

Abstract
Plasminogen activators (PAs) and matrix metalloproteinases (MMPs) are considered to play an important role in the pathogenesis of multiple sclerosis. Experimental autoimmune encephalomyelitis (EAE) is widely used as an animal model of multiple sclerosis. Whereas several studies have addressed the expression of various MMPs and their inhibitors in the pathogenesis of EAE, the expression of the molecules of the PA system during EAE has not been reported previously. The present study was undertaken to investigate the expression of the molecules of the PA system (tPA, uPA, PAI-1, uPAR, LRP), as well as several members of the MMP family and their inhibitors in the course of actively induced EAE in BALB/c mice. During clinical EAE, the PA system was up-regulated in the central nervous system at several levels. Induction of expression of tPA and PAI-1 transcripts was detected in activated astrocytes in the white matter. Inflammatory cells expressed uPA receptor, uPAR. In situ zymography demonstrated the presence of increased tPA and uPA activities in the areas of the inflammatory damage. Accumulation of fibrin, fibronectin, and vitronectin immunoreactivity was seen in perivascular matrices of symptomatic animals. In addition, transcription of MT1-MMP and metalloelastase (in inflammatory cells), and TIMP-1 (in activated astrocytes) was induced during EAE. Increased gelatinolytic activity was detected at the sites of inflammatory cell accumulation by in situ zymography of fluorescently labeled gelatin; substrate gel zymography identified the up-regulated gelatinolytic activity as gelatinase B. Overall, our study demonstrates concurrent induction of PA and MMP systems during active EAE, supporting further the concept that the neuroinflammatory damage in EAE involves altered balance between multiple extracellular proteases and their inhibitors.
Behavior of cells depends critically on their interactions with the surrounding microenvironment: extracellular matrix (ECM), other cells, and different regulatory molecules. 1 Extracellular proteolysis represents a potent and irreversible mechanism of modulating all these interactions, and of remodeling structural components of the tissue. Regulated extracellular proteolysis is critical for physiological processes. Importantly, it is also involved in a number of pathological conditions such as tumor invasion, inflammation, tissue repair, and excitoxicity. 2,3 The two best-characterized groups of extracellular proteolytic enzymes are the plasminogen activators/plasmin (PA) system 4 and matrix metalloproteinases (MMPs). 5 Proteases of both families are secreted as inactive precursors along with their inhibitors (PA inhibitors, PAIs, and tissue inhibitors of metalloproteinases, TIMPs), to ensure stringent regulation of potentially deleterious proteolytic activity. Rather than being independent entities, the PA and MMP systems interact; plasmin has been demonstrated to act as a physiologically relevant activator of pro-MMPs. 6-8
Extracellular proteolytic enzymes are implicated as pathogenic factors in demyelinating neuroinflammatory disorders such as multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). In particular, the role of MMPs in inflammatory demyelination is well established, and it is supported by several lines of correlative and functional evidence. Increased levels of MMPs have been found in the cerebrospinal fluid in MS and EAE, and these enzymes are up-regulated in situ, in patterns suggestive of an active role in inflammation and demyelination. 9-12 Furthermore, inhibition of metalloproteinases in EAE has been shown to suppress the development of clinical EAE in a dose-dependent way, and to restore the damaged blood-brain barrier in the clinical phase of the disease. 13-16 On the other hand, the data on the involvement of the PA system in the pathogenesis of EAE are more fragmentary. In early studies, the nonspecific inhibitor of plasmin, tranexamic acid, was shown to suppress development of EAE. 17 Later, in situ up-regulation of tPA and PAI-1 in MS was demonstrated. 11,18 Our finding of elevated tPA and PAI-1 antigen levels in the cerebrospinal fluid of MS patients 19 further supports the involvement of the PA system in inflammatory demyelination. It is likely that the pathophysiological process of evolution of MS involves a concerted action of several enzymes of different proteolytic pathways. Mapping the spatiotemporal expression patterns of multiple genes encoding ECM proteinases is important for better understanding of the mechanisms of pathogenesis of EAE, and may lead to novel therapeutic strategies of selectively interfering with the action of critical proteolytic enzymes.
The present study was designed to describe the patterns of expression and activity of proteinases of the plasminogen activation and MMP pathways, as well as their regulatory molecules (inhibitors, receptors) in the central nervous system (CNS) during acute EAE. We show that the plasminogen activation system is induced during clinical EAE in critical locations of tissue damage and neuroinflammation. The partially overlapping expression patterns of proteinases of PA and MMP families suggest the existence of a functional cooperation between these two major systems of extracellular proteolysis in the pathogenesis of EAE.
Materials and Methods
Induction of EAE
BALB/c AnNHsd (H-2d) (Harlan Laboratories, IN) mice were bred and maintained in the animal facility of Haartman Institute of the University of Helsinki. The protocol of experimental manipulation of the animals was approved by the Bioethics Committee of Southern Finland. Antigen/adjuvant emulsion consisted of a lyophilized mouse spinal cord homogenate (3 mg/ml), suspended in phosphate-buffered saline (PBS), pH 7.4, and emulsified at 1:1 ratio (v/v) in complete Freund’s adjuvant (Sigma, St. Louis, MO) by sonication. EAE was induced in 6- to 10-week-old female mice by injection of the sonicated emulsion into hind footpads, followed by pertussis toxin (List Laboratories, Campbell, CA) administration 24 hours later, according to the protocol developed by Määttä and colleagues. 20 The protocol leads to robust induction of clinical EAE on days 11 to 14 after immunization. The clinical status of the animals was monitored daily, and it was scored as follows: 0, no neurological symptoms; 1, tail atony; 2, slight hind limb paralysis; 3, severe hind limb paralysis; 4, moribund; and 5, dead. EAE was induced in four independent experiments in groups of 20 animals (10 induced and 10 untreated). Brains and spinal cords were collected from symptomatic animals on days 11 to 15 after immunization, as well as from untreated littermates. For each animal, a part of the CNS tissue was snap-frozen in liquid nitrogen, and the rest fixed with freshly prepared 4% paraformaldehyde in PBS overnight at 4°C. The fixed tissues were dehydrated, paraffin-embedded, and sectioned at 6 μm. Serial sections were mounted on Super Frost Plus microscope slides (Menzel-Gläser, Braunschweig, Germany).
In Situ Hybridization
S35-UTP-labeled RNA probes were prepared as described previously. 21 The following probes were used: tPA, 22 uPA, 23 PAI-1, 24 uPAR, 25 LRP, 26 MT1-MMP, 27 gelatinase A, gelatinase B, TIMP-1, TIMP-2, TIMP-3, 28 and metalloelastase. 29 All probes have been used in in situ hybridization analysis in published studies and have been tested for specificity. The linearized plasmids containing cDNA inserts were transcribed in vitro to generate both antisense (hybridizing) and sense (control) riboprobes. The labeled RNAs were reduced to ∼100 bases by alkaline hydrolysis, neutralized, extracted with phenol-chloroform, purified from unincorporated nucleotides by filtration through Sephadex G-50 column (Roche Molecular Biochemicals, Mannheim, Germany), and ethanol precipitated. In situ hybridization was performed essentially as described previously. 30 After hybridization, the slides were dipped into Kodak NTB2 emulsion (Eastman-Kodak, Rochester, NY), exposed at 4°C for 1 to 3 weeks, and after standard development procedures stained with toluidine blue and mounted in DPX mountant. In some cases, coating with the emulsion was preceded by immunohistochemistry with cell-type-specific antibodies. Slides were observed and photographs taken using an Olympus BX-50 microscope.
In Situ Zymography of Plasminogen Activators
Ten-μm cryosections on microscope slides were brought at room temperature; overlaid with a solution containing dry milk, plasminogen, and agar; and incubated in a humid chamber at 37°C as described previously. 22 To distinguish between the two PAs, a specific inhibitor of uPA, amiloride, 38 was included in some reaction mixtures at a final concentration of 0.5 mmol/L. Zymography on sections of kidneys from uPA−/− and tPA−/− animals 31 was used to titrate the amiloride concentration that was sufficient to inhibit uPA completely while only slightly affecting tPA activity. The caseinolytic areas were observed and microphotographed with an Olympus BX-50 microscope under dark-field illumination.
In Situ Zymography of Gelatinases
For detection of gelatinase activities on cryosections we used a novel in situ gelatinolysis assay. 32 The assay is based on the increase of fluorescence of intramolecularly quenched fluorescein isothiocyanate-labeled DQ-gelatin (Molecular Probes, Eugene, OR) on proteolytic cleavage.
Cryosections on Super Frost Plus microscope slides were equilibrated to room temperature, and overlaid with a mixture of 20 μmol/L of DQ-gelatin in 0.05 mol/L Tris-HCl, 0.15 mol/L NaCl, 5 mmol/L CaCl2, and 0.2 mmol/L NaN3, pH 7.6). As a difference from the original protocol, we included in the overlay mixture 1 μg/ml of 4.6-diamidino-2-phenylindole fluorescent nuclear stain, 0.03% Triton X-100, and 0.5% low-melting agarose. Five mmol/L of 1,10-phenanthroline, inhibitor of metalloproteinases, were included in some overlay mixtures.
Immunohistochemistry
Immunohistochemistry was performed on 6-μm paraffin sections and 10-μm cryosections; in the case of paraffin sections, epitope unmasking was performed by treatment of the sections with 0.05% trypsin at 37°C for 10 minutes.
The following primary antibodies were used: rabbit anti-human fibrinogen (DAKO, Glostrup, Denmark), rabbit anti-human fibronectin (DAKO), goat anti-mouse vitronectin (Santa Cruz Biotechnology, Santa Cruz, CA), rat monoclonal anti-mouse Ly-6G (Caltag, Burlingame, CA), rabbit anti-cow glial fibrillary acidic protein (GFAP, DAKO). Lectin histochemistry with biotinylated Lycopersicon esculentum lectin (Sigma, St. Louis, MO) was used for visualization of macrophages/microglia. The secondary reagents used were peroxidase-conjugated anti-species-specific antibodies from DAKO.
Zymography on Polyacrylamide Gels
Brain/spinal cord extracts were analyzed by substrate zymography of polyacrylamide gels (adopted with modifications from Behrendtsen and colleagues 33 ). Tissues were directly solubilized in nonreducing gel-loading buffer, and equal loading of the lanes was assessed by Coomassie Blue staining of the gel. Human fibrosarcoma HT-1080-conditioned medium (positive control of gelatinolysis) was prepared by incubating the cells for 48 hours in serum-free medium. Samples were electrophoresed on nonreducing 10% polyacrylamide-sodium dodecyl sulfate (SDS) gels, co-polymerized with substrate (for gelatinases 1 mg/ml of type B collagen; for PAs 2 mg/ml of nonfat dry milk and 7.5 μg/ml of human plasminogen). After electrophoresis, gels were washed twice for 15 minutes in 2.5% Triton X-100, incubated in 10 mmol/L of CaCl2 and 50 mmol/L of Tris, pH 7.6, for 24 to 40 hours at 37°C and stained with Coomassie Blue. The areas of proteolytic activity appeared as clear areas in the background of blue staining.
Semiquantitative Reverse Transcriptase-Polymerase Chain Reaction for uPA
cDNA synthesis from total RNA of the spinal cords was performed as described previously by Määttä and colleagues. 34 Polymerase chain reaction amplification mixture contained 3.5 mmol/L of MgCl2 for uPA and 2.5 mmol/L of MgCl2 for GAPDH, as well as 0.2 mmol/L of dNTP, 1 μmol/L of each primer, 1× polymerase chain reaction buffer (Fermentas, Vilnius, Lithuania) and 2.5 U/100 μl of Taq DNA polymerase (Fermentas). For GAPDH, commercial primers were used (22 cycles, 30 seconds at 95°C, 30 seconds at 60°C, 30 seconds at 72°C) (TaqMan Rodent GAPDH control kit; Applied Biosystems, Warrington, UK). uPA primers were: 5′-TTACTGCAGGAACCCTGACAAC-3′ (forward) and 5′-GACAAACTGCCTTAGGCCAATC-3′ (reverse). Cycling parameters for uPA were: 32 cycles, 30 seconds at 95°C, 10 seconds at 62°C, 10 seconds at 72°C.
For amplification dilution series of cDNAs were used as templates. Samples were run on agarose gels (45 minutes, 75 V, 1× Tris-acetate EDTA running buffer (TAE)), and images of ethidium bromide-stained gels were captured and analyzed using Gel-Doc 2000 system (Bio-Rad, Richmond, CA). The obtained values for uPA were adjusted relative to the GAPDH mRNA levels and expressed in arbitrary units (AU).
Data Analysis
Spinal cords and brains from 40 mice were sectioned serially and subjected to in situ hybridization and immunohistochemistry. In situ hybridization and immunostaining experiments were repeated at least twice with the same findings. Zymography [in situ and polyacrylamide gel electrophoresis (PAGE)] was repeated three times with similar outcomes. In situ hybridization signal was quantified by KS400 image analyzer (Zeiss, Oberkochen, Germany). Confidence interval (CI) for observed values was calculated with normality assumption.
Results
Induction of EAE in BALB/c Mice and Histological Analysis of CNS Lesions
EAE was induced in BALB/c mice by immunization with sonicated mouse whole spinal-cord extract as described earlier. 35 This protocol leads to highly penetrant acute monophasic disease at days 11 to 14 after immunization, with clinical disease appearing mainly as ascending paralysis. Histological analysis of luxol fast blue/cresyl violet-stained sections of brains and spinal cords of mice with grades 3 and 4 EAE demonstrated numerous foci of inflammatory cell accumulation (Figure 1, A and B) ▶ . Perivascular inflammatory cell infiltrates were observed in all levels of the spinal cord (Figure 1B) ▶ , as well as in pons and medulla (data not shown); meningeal inflammation was present in spinal cord and to a lesser extent in pons/medulla (data not shown). In accordance with the clinical course of the disease, the degree of inflammation decreased rostrally along the spinal cord, and the most prominent inflammatory reaction was present at sacral and lumbar levels, with some predilection for dorsal root entry zones.
Figure 1.
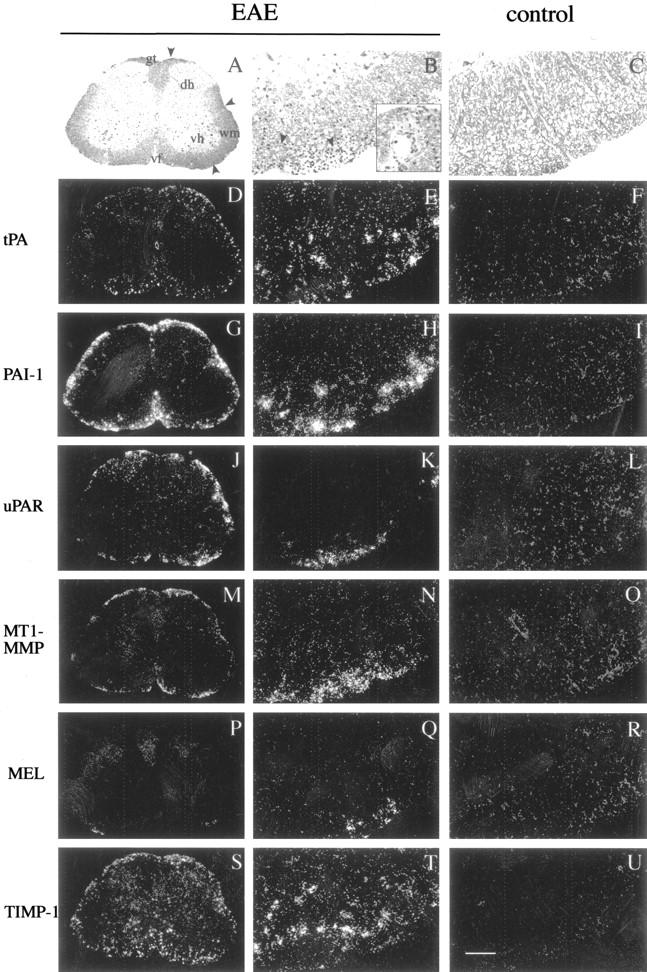
Expression of transcripts of tPA, PAI-1, uPAR, MT1-MMP, metalloelastase (MEL), and TIMP-1 in transversal sections of lumbar spinal cord of mouse with grade 3 EAE (columns 1 and 2) and lumbar spinal cord of control mouse (column 3). A to C: Bright-field images of luxol fast blue/cresyl violet-stained spinal cord sections demonstrate the presence of inflammatory cell deposits (arrowheads) at sites of immune cell accumulation in EAE (A and B) and not in nonsymptomatic spinal cord (C). Inset in B shows a higher magnification view with infiltrating perivascular inflammatory cells. During clinical EAE, tPA is up-regulated in scattered cells in the periphery of the white matter (D and E), whereas its expression in control spinal cord was under the same conditions below the detection limit (F). High levels of PAI-1 transcripts were detected in glial cells adjacent to the inflammatory lesions in spinal cord of symptomatic animals (G and H), whereas control CNS appeared negative (I). Prominent expression of transcripts of uPAR was observed in perivascular/meningeal inflammatory cells (J and K), whereas control spinal cord was negative for the expression of uPAR (L). MT1-MMP was similarly induced in the inflammatory cells (M and N), albeit at a lower level. Metalloelastase expression was not detectable in the control spinal cord (R); its expression was induced at EAE in a small subset of inflammatory cells (P and Q). Widespread expression of TIMP-1 transcripts was detected throughout the white matter of EAE spinal cord (S and T), whereas control spinal cord appeared negative for expression of TIMP-1 (U). Integrity of the RNA in the control tissue sections was demonstrated by positive hybridization signal for LRP, TIMP-2, and TIMP-3 (data not shown). Abbreviations: gt, gracile tract; dh, dorsal horn; wm, white matter; vh, ventral horn; vf, ventral (anterior) fissure. Scale bars: 220 μm (column 1); 45 μm (columns 2 and 3).
Immunostaining with Ly-6G antibody indicated the prevalence of granulocytes among infiltrating immune cells (data not shown), as expected for this model. 35 Inflammation was accompanied by damage of the blood-brain barrier, as judged by the appearance of diffuse immunoreactivity in perivascular areas using anti-mouse Ig antibody, whereas in control CNS Ig-labeling was present only in blood vessels/capillaries (data not shown).
Differential Expression of the Molecules of PA System in EAE
In Situ Hybridization
The tissue distribution of transcripts of tPA, uPA, uPAR, PAI-1, and LRP was determined by in situ hybridization analysis. In some cases, the cellular localization of the transcripts was further analyzed by immunostaining the in situ-hybridized slides with cell-type-specific reagents (anti-GFAP for astrocytes and L. esculentum lectin for macrophages/microglia). Serial sections of matched regions of control and EAE (grades 3 and 4) CNS were positioned adjacent on the same slide series, to ensure identical treatment of the tissue sections, and to allow direct comparison of the expression patterns/levels in normal and symptomatic CNS.
In the spinal cords of untreated mice, the expression of tPA transcripts was below the detection limit (Figure 1F) ▶ , or only weak expression was seen in scattered glial cells in the white matter (data not shown). In the mice symptomatic for EAE, tPA transcripts were prominently up-regulated in the periphery of white matter in cells having morphological features of hypertrophic astrocytes (Figure 1, D and E) ▶ . In the cervical spinal cord, tPA expression was limited to cells adjacent to sites of inflammatory cell accumulation (data not shown), whereas in the lumbar spinal cord tPA expression appeared less focalized (Figure 1, D and E) ▶ . tPA-expressing cells were unequivocally identified as astrocytes by double-labeling experiments by in situ hybridization with tPA probe, followed by immunostaining with the astrocyte-marker GFAP or lectin histochemistry with L. esculentum lectin (for detection of macrophages/microglia). High levels of tPA transcripts were present mainly in GFAP-positive astrocytes (Figure 2B) ▶ but not microglia (data not shown). In addition to spinal cord, tPA expression was also up-regulated in the brain of the mice with clinical EAE, in particular in the brainstem and pons (Figure 2C ▶ and data not shown). Expression of tPA transcripts in the sections of lumbar spinal cords was quantified by computerized image analysis (Table 1) ▶ .
Figure 2.
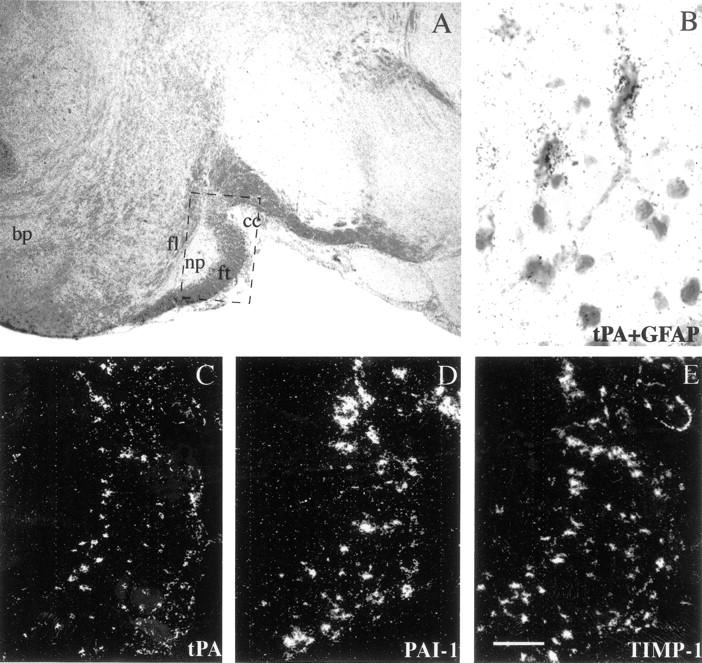
Expression of tPA, PAI-1, and TIMP-1 genes in the pons of BALB/c mice with acute grade 4 EAE. A: Low magnification bright-field image of luxol fast blue/cresyl violet-stained section; dashed box indicates the region shown at higher magnification in microphotographs (C to E). C to E: tPA, PAI-1, and TIMP-1 are expressed by the scattered cells in transversal fibers of the white matter of the pons, with pontine nuclei scoring negative. B: Combined in situ hybridization analysis/glial fibrillary acidic protein immunostaining, demonstrating that tPA is expressed by hypertrophic astrocytes. Abbreviations: ft, fibrae transversae pontis; fl, fibrae longitudinales pontis; np, nuclei pontis; ml, crus cerebri; bp, basis pontis. Scale bars: 350 μm (A); 20 μm (B), 90 μm (C to E).
Table 1.
Quantitative Comparison of Radioactive In Situ Hybridization Signal tPA, PAI-1, and TIMP-1
| Gene | Number of pairs analyzed | Fold induction (mean) | 95% confidence interval |
|---|---|---|---|
| tPA | 12 | 8.3 | 4.8–11.8 |
| PAI-1 | 9 | 16.9 | 8.9–24.9 |
| TIMP-1 | 11 | 8.9 | 4.8–13.1 |
In situ hybridized slides were viewed under epipolarized light to highlight deposited silver grains and images were captured with CCD camera. KS400 image analyzer (Zeiss, Germany) was used for thresholding and calculation of total image area occupied by silver grains. To avoid reproducibility problems due to slide-to-slide variation (differences in emulsion thickness, hybridization conditions, etc.) pairs of series of sections of control and EAE sections of spinal cords treated on the same slides were compared. White matter was analyzed in the area adjacent to the ventral white column. For each EAE/control pair eight × 20 fields (four control and four EAE) were analyzed. After background subtraction, fold increase in silver grain area was estimated. CI was calculated with normality assumption.
The expression of transcripts of the other PA, uPA, remained below the detection limit of the in situ hybridization analysis and Northern blot hybridization (data not shown). As a control, we tested the uPA probe on sections of day 13.5 mouse placenta after fertilization, in which the uPA-antisense riboprobe labeled spongiotrophoblasts and uterine epithelial cells, as expected. 21 Semiquantitative reverse transcriptase-polymerase chain reaction with uPA-specific primers was performed on 12 spinal cord samples to estimate possible changes in uPA transcripts during EAE. uPA mRNA levels appeared after normalization with GAPDH to be similar in control and EAE spinal cords (control mean, 29.8 AU; CI, 20.8 to 38.8; EAE mean, 25.8 AU; CI, 14.8 to 36.8).
PAI-1 expression was not detectable in normal CNS by in situ hybridization (Figure 1I) ▶ . During acute EAE high levels of PAI-1 transcripts were up-regulated in hypertrophic astrocytes adjacent to the lesions in the periphery of the white matter of the spinal cord (Figure 1, G and H) ▶ . PAI-1 expression was induced also in brainstem and pons (Figure 2D ▶ and data not shown).
uPAR is a specific pleiotropic high-affinity receptor of uPA, which focalizes plasmin generation to the cell surface, and participates in cell adhesion and signal transduction. uPAR transcripts were not detectable in the CNS of control mice (Figure 1L) ▶ . In spinal cords of mice symptomatic for EAE, uPAR transcripts were present in the immune cells in the inflammatory lesions (Figure 1, J and K) ▶ .
LRP acts together with uPAR in clearance of uPA-inhibitor complexes. This pleiotrophic molecule interacts with and internalizes also other ligands, such as proteolytic enzymes and apolipoprotein E-rich chylomicron remnants. 36 Ubiquitous expression of LRP was present in different subtypes of neurons throughout the control CNS (data not shown), as described earlier. 37 There were no qualitative changes in the expression pattern of LRP during the acute phase of EAE (data not shown).
Zymographic Analysis of PA Activity
In situ zymography was used to assay the proteolytic activities of PAs on cryosections of spinal cord. In control lumbar spinal cord sections, weak plasminogen-dependent caseinolytic activity was present at the dorsal root entry zone (Figure 3a, A) ▶ . After the same time of incubation, prominent caseinolysis was seen over sections of EAE spinal cord (Figure 3a, D) ▶ ; in addition to the dorsal horn, PA activity was also detected in the area of the central spinal canal. Also, patchy PA activity appeared along the meninges. An inhibitor of uPA, amiloride, 38 was used to differentiate between the activities of uPA and tPA. In sections of control spinal cord, amiloride had no effect on caseinolysis (Figure 3a, B) ▶ , indicating a tPA dependence of the plasmin-generating activity. In sections of spinal cord of animals with EAE, the inclusion of amiloride in the overlay mix seemed to reduce caseinolysis (Figure 3a, E) ▶ . Similarly to lumbar spinal cord sections, elevated PA activity was seen also on cryosections of cervical spinal cord of affected animals as compared to control mice (data not shown).
Figure 3.
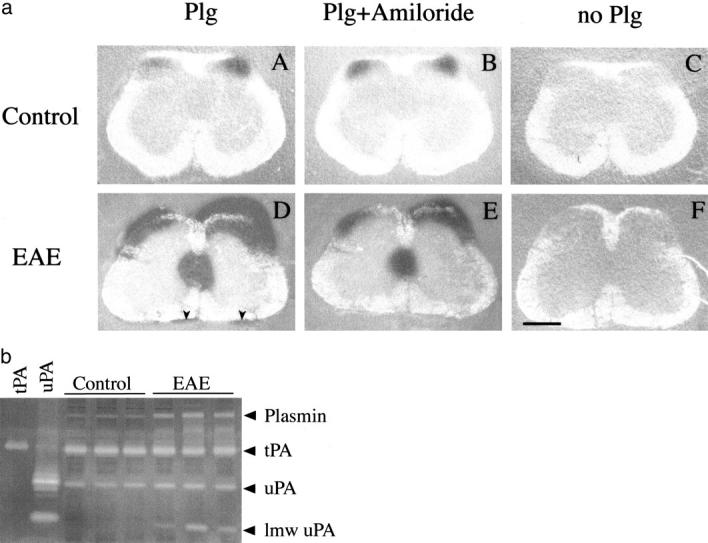
Zymographic analysis of PAs in lumbar spinal cords of mice with grade 3 EAE and control mice. a: In situ zymography of PAs on cryosections of normal lumbar spinal cord (top row) and spinal cord of mice with grade 3 EAE. A: Control spinal cord sections exhibit a weak plasminogen-dependent caseinolytic activity in the area of dorsal root exit zone; B: the activity is not affected by inhibition of uPA. D: After the same time of incubation at 37°C, EAE spinal cord sections show strong caseinolysis in the periphery of the white matter, in particular in the gracile tract, along meninges and in the central canal area. E: Amiloride appears to block caseinolysis along meninges and reduce it in other locations. C and F: Omission of plasminogen in the overlay mixture abolishes caseinolysis in both control and EAE spinal cord. Scale bar, 280 μm. b: SDS-PAGE zymography of the PAs in lumbar spinal cord extracts of three untreated mice and three mice with grade 3 EAE. As tPA and uPA standards, urine from uPA and tPA null mice was used. Control spinal cords exhibited uPA, tPA, and plasmin activities. Caseinolytic activities corresponding to low molecular weight uPA (lmw uPA), plasmin and PA-inhibitors complexes (migrating between tPA and plasmin activity) appear elevated at EAE.
Zymography on SDS-PAGE substrate gel was applied for molecular analysis of PAs. Four major caseinolytic bands corresponding to low-molecular weight uPA (LMW-uPA, a 28-kd fragment of uPA derived from 46-kd molecule by limited proteolysis), uPA, tPA, and plasmin were detectable (Figure 3b) ▶ . As controls of uPA and tPA activities, urine of tPA and uPA null mice was used. Plasmin was identified on the basis of its molecular weight, and independence of the corresponding caseinolytic band from the presence of plasminogen in the gel.
Control spinal cord extract contained tPA and uPA activities, in addition weak plasmin activity was present. In extracts of spinal cords of mice symptomatic for EAE, plasmin activity was elevated, and a caseinolytic band of apparent molecular weight of 28 kd, corresponding to LMW-uPA, was detected. LMW-uPA In EAE extracts, elevated diffuse high-molecular weight bands of plasminogen-dependent caseinolysis were observed. These correspond likely to PA-inhibitor complexes, in which PAs have regained enzymatic activity because of activation in SDS sample buffer. Activities of full-length PAs appeared similar in control and diseased spinal cords.
Modulated Expression of Matrix Metalloproteinases and TIMPs in EAE
In Situ Hybridization
In situ hybridization was used to analyze the expression of transcripts of gelatinase A, gelatinase B, metalloelastase, membrane type-1 MMP (MT1-MMP), and their inhibitors TIMP-1, TIMP-2, and TIMP-3. Widespread expression of TIMP-2 and -3 (in neurons and choroid plexus, respectively) was detected in control CNS as described earlier 12 (data not shown). There were no clear changes in the expression patterns and levels of TIMP-2 and -3 during EAE as compared to control CNS.
Sections from control spinal cords and brains were negative for the presence of MT1-MMP, gelatinases A and B, metalloelastase, and TIMP-1 transcripts (Figure 1 ▶ ; O, R, and U, and data not shown). In EAE, up-regulation of transcripts of MT1-MMP, metalloelastase and TIMP-1 was detected in the areas of neuroinflammation and tissue damage. MT1-MMP expression was detected in inflammatory cells in all levels of spinal cord (Figure 1, M and N) ▶ . Metalloelastase was expressed by a small subset of inflammatory cells in the same locations (Figure 1, P and Q) ▶ . Prominent induction of TIMP-1 expression was observed by activated hypertrophic astrocytes in the white matter juxtaposing the inflammatory cells, both in the spinal cord (Figure 1, S and T ▶ , and Table 1 ▶ ) and brain (Figure 2E) ▶ , as observed previously. 12
Gelatinase B has been suggested to be a key factor in the progression of inflammatory demyelination. However, transcripts of gelatinase B were not detectable by in situ hybridization in our model of EAE (data not shown). To verify the negative result, gelatinase probes were tested on sections of murine placenta, and they labeled decidual cells (gelatinase A) and giant trophoblasts (gelatinase B), as expected. 39
Zymography of Gelatinases
Neutrophils can release presynthesized gelatinases from secretory granules of their abundant cytoplasm, as part of a program of tissue invasion by these cells. 40 Compared to de novo protein synthesis, the strategy of using exocytosis of presynthesized enzyme is thought to enable a more robust increase in extracellular proteolysis. 41
A modified fluoresceinated gelatin-based assay was used for the localization of gelatinolytic activity on tissue sections. 32 Tissue sections were overlaid with a mixture containing DQ-gelatin and the fluorescent nuclear counterstain 4.6-diamidino-2-phenylindole. Strong fluorescence of DQ-gelatin, indicating proteolytic cleavage, appeared along the meninges, around blood vessels and in the parenchyma of the areas of inflammatory cell invasion in animals symptomatic for EAE (Figure 4a, A) ▶ , but not in control animals (Figure 4a, D) ▶ . In situ gelatinolysis was reduced in the presence of the metalloproteinase inhibitor 1,10-phenanthroline in the overlay (Figure 4a, B) ▶ .
Figure 4.
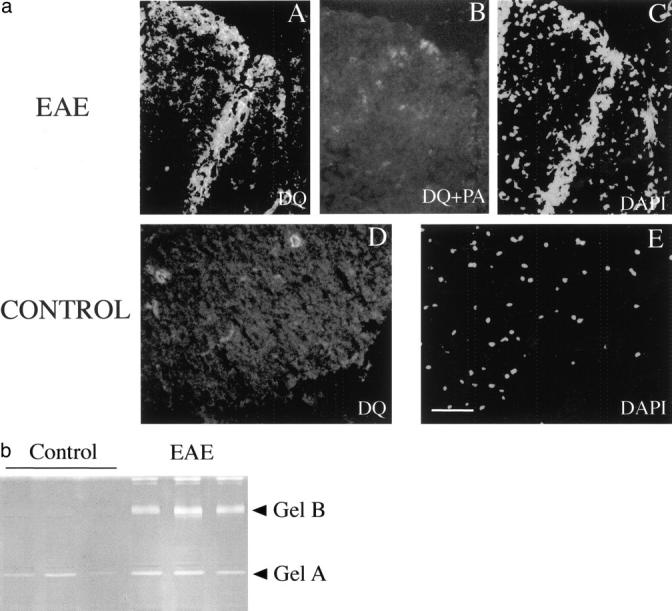
Zymographic analysis of gelatinases in the lumbar spinal cord extract of the naïve mice and mice with grade 3 EAE. a: In situ zymography of gelatinases using DQ-gelatin. A and C: Increased green fluorescence indicating the cleavage of the DQ-gelatin is present in the areas of accumulation of inflammatory cells. B: Inhibition of metalloproteinases by 1,10-phenanthroline (PA) reduces the cleavage of the DQ-gelatin. C and E: Nuclear staining with 4.6-diamidino-2-phenylindole of the same section shown on A, B, and C. D: In control spinal cord sections fluorescence was observed occasionally over blood capillaries. Scale bar, 70 μm. B: SDS-PAGE zymography of gelatinases in the lumbar spinal cord extracts of three naïve mice and three symptomatic mice with grade 3 EAE. Note that whereas gelatinase A is constitutively expressed, gelatinase B is up-regulated during EAE. Upper high molecular weight complexes correspond to gelatinase B dimers.
SDS-PAGE zymography was used to analyze the presence of gelatinolytic enzymes in tissue extracts of different levels of spinal cord and brain (Figure 4b ▶ and data not shown). As a positive control for gelatinolysis, conditioned medium of HT-1080 fibrosarcoma cells was used (data not shown). Tissue extracts of both control and EAE brains and spinal cords contained gelatinolytic activities migrating at ∼65 kd and 95 kd, corresponding precursor forms of gelatinase A and B, respectively. Although gelatinase A was constitutively expressed in all samples, gelatinase B was strongly up-regulated during EAE in lumbar and cervical spinal cord, and it was elevated in brain extract. Also, large complexes migrating at ∼200 kd and corresponding to gelatinase B dimers 42 were detected in the extracts of spinal cord of animals with clinical EAE. Gelatinolytic activity was completely blocked by inclusion of 1,10-phenanthroline in the incubation solution (data not shown) confirming that the observed gelatinolysis was because of metalloproteinases.
Differential Regulation of ECM Proteins at EAE
In EAE and MS, increased blood-brain-barrier permeability has been associated with accumulation of ECM components around blood vessels. 43-45 Also noninflammatory conditions of the CNS resulting in vascular cell injury have been shown to lead to fibronectin deposition. The ECM deposition is presumably brought about by extravasation from the plasma. Newly deposited matrices can act as substrate for inflammatory cell adhesion and hence influence the extent of tissue infiltration. Extracellular proteolysis represents a major mechanism regulating ECM turnover, hence we decided to study the localization of fibrin(ogen), fibronectin, and vitronectin immunoreactivities in the spinal cords of mice with acute EAE and their naïve littermates. Fibrin and fibronectin could be localized in paraffin-embedded tissue sections, whereas our vitronectin antibody was suitable for cryosections only. In the spinal cords of mice with clinical EAE, fibrin along with fibronectin were deposited in the perivascular areas and along the meninges at the sites of inflammatory damage (Figure 5, B and D) ▶ , the deposits were particularly prominent in the dorsal aspect of the spinal cord. In the untreated littermates, no extravascular fibrin and fibronectin were observed, and the immunoreactivities were exclusively limited to the vasculature. Vitronectin gave a similar staining pattern, with increased immunoreactivity in perivascular matrices and along the meninges in mice symptomatic for EAE (Figure 5F) ▶ . Similar immunostaining results were obtained in animals that were perfused with PBS before tissue collection.
Figure 5.
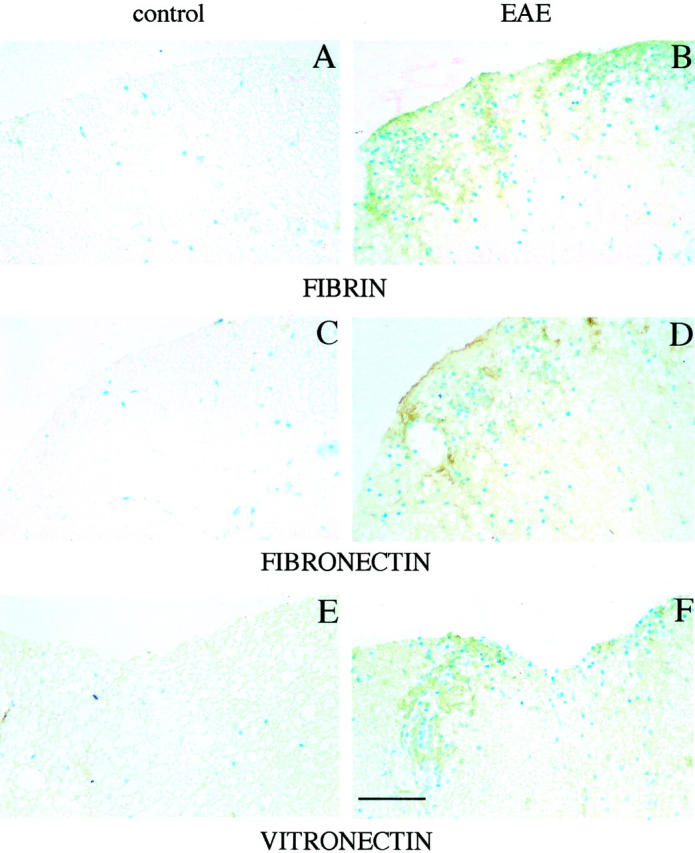
Localization of immunoreactivities of fibrin (A and B), fibronectin (C and D), and vitronectin (E and F) in the lumbar spinal cord of control mouse and of mouse with of grade 3 EAE. Note increased immunoreactivity in the perivascular matrix and along the meninges for all of the four antigens in EAE spinal cords (B, D, and F) and not in control spinal cords (A, C, and E). In contrast, laminin immunoreactivity remained unchanged (not shown). Scale bar, 280 μm.
Discussion
Extracellular proteolysis is involved in many aspects of acute and chronic inflammatory brain diseases. The role of MMPs in the pathogenesis of EAE has been the subject of several studies, 46,47 whereas no molecular analysis of the PA system in EAE has been performed. Importantly, PAs are relevant to several aspects of neurophysiology, such as neuronal cell migration, learning, excitotoxicity, and degradation of amyloid-β aggregates. 48-51 The concept that a single proteolytic enzyme is critical for the development of such a complex phenotype as EAE is probably flawed, and it is likely that there exists a network of functionally redundant proteases involved in EAE progression. For example, it was recently demonstrated using gene-inactivated mice that gelatinase B (for a long time considered a key proteinase in the pathogenesis of EAE) is not required for the development of EAE in adult mice. 42 In our study, we demonstrate that during acute EAE both families of extracellular proteases are coordinately induced. We show that during the clinical phase of EAE, several proteases (tPA, MT1-MMP, metalloelastase, gelatinase-B) and functionally related regulatory molecules (uPAR, PAI-1, TIMP-1) are up-regulated, with expression patterns coinciding with the sites of tissue damage and neuroinflammation.
It has been suggested that in MS the PA/Plg/PAI-1 axis constitutes a rate-limiting step of extracellular proteolysis. 46 Our data indicate that both uPA and tPA-mediated plasminogen activation is boosted in the course of EAE.
The expression of tPA and its inhibitor PAI-1 is prominently up-regulated in hypertrophic astrocytes in the white matter in the proximity of inflammatory lesions, and the expression patterns of the two molecules overlap partially. The co-expression of the protease and its inhibitor is probably required to ensure a stringent regulation of plasmin generation by these CNS-resident cells. On the other hand uPA/uPAR membrane complexes could act as adhesion receptors and promote motility of inflammatory cells on newly deposited vitronectin matrix. PAI-1, secreted by adjacent astrocytes, would act to disrupt both processes.
What could be the physiological significance of the increased generation of plasmin in inflammatory lesions during EAE? Plasmin is a broad-spectrum proteinase that is able to cleave a number of molecules of ECMs, eg, fibrin and fibronectin. 52 Deposition of fibrin has been reported in demyelinated plaques in MS 53 and EAE. 54 Our study demonstrates deposition of fibrin(ogen), fibronectin, and vitronectin immunoreactivities in the areas of perivascular inflammation during clinical EAE. These newly deposited local matrices may drive the inflammatory response, serving as substrates for immune cell adhesion and invasion; in addition, the matrices can modulate the profile of secreted cytokines and growth factors of adhering cells. 55,56
Catalytic activity of tPA is induced on its interaction with fibrin, and of the two PAs, tPA is thought to function primarily in the fibrinolytic pathway. 57 tPA/plasmin-mediated fibrinolysis has been demonstrated to reduce axonal damage and demyelination after sciatic nerve injury in mice. 58 Systemic defibrination of Lewis rats by batroxobin (a thrombin-like enzyme derived from the venom of the snake Bothrops atrox) was shown to suppress EAE. 59 Our analysis demonstrates that during acute EAE tPA is expressed by activated astrocytes in the proximity of fibrin deposits. Collectively, this evidence suggests that plasmin-mediated clearance of fibrin may limit the extent of immune infiltration and neuroinflammation. However, during excitotoxin-induced neuronal cell death tPA-mediated plasminogen activation was demonstrated to promote neuronal degeneration, presumably through laminin degradation. 60 It can be speculated that also during EAE, tPA could mediate proteolysis of matrix proteins other than fibrin. The role of tPA-mediated fibrinolysis in the pathogenesis of EAE may be clarified by experiments with mice genetically deficient in PAs and fibrinogen. 61
In addition to its involvement in ECM metabolism, the PAs/plasmin system may participate in the pathogenesis of EAE by regulating the bioavailability of different growth factors and proinflammatory cytokines. 62,63 Transforming growth factor (TGF)-β is known to be an important regulator of self-tolerance, 64 and administration of TGF-β protects mice from EAE and collagen-induced adjuvant arthritis. 65 Microglia and astrocytes produce TGF-β in a biologically inactive form, and astrocyte-derived tPA has been demonstrated to mediate activation of latent TGF-β, leading to down-regulation of nitric oxide release by microglia. 66 High levels of nitric oxide are toxic to neurons and oligodendrocytes and may play a role in different neuropathologies, including MS. 67 The system seems to be feedback-controlled; as it is known, that in astrocytes PAI-1 is regulated by TGF-β1 and basic fibroblast growth factor. 68
MS is a multifactorial disease, and its clinical and pathological outcome may be reached through different etiological pathways. For a better understanding of the different variants of the disease and common mechanisms in its pathogenesis, it is important to study the expression of candidate pathogenic factors in various models of EAE using different animal species, genetic backgrounds and methods of disease induction. The expression of MMPs has not been analyzed previously in actively induced EAE in BALB/c mice. It seems that the profile of expression of MMPs in our model generally parallels that seen in other mouse EAE variants. 10,12 The expression of MMPs/TIMPs in EAE in BALB/c mice seems to follow the dichotomic pattern previously observed by Pagenstecher and colleagues, 12 with inflammatory cells producing MMPs (MT1-MMP, metalloelastase, and gelatinase B), and glial induction of the endogenous inhibitor TIMP-1. As a variation of the theme, in our model the up-regulation of gelatinase B in inflammatory lesions does not correlate with transcriptional induction of the proteinase, but it seems to involve recruitment of leukocytes with preformed gelatinase stores and degranulation. Our study is the first demonstration of the in situ expression of MT1-MMP by inflammatory cells in EAE lesions. Expression of membrane-type MMPs confers cells with the ability to activate MMP-2, MT-MMPs also act as ECM-remodeling agents on their own rights. 69,70 Of the MT-MMPs, MT1-MMP seems to have the widest range of substrates, and it has been recently demonstrated to act as a highly efficient fibrinolysin. 71
In conclusion, our analysis demonstrates the parallel up-regulation of two key systems of extracellular proteolysis—the PA system and MMPs—during actively induced EAE in mice. Regulated expression of the components of PA system warrants inhibitors/knockout-based functional studies of the effect of inhibition of the PA system on the progression of inflammatory demyelination. The concurrent induction of the two systems in EAE suggests that treatments aiming at modulating the expression of both PAs and MMPs simultaneously may prove more efficient than therapeutic strategies focusing on a single system of extracellular proteolysis.
Acknowledgments
We thank Jorma Määttä for help with setting up the mouse model of EAE used in this study, Daniela Talarico for critical reading of the manuscript, Tor Ny for providing plasmids containing MT1-MMP and metalloelastase cDNAs, and Hanna Oksanen for help with statistical analysis.
Footnotes
Address reprint requests to Tambet Teesalu, Ph.D., Haartman Institute, University of Helsinki, POB 21, FIN-00014, Helsinki, Finland. E-mail:tambet.teesalu@helsinki.fi.
Supported by the Finnish Academy and the Finnish Cancer Societies.
References
- 1.Werb Z: ECM and cell surface proteolysis: regulating cellular ecology. Cell 1997, 91:439-442 [DOI] [PubMed] [Google Scholar]
- 2.Matrisian LM, Hogan BLM: Growth factor-regulated proteases and extracellular matrix remodeling during mammalian development. Curr Top Dev Biol 1990, 24:219-257 [DOI] [PubMed] [Google Scholar]
- 3.Birkedal-Hansen H: Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol 1995, 7:728-735 [DOI] [PubMed] [Google Scholar]
- 4.Andreasen PA, Kjoller L, Christensen L, Duffy MJ: The urokinase-type plasminogen activator in cancer metastasis: a review. Int J Cancer 1997, 72:1-22 [DOI] [PubMed] [Google Scholar]
- 5.Jones JL, Walker RA: Control of matrix metalloproteinase activity in cancer. J Pathol 1997, 183:377-379 [DOI] [PubMed] [Google Scholar]
- 6.Werb Z, Mainardi CL, Vater CA, Harris EDJ: Endogenous activation of latent collagenase by rheumatoid synovial cells. Evidence for a role of plasminogen activator. N Engl J Med 1977, 296:1017-1023 [DOI] [PubMed] [Google Scholar]
- 7.Baramova EN, Bajou K, Remacle A, L’Hoir C, Krell HW, Weidle UH, Noel A, Foidart JM: Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett 1997, 405:157-162 [DOI] [PubMed] [Google Scholar]
- 8.Lijnen HR, Van Hoef B, Lupu F, Moons L, Carmeliet P, Collen D: Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Aterioscler Thromb Vasc Biol 1998, 18:1035-1045 [DOI] [PubMed] [Google Scholar]
- 9.Gijbels K, Masure S, Carton H, Opdenakker G: Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological diseases. J Neuroimmunol 1992, 41:29-34 [DOI] [PubMed] [Google Scholar]
- 10.Gijbels K, Proost P, Masure S, Carton H, Biliau A, Opdenakker G: Gelatinase B is present in the cerebrospinal fluid during experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J Neurosci Res 1993, 36:431-440 [DOI] [PubMed] [Google Scholar]
- 11.Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J: The expression of tissue type plasminogen activator, matrix metalloproteinases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol 1996, 55:1194-1204 [DOI] [PubMed] [Google Scholar]
- 12.Pagenstecher A, Stalder AK, Kincaid CL, Shapiro SD, Campbell IL: Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am J Pathol 1998, 152:729-741 [PMC free article] [PubMed] [Google Scholar]
- 13.Gijbels K, Galardy RE, Steinman L: Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteinases. J Clin Invest 1994, 94:2177-2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norga K, Paemen L, Masure S, Dillen C, Heremans H, Billiau A, Carton H, Cuzner L, Olsson T, Van Damme J, Opdenakker G: Prevention of acute autoimmune encephalomyelitis and abrogation of relapses in murine models of multiple sclerosis by the protease inhibitor D-penicillamine. Inflamm Res 1995, 44:529-534 [DOI] [PubMed] [Google Scholar]
- 15.Hewson AK, Smith T, Leonard JP, Cuzner ML: Suppression of experimental allergic encephalomyelitis in the Lewis rat by the matrix metalloproteinase inhibitor Ro31–9790. Inflamm Res 1995, 44:345-349 [DOI] [PubMed] [Google Scholar]
- 16.Clements JM, Cossins JA, Wells GMA, Corkill DJ, Helfrich K, Wood M, Pigott R, Stabler G, Ward GA, Gearing AJH, Miller KM: Matrix metalloproteinase expression during experimental autoimmune encephalomyelitis and effects of a combined matrix metalloproteinase and tumor necrosis factor-alpha inhibitor. J Neuroimmunol 1997, 74:85-94 [DOI] [PubMed] [Google Scholar]
- 17.Brosnan CF, Cammer W, Norton WT, Bloom BR: Proteinase inhibitors suppress the development of experimental allergic encephalomyelitis. Nature 1980, 285:235-237 [DOI] [PubMed] [Google Scholar]
- 18.Akenami FOT, Siren V, Weissman M, Koskiniemi M, Vaheri A: Tissue plasminogen activator gene expression in multiple sclerosis brain tissue. J Neurol Sci 1999, 165:71-76 [DOI] [PubMed] [Google Scholar]
- 19.Akenami FOT, Koskiniemi M, Mustjoki S, Siren V, Färkkila M, Vaheri A: Plasma and cerebrospinal fluid activities of tissue plasminogen activator, urokinase and PAI-1 in multiple sclerosis. Fibrinolysis Proteolysis 1997, 11:109-113 [Google Scholar]
- 20.Määttä JA, Erälinna JP, Röyttä M, Salmi AA, Hinkkanen AE: Physical state of the neuroantigen in adjuvant emulsion determines encephalogenic status in the Balb/C mouse. J Immunol Methods 1996, 190:133-141 [DOI] [PubMed] [Google Scholar]
- 21.Teesalu T, Blasi F, Talarico D: Embryo implantation in the mouse: fetomaternal coordination in the pattern of expression of uPA, uPAR, PAI-1, and α2MR/LRP genes. Mech Dev 1996, 56:103-116 [DOI] [PubMed] [Google Scholar]
- 22.Sappino AP, Huarte J, Vassalli JD, Belin D: Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J Clin Invest 1991, 87:962-970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belin D, Vassalli JD, Combepine C, Godeau F, Nagamine Y, Reich E, Kocher HP, Duvoisin RM: Cloning, nucleotide sequencing and expression of cDNAs encoding mouse urokinase-type plasminogen activator. Eur J Biochem 1985, 148:225-232 [DOI] [PubMed] [Google Scholar]
- 24.Prendergast GC, Diamond LE, Dahl D, Cole MD: The c-myc-regulated gene mrl encodes plasminogen activator inhibitor 1. Mol Cell Biol 1990, 10:1265-1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristensen P, Eriksen J, Blasi F, Danø K: Two alternatively spliced mouse urokinase receptor mRNAs with different histological localization in the gastrointestinal tract. J Cell Biol 1991, 115:1763-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Leuven F, Stas L, Raymakers L, Overbergh L, De Strooper B, Hilliker C, Lorent K, Fias E, Umans L, Torrekens S, Serneels L, Moechars D, Van den Berghe H: Molecular cloning and sequencing of the murine alpha-2-macroglobulin receptor cDNA. Biochim Biophys Acta 1993, 1173:71-74 [DOI] [PubMed] [Google Scholar]
- 27.Liu K, Oloffson JI, Wahlberg P, Ny T: Distinct expression of gelatinase A (MMP-2), collagenase-3 (MMP-13), membrane type MMP1 (MMP-14), and tissue inhibitor of MMPs type 1 mediated by physiological ligands during formation and regression of the rat corpus luteum. Endocrinology 1999, 140:5330-5338 [DOI] [PubMed] [Google Scholar]
- 28.Alexander CM, Hansell EJ, Behrendtsen O, Flannery ML, Kishnani NS, Hawkes SP, Werb Z: Expression and function of matrix metalloproteinases and their inhibitors at the maternal-embryonic boundary during mouse embryo implantation. Development 1996, 122:1723-1736 [DOI] [PubMed] [Google Scholar]
- 29.Shapiro SD, Griffin GL, Gilbert DJ, Jenkins NA, Copeland NG, Welgus HG, Senior RM, Ley TJ: Molecular cloning, chromosomal localization, and bacterial expression of a murine macrophage metalloelastase. J Biol Chem 1992, 267:4664-4671 [PubMed] [Google Scholar]
- 30.Wilkinson DG, Bailes JA, McMahon AP: Expression of the proto-oncogene int-1 is restricted to specific neural cells in the developing mouse embryo. Cell 1987, 50:79-88 [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC: Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994, 368:419-424 [DOI] [PubMed] [Google Scholar]
- 32.Missing.
- 33.Behrendtsen O, Alexander CM, Werb Z: Metalloproteases mediate extracellular matrix degradation by cells from mouse blastocyst outgrowths. Development 1992, 114:447-456 [DOI] [PubMed] [Google Scholar]
- 34.Määttä JA, Sjöholm UR, Nygårdas PT, Salmi AA, Hinkkanen AE: Neutrophils secreting tumor necrosis factor alpha infiltrate the CNS of Balb/c mice with EAE. J Neuroimmunol 1998, 90:162-175 [DOI] [PubMed] [Google Scholar]
- 35.Määttä JA, Erälinna J-P, Röyttä M, Salmi AA, Hinkkanen AE: Physical state of the antigen/adjuvant emulsion determines encephalogenicity in Balb/c mice. J Immunol Methods 1996, 190:133-141 [DOI] [PubMed] [Google Scholar]
- 36.Krieger M, Herz J: Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein. Annu Rev Biochem 1994, 63:601-637 [DOI] [PubMed] [Google Scholar]
- 37.Bu G, Maksymovitch EA, Nerbonne JM, Schwartz AL: Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem 1994, 269:18521-18528 [PubMed] [Google Scholar]
- 38.Vassalli J-D, Belin D: Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett 1987, 214:187-191 [DOI] [PubMed] [Google Scholar]
- 39.Teesalu T, Masson R, Basset P, Blasi F, Talarico D: Expression of matrix metalloproteinases during murine chorioallantoic placenta formation. Dev Dyn 1999, 214:248-258 [DOI] [PubMed] [Google Scholar]
- 40.Kjeldsen L, Bjerrum OW, Askaa J, Borregaard N: Subcellular localization and release of human neutrophil gelatinase, confirming the existence of separate gelatinase-containing granules. Biochem J 1992, 287:603-610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonh A, Tuszynski G: The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res 2001, 7:14-23 [DOI] [PubMed] [Google Scholar]
- 42.Dubois B, Masure S, Hurtenbach U, Paemen L, Heremans H, van den Oord J, Sciot R, Meinhardt T, Hämmerling G, Opdenakker G, Arnold B: Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest 1999, 104:1507-1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butter C, Healey DG, Agha N, Wicox CE, Antoniou AV, Turk JL: The localization of fibrin, fibronectin and class II major histocompatibility complex antigen in the spinal cord in chronic relapsing experimental allergic encephalomyelitis. J Neuroimmunol 1989, 22:11-17 [DOI] [PubMed] [Google Scholar]
- 44.Sobel RA, Schneeberger EE, Colvin RB: The immunopathology of acute experimental allergic encephalomyelitis. A light microscopic and ultrastructural immunohistochemical analysis of fibronectin and fibrinogen. Am J Pathol 1988, 134:547-558 [PMC free article] [PubMed] [Google Scholar]
- 45.Inoue A, Koh C-S, Yamazaki M, Yanagisawa N, Ishihara Y, Kim SK: Fibrin deposition in the central nervous system correlates with the degree of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Neuroimmunol 1997, 77:185-194 [DOI] [PubMed] [Google Scholar]
- 46.Cuzner ML, Opdenakker G: Plasminogen activators and matrix metalloproteinases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol 1999, 94:1-14 [DOI] [PubMed] [Google Scholar]
- 47.Hartung H-P, Kieseier BC: The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J Neuroimmunol 2000, 107:140-147 [DOI] [PubMed] [Google Scholar]
- 48.Seeds NW, Williams BL, Bickford BC: Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science 1995, 270:1992-1994 [DOI] [PubMed] [Google Scholar]
- 49.Tsirka SE, Rogove AD, Strickland S: Tissue plasminogen activator and neuronal cell death. Nature 1996, 384:123-124 [DOI] [PubMed] [Google Scholar]
- 50.Seeds NW, Basham ME, Haffke SP: Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci USA 1999, 96:14118-14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker HM, Kihiko M, Cladwell JN, Wright S, Kawarabayashi T, Price D, Walker D, Scheff S, McGillis JP, Rydel RE, Estus S: The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci 2000, 20:3937-3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwaan HC: The plasminogen-plasmin system in malignancy. Cancer Metastasis Rev 1992, 11:291-311 [DOI] [PubMed] [Google Scholar]
- 53.Claudio L, Raine CS, Brosnan CF: Evidence of persistent blood-brain barrier abnormalities in chronic progressive multiple sclerosis. Acta Neuropathol 1995, 90:228-238 [DOI] [PubMed] [Google Scholar]
- 54.Patterson PY, Koh C-S, Kwaan HC: Role of the clotting system in the pathogenesis of neuroimmunologic disease. Fed Proc 1987, 46:91-96 [PubMed] [Google Scholar]
- 55.Formica S, Roach TI, Blackwell MJ: Interaction with extracellular matrix proteins influences Lsh/Ity/Bcg (candidate Nramp) gene regulation of macrophage priming/activation for tumor necrosis factor-alpha and nitrite release. Immunology 1994, 82:42-50 [PMC free article] [PubMed] [Google Scholar]
- 56.Perez RL, Roman J: Fibrin enhances the expression of Il-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J Immunol 1995, 154:1879-1887 [PubMed] [Google Scholar]
- 57.van Zonnenweld A-J, Veerman H, MacDonald M, van Mourik J, Pannekoek H: Structure and function of human tissue-type plasminogen activator. J Cell Biochem 1986, 32:169-178 [DOI] [PubMed] [Google Scholar]
- 58.Akassoglou K, Kombrinck KW, Degen JL, Strickland S: Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol 2000, 149:1157-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inoue A, Koh CS, Shimada K, Yanagisawa N, Yoshimura K: Suppression of cell-transferred experimental autoimmune encephalomyelitis in defibrinated Lewis rats. J Neuroimmunol 1996, 71:131-137 [DOI] [PubMed] [Google Scholar]
- 60.Chen Z-L, Strickland S: Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997, 91:917-925 [DOI] [PubMed] [Google Scholar]
- 61.Carmeliet P, Collen D: Targeted gene manipulation and transfer of the plasminogen and coagulation systems in mice. Fibrinolysis 1996, 10:195-213 [DOI] [PubMed] [Google Scholar]
- 62.Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM: Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J 1992, 11:4825-4833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB: Characterization of the activation of latent TGF-beta by co-cultures of pericytes and smooth muscle cells: a self-regulating system. J Cell Biol 1990, 111:757-763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shull M, Ormsby I, Kier AB, Pawlowski S, Diebold R, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T: Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359:693-699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santambrogio L, Hochwald GM, Leu CH, Thorbecke GJ: Antagonistic effects endogenous and exogenous TGF-β and TNF on autoimmune diseases in mice. Immunopharmacol Immunotoxicol 1993, 15:461-478 [DOI] [PubMed] [Google Scholar]
- 66.Vincent VAM, Löwik CWGM, Verheijen JH, De Bart ACW, Tilders FJH, Van Dam A-M: Role of astrocyte-derived tissue-type plasminogen activator in the regulation of endotoxin-stimulated nitric oxide production by microglial cells. Glia 1998, 22:130-137 [PubMed] [Google Scholar]
- 67.Bagasra O, Michaels FH, Zheng YM, Bobroski LE, Spitsin SV, Fu ZF, Tawadros R, Koprowski H: Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc Natl Acad Sci USA 1995, 92:12041-12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Treichel JA, Reddington M, Kreutzberg GW: Regulation of plasminogen activator inhibitor-1 mRNA accumulation by basic fibroblast growth factor and transforming growth factor-beta in cultured rat astrocytes. J Neurochem 1998, 71:1944-1952 [DOI] [PubMed] [Google Scholar]
- 69.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M: A matrix metalloproteinase expressed on the cell surface of invasive tumor cells. Nature 1994, 370:61-65 [DOI] [PubMed] [Google Scholar]
- 70.Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ: Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J Cell Biol 2000, 149:1309-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bini A, Wu D, Schnuer J, Kudryk BH: Characterization of stromelysin 1 (MMP-3), matrilysin (MMP-7), and membrane type 1 matrix metalloproteinase (MT1-MMP) derived fibrin(ogen) fragments D-dimer and D-like monomer: NH2-terminal sequences of late-stage digest fragments. Biochemistry 1999, 38:13928-13936 [DOI] [PubMed] [Google Scholar]