 To determine whether activation of HSCs may contribute to the excessive ECM accumulation observed in Plg0 livers, we determined the number of α-SMA-stained HSCs following CCl4. Plg0 mice treated with CCl4 had a significantly increased number of activated HSCs in both periportal and pericentral regions compared to Plg+ mice treated with CCl4 at 6 weeks (p < 0.004) (Table 1) ▶ . HSCs within the unaffected parenchyma did not display α-SMA staining but could be identified with anti-desmin antibody, suggesting that activation of HSCs was restricted to zones of injury (data not shown). Notably, desmin staining also showed that the total number of HSCs in the zone of injury did not change between genotypes (Table 1) ▶ . These data suggest that plasminogen deficiency leads to persistent activation of HSCs within the diseased microenvironment, which may establish an ongoing state of matrix production and further impair the defective liver repair of Plg0 mice.
Table 1.
Number of α-SMA- and Desmin-Stained HSCs in Livers of Plg+ and Plg0 Mice after 6 Weeks of CCl4 (mean ± SD)
| Immunostaining | Lobular region | Plg+ livers | Plg0 livers |
|---|---|---|---|
| Anti-α-SMA | Periportal | 0.55 ± 0.42* | 1.53 ± 0.4* |
| Pericentral | 4.43 ± 3.85** | 12.33 ± 1.66** | |
| Anti-desmin | Periportal | 6.96 ± 2.2 | 6.03 ± 3 |
| Pericentral | 8.96 ± 2.68 | 12.2 ± 0.46 |
*p = 0.003.
**p = 0.001.
Discussion
Our studies demonstrate that plasminogen is critical to the hepatic repair/remodeling following a chronic toxic injury. Mice lacking this key protease fail to clear necrotic tissue (particularly in the pericentral regions), repopulate, and organize the diseased areas by regenerating hepatocytes. As a consequence of chronic injury, livers of Plg0 mice have marked derangement in the lobular architecture, with progressive accumulation of eosinophilic material within injured zones that leads to a pale gross appearance as early as one week from the onset of toxic injury. Despite the diseased appearance, livers of Plg0 mice have an appropriate hepatocellular proliferation in response to the chronic toxic insult and maintain a normal synthetic function. These data suggest that the primary consequences of impaired plasminogen-mediated proteolysis are defective clearance of necrotic cells and a disruption of the balance between proteolytic clearance and production of hepatic matrix.
The prominent and persistent defect in the clearance of necrotic tissues following chronic toxic injury in plasminogen-deficient mice is qualitatively similar to the defect previously reported following an acute injury. 5 Specific to chronic injury, however, is the progressive retention of necrotic cells and accumulation of matrix components that result in a greater increase in liver mass. In extra-hepatic tissues, fibrin accumulation is the main mechanism of defective wound healing in the plasminogen-deficient state, and accumulation of fibrin appears to form a physical barrier to the migration of epithelial cells from the wound edges. 7,9 The removal of fibrin completely rescued plasminogen-deficient mice from defective wound healing in both skin and cornea. 12 Although fibrin is an important component of the provisional hepatic matrix following an injury, 19 the findings presented here show that genetically superimposed loss of fibrinogen in plasminogen-deficient mice does not correct the reparative failure in the liver. These data point to the existence of one or more fibrin-unrelated targets for plasmin-mediated proteolysis that are important for hepatic repair within cellular and extracellular matrices.
Plasminogen may be involved in the clearance of cellular and extracellular components either directly or through activation of MMPs or growth factors. 20 MMPs are directly involved in matrix degradation and have often been proposed to be linked to the plasminogen activator/plasminogen system of proteases. 21,22 Plasminogen has been shown to directly activate MMPs in vitro. 4 Impairment in plasminogen activation has been suggested to result in impaired activation of MMP-9 and defective scar formation in a model of acute myocardial infarction in vivo. 8 In the liver, we did not find that activation of MMP-9 is impaired in the absence of plasminogen despite the obvious deposition of collagen-like fibrillary material in the areas of abnormal repair. Nevertheless, the available data do not exclude the possibility that plasminogen is required for activation of other pro-MMPs that may be involved in liver regeneration and repair. 1,23,24
The prominent activation of collagen-producing HSCs in the livers of CCl4-treated Plg0 mice suggests that production, rather than proteolytic matrix turnover, may be primarily responsible for the observed accumulation of fibrillary material in these mice. In addition to impaired proteolytic removal of ECM components in livers of Plg0 mice, matrix accumulation also results from increased production, as demonstrated by an increase in the activation of HSCs. On injury, HSCs proliferate, display a myofibroblast phenotype, and produce matrix substrates. 2,3 This response to injury was not affected by the plasminogen status, but the absence of plasminogen clearly led to a more pronounced activation of HSCs within the injured microenvironment. Although the current studies do not formally determine which molecular factor(s) direct the phenotype of HSCs, one hypothesis consistent with the current findings is that matrix and cellular debris in injured centrilobular areas provide signals that favor activation of HSCs and persistent biosynthesis of collagen-rich matrices. As a consequence, the progressive accumulation of matrix substrates in livers of Plg0 mice results from the combined effects of impaired proteolysis and increased matrix production.
Plasminogen-deficiency provides a valuable in vivo model for studies concerning the role of impaired proteolysis in hepatic matrix production and clearance following chronic liver injury. The molecular mechanisms regulating plasmin-driven matrix proteolysis outside the context of fibrin degradation have not yet been uncovered. Future studies quantifying the impact of plasminogen deficiency on whole tissue proteolysis and the identification of remaining matrix components that trigger and maintain activation of HSCs will be important to define the direct role of plasmin-mediated proteolysis on fibrogenesis. Here, our studies clearly show that plasminogen deficiency leads to a significant accumulation of matrix components after a chronic injury. Since excessive accumulation of matrix elements plays an integral role in this abnormal liver repair, Plg0 mice may be useful in addressing whether therapeutically targeted plasmin-mediated proteolysis can correct alterations in matrix homeostasis.
Acknowledgments
We thank Alicia Emley for assistance with illustrations and Dr. William Balistreri for insightful review of the manuscript.
Footnotes
Address reprint requests to Jorge A. Bezerra, Division of Pediatric Gastroenterology, Hepatology, and Nutrition, Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229-3039. E-mail: jorge.bezerra@chmcc.org.
Supported by National Institutes of Health training grant DK 07727 (J.F.P.), HL 47826 (J.L.D.), and DK 55710 (J.A.B).
Current address for John F. Pohl is the Division of Pediatric Gastroenterology, Scott and White Memorial Hospital, Texas A & M University Health Sciences Center, Temple, TX 76508, and the current address for Hector Melin-Aldana is the Division of Pathology, Children’s Memorial Hospital, Northwestern University, Chicago, IL 60614.
References
- 1.Kim TH, Mars WM, Stolz DB, Petersen BE, Michalopoulos GK: Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997, 26:896-904 [DOI] [PubMed] [Google Scholar]
- 2.Li D, Friedman SL: Liver fibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy. J Gastroenterol Hepatol 1999, 14:618-633 [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL: Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis: mechanisms and treatment strategies. N Engl J Med 1993, 328:1828-1835 [DOI] [PubMed] [Google Scholar]
- 4.Lijnen HR, Collen D: Matrix metalloproteinase system deficiencies and matrix degradation. Thromb Haemost 1999, 82:837-845 [PubMed] [Google Scholar]
- 5.Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, Degen JL: Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver. Proc Natl Acad Sci USA 1999, 96:15143-15148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bugge TH, Flick MJ, Daugherty CC, Degen JL: Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 1995, 9:794-807 [DOI] [PubMed] [Google Scholar]
- 7.Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K: Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 1996, 2:287-292 [DOI] [PubMed] [Google Scholar]
- 8.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P: Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 1999, 5:1135-1142 [DOI] [PubMed] [Google Scholar]
- 9.Drew AF, Schiman HL, Kombrinck KW, Bugge TH, Degen JL, Kaufman AH: Persistent corneal haze after excimer laser photokeratectomy in plasminogen-deficient mice. Invest Ophthalmol Vis Sci 2000, 41:67-72 [PubMed] [Google Scholar]
- 10.Creemers E, Cleutjens J, Smits J, Heymans S, Moons L, Collen D, Daemen M, Carmeliet P: Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol 2000, 156:1865-1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmeliet P, Collen D: Development and disease in proteinase-deficient mice: role of the plasminogen, matrix metalloproteinase, and coagulation system. Thromb Res 1998, 91:255-285 [DOI] [PubMed] [Google Scholar]
- 12.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ, Degen JL: Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell 1996, 87:709-719 [DOI] [PubMed] [Google Scholar]
- 13.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL: Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev 1995, 9:2020-2033 [DOI] [PubMed] [Google Scholar]
- 14.Tamayo RP: Is cirrhosis of the liver experimentally produced by CCl4 an adequate model of human cirrhosis? Hepatology 1983, 3:112-120 [DOI] [PubMed] [Google Scholar]
- 15.Brenner DA, Veloz L, Jaenisch R, Alcorn JM: Stimulation of the collagen alpha 1 (I) endogenous gene and transgene in carbon tetrachloride-induced hepatic fibrosis. Hepatology 1993, 17:287-292 [PubMed] [Google Scholar]
- 16.Yazigi NA, Carrick TL, Bucuvalas JC, Schmidt CS, Balistreri WF, Bezerra JA: Expansion of transplanted hepatocytes during liver regeneration. Transplantation 1997, 64:816-820 [DOI] [PubMed] [Google Scholar]
- 17.Gendimenico GJ, Bouquin PL, Tramposch KM: Diphenylamine-colorimetric method for DNA assay: a shortened procedure by incubating samples at 50°C. Anal Biochem 1988, 173:45-48 [DOI] [PubMed] [Google Scholar]
- 18.Brown PD, Bloxidge RE, Anderson E, Howell A: Expression of activated gelatinase in human invasive breast carcinoma. Clin Exp Metastasis 1993, 11:183-189 [DOI] [PubMed] [Google Scholar]
- 19.Neubauer K, Knittel T, Armbrust T, Ramadori G: Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology 1995, 108:1124-1135 [DOI] [PubMed] [Google Scholar]
- 20.Vassalli JD, Sappino AP, Belin D: The plasminogen activator/plasmin system. J Clin Invest 1991, 88:1067-1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy G, Docherty AJ: The matrix metalloproteinases and their inhibitors. Am J Respir Cell Mol Biol 1992, 7:120-125 [DOI] [PubMed] [Google Scholar]
- 22.Okazaki I, Watanabe T, Hozawa S, Arai M, Maruyama K: Molecular mechanism of the reversibility of hepatic fibrosis: with special reference to the role of matrix metalloproteinases. J Gastroenterol Hepatol 2000, 15(Suppl):D26-D32 [DOI] [PubMed] [Google Scholar]
- 23.Haruyama T, Ajioka I, Akaike T, Watanabe Y: Regulation and significance of hepatocyte-derived matrix metalloproteinases in liver remodeling. Biochem Biophys Res Commun 2000, 272:681-686 [DOI] [PubMed] [Google Scholar]
- 24.Kim TH, Mars WM, Stolz DB, Michalopoulos GK: Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology 2000, 31:75-82 [DOI] [PubMed] [Google Scholar]