

Abstract
Despite the growing awareness of intraductal papillary-mucinous neoplasms (IPMNs) of the pancreas among clinicians, the molecular features of IPMNs have not been well characterized. Previous reports suggest that inactivation of the STK11/LKB1, a tumor-suppressor gene responsible for Peutz-Jeghers syndrome (PJS), plays a role in the pathogenesis of gastrointestinal hamartomas as well as several cancers, including pancreatic adenocarcinoma. Using polymerase chain reaction amplification of five microsatellite markers from the 19p13.3 region harboring the STK11/LKB1 gene, we analyzed DNA from 22 IPMNs for loss of heterozygosity (LOH). LOH at 19p13.3 was identified in 2 of 2 (100%) IPMNs from patients with PJS and 5 of 20 (25%) from patients lacking features of PJS (7 of 22, 32% overall). Sequencing analysis of the STK11/LKB1 gene in these IPMNs with LOH revealed a germline mutation in one IPMN that arose in a patient with PJS and a somatic mutation in 1 of the 20 sporadic IPMNs. None of the 22 IPMNs showed hypermethylation of the STK11/LKB1 gene. These results suggest that the STK11/LKB1 gene is involved in the pathogenesis of some IPMNs.
Intraductal papillary-mucinous neoplasms (IPMNs) of the pancreas are a distinct clinicopathological entity, characterized by dilated pancreatic ducts and ductules that are lined by papillary proliferations of tall columnar mucin-producing neoplastic epithelial cells. 1-5 The neoplastic epithelium in IPMNs shows a spectrum of cytologic and architectural atypia ranging from benign (adenoma) to malignant (carcinoma), and some neoplasms are associated with an invasive adenocarcinoma. 4 The prognosis of IPMNs is generally better than that of ductal adenocarcinoma of the pancreas; however, the recurrence rate is relatively high even after curative resection. 3,6-9 A better understanding of molecular alterations underlying the development of IPMNs may provide insights into the distinctive phenotype of this neoplasm and it may help predict the underlying malignant potential and the risk of recurrence.
In contrast to the substantial progress in our understanding of the molecular genetics of conventional pancreatic ductal adenocarcinoma, 10 relatively little is known about the genetic events that occur in IPMNs. Genetic alterations in IPMNs have been reported to involve activating point mutations in the K-ras oncogene and overexpression of HER-2/neu (c-erbB2) gene product, 11-13 although the frequency of these changes is controversial. Fujii et al 14 analyzed 13 IPMNs and found frequent loss of heterozygosity (LOH) at several chromosomal loci including 6q (54%), 8p (31%), 9p (62%), 17p (38%), and 18q (38%), suggesting that inactivation of the p16 (at chromosome 9p), p53 (at 17p), and DPC4 (at 18q) tumor-suppressor genes may occur in these neoplasms. However, biallelic inactivation of these tumor-suppressor genes occur less frequently in IPMNs than it does in ductal adenocarcinomas. 15 For example, mutations in the p53 tumor-suppressor gene were detected in only 8% of IPMNs by Sessa et al, 16 and recently, Iacobuzio-Donahue et al 17 examined a large series of IPMNs immunohistochemically for the expression of Dpc4 protein, a marker of DPC4 gene status, 18 and found almost no loss of expression. In support of this finding, Inoue et al 19 reported no inactivating mutation of DPC4/SMAD4 gene in 18 IPMNs. Therefore, targeted inactivation of other tumor-suppressor loci may be involved in tumorigenesis of IPMNs.
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant disorder characterized by mucocutaneous pigmentation, hamartomatous polyposis, and predisposition to various neoplasms. 20 A susceptibility locus for PJS was found on 19p13.3 by linkage analysis, and subsequently, germline mutations in the STK11/LKB1 gene were identified in most PJS patients. 21-23 The STK11/LKB1 gene encodes a serine/threonine kinase that has growth-suppressing activity. 24 Furthermore, a recent study has shown that this gene product associates with p53 and regulates p53-dependent apoptosis. 25 Patients with PJS have an elevated risk of malignancy, most commonly affecting the gastrointestinal tract, pancreas, breast, testis, and ovary. 26-28 Germline and somatic mutations in the STK11/LKB1 gene have been reported in a subset of pancreatic adenocarcionomas. 29 We recently observed two patients with PJS who developed IPMNs. This association led us to investigate the role of the STK11/LKB1 gene in IPMNs. In the present study, we analyzed DNA from a total of 22 IPMNs for LOH in the 19p13.3 region, for the mutations in the STK11/LKB1 gene, and for promoter region hypermethylation in the STK11/LKB1 gene.
Materials and Methods
Tissue Specimens, Microdissection, and DNA Extraction
The archives of The Johns Hopkins Hospital were searched for IPMNs. Hematoxylin and eosin-stained slides from each case were reviewed and the IPMNs were classified according to recently established criteria. 30 A total of 22 IPMNs were selected based on the availability of sufficient quantities of tumor and matched normal tissue. Five-μm sections were prepared from the selected paraffin blocks and deparaffinized by routine techniques. IPMNs and adjacent normal tissues were needle-dissected under direct visualization as has been previously described. 31 Microdissection was performed to obtain a neoplastic cellularity of ∼70 to 100%. Several IPMNs were associated with infiltrating adenocarcinoma, but only the intraductal component of these IPMNs was selectively dissected for analysis. DNA was extracted from the microdissected tissue with the use of 200 μg/ml of proteinase K (USB, Cleveland, OH) and 0.5% Tween 20 (Sigma Chemical Co., St. Louis, MO) as previously described. 32
LOH Analysis
LOH at the STK11/LKB1 locus was determined using five microsatellite markers, D19S886, D19S565, D19S591, D19S549, and D19S216 (Research Genetics, Huntsville, AL). As control markers, D5S346 (for 5q LOH) and TH (for 11p LOH) were used. DNA templates were amplified using initial denaturation step of 95°C for 5 minutes, followed by 30 cycles of 94°C for 30 seconds, 57°C for 20 seconds, and 72°C for 40 seconds, and by a final extension step at 72°C for 7 minutes. Polymerase chain reaction (PCR) products were resolved by electrophoresis and visualized by autoradiography. Allelic loss was considered to be present if the intensity of one of the alleles from the tumor showed more than a 50% reduction compared to the corresponding normal tissue. LOH was confirmed by either the demonstration of allelic loss at multiple markers or, in cases in which only one informative marker was available, by repeating the PCR amplification of that marker. Cases were scored as not informative when only a single allele was present in both tumor and matched normal tissue.
Direct Sequencing
Exons 1–9 of the STK11/LKB1 gene were amplified from genomic DNA using the primer sets described in previously published reports. 23,29,33 The PCR products were treated with exonuclease I and shrimp alkaline phosphatase (USB), and were subjected to cycle-sequencing (SequiTherm Excel II, Epicentre Technologies, Madison, WI). Products of the sequencing reactions were resolved by electrophoresis and visualized by autoradiography.
Methylation-Specific PCR
Methylation status of the 5′ CpG island of the STK11/LKB1 was determined by methylation-specific PCR (MSP) as described previously. 34 Briefly, 1 μg of genomic DNA was treated with sodium bisulfite for 16 hours at 50°C. After purification, 2 μl of modified DNA were amplified using primers specific for either the methylated or for the unmethylated DNA under the conditions as follows: 95°C for 3 minutes; then 40 cycles of 95°C for 15 seconds; 60°C (for unmethylated reaction) or 64°C (methylated) for 30 seconds, and 72°C for 30 seconds; and a final extension of 3 minutes at 72°C. Primers used for the unmethylated reaction were 5′-AATGTTTTGTTGTGGATGATTG-3′ (sense) and 5′-CAACAACCACCTTAAAAATCAC-3′ (antisense) and for the methylated reaction were 5′-CGATCGAGCGGATTTTTCG-3′ (sense) and 5′-CGCTCGAACAAACGTTTACG-3′ (antisense). PCR products were separated in 3% agarose gels. DNA extracted from normal pancreatic tissue and treated in vitro by SssI methylase (New England Biolabs, Beverly, MA) was used as a positive control for methylated alleles.
Results
Clinicopathological Characteristics
The present series included 13 men and 9 women with a mean age of 67 (range, 36 to 81). The maximum diameter of the neoplasms ranged from 1.5 to 9.0 cm (mean, 4.1 cm). Histologically, all IPMNs were characterized by having tall, columnar, mucin-containing neoplastic epithelium with or without papillary proliferations (Figure 1A) ▶ . The intraductal components of these neoplasms were classified as adenoma in 1 (4%), borderline in 5 (23%), and carcinoma in situ in 16 (73%). Twelve (55%) of the IPMNs were associated with an infiltrating adenocarcinoma.
Figure 1.
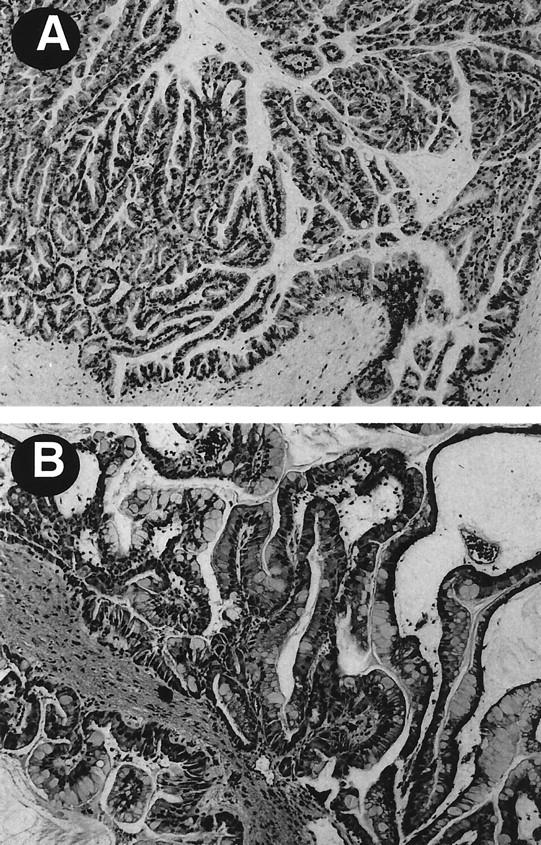
A: IPMN from a patient without PJS showing papillary proliferations of severely dysplastic epithelial cells, lining the dilated pancreatic duct. B: IPMN from a patient with PJS with moderate epithelial atypia (borderline).
Among the 22 cases, 2 patients were diagnosed as PJS based on the clinicopathological findings. One was a 36-year-old man with PJS who underwent pancreaticoduodenectomy after a screening endoscopic and ultrasound study revealed an asymptomatic cystic neoplasm in the head of the pancreas. 35 Intraoperative polypectomy from the distal ileum also revealed a hamartomatous (Peutz-Jeghers) polyp. Histological examination of the cystic neoplasm of the pancreas revealed a borderline IPMN (Figure 1B) ▶ . The second patient with PJS was a 50-year-old woman who underwent pancreaticoduodenectomy for a pancreatic mass. Histological examination of the resected specimen revealed an IPMN with focal high-grade epithelial dysplasia (carcinoma in situ) and an associated infiltrating pancreatic adenocarcinoma. The patient also had a number of hamartomatous polyps in the duodenum and jejunum.
LOH at the STK11/LKB1 Locus in IPMNs
DNA samples from 22 IPMNs were evaluated for LOH at the STK11/LKB1 locus using five polymorphic microsatellite markers. These markers span approximately 4.0 Mb within chromosome 19p13.3 and D19S886 is localized closest to the STK11/LKB1 (approximately 190 kb telomeric). The results of the LOH analysis are summarized in Figure 2 ▶ . Allelic loss of 19p13.3 markers was observed in 2 of 2 (100%) IPMNs from the patients with PJS (cases 1 and 2) and 5 of 20 (25%) from patients lacking features of PJS. Two samples (cases 1 and 14) showed LOH with only one marker, whereas other five showed LOH with two or more adjacent markers. Thus, we identified allele loss at the STK11/LKB1 locus in 7 of 22 (32%) IPMNs. In two PJS-associated IPMNs, LOH was seen exclusively with markers at 19p13.3 but not with two markers (D5S346 and TH) mapped to other chromosomal loci (Figure 3) ▶ . Of the seven IPMNs with LOH at 19p13.3, three (cases 1, 2, and 14) showed LOH boundaries that localized to the STK11/LKB1 locus.
Figure 2.

Deletion map of chromosomal region 19p13.3 in IPMNs. STK11/LKB1 is located at approximately 190 kb centromeric side from D19S886.
Figure 3.

LOH analysis in a representative IPMN. A PJS-associated IPMN (case 1) exhibits LOH at D19S565 (arrow), but not at D5S346.
Germline and Somatic Mutations of the STK11/LKB1 Gene in IPMNs
To further validate the STK11/LKB1 as a genetic target in IPMNs, we analyzed the seven IPMNs which displayed LOH at 19p13.3 for somatic mutations in the STK11/LKB1 gene. Direct sequencing of the complete coding sequence and exon/intron boundaries revealed one non-sense mutation in exon 2 and one frameshift mutation in exon 5 (two of 22, 9% overall). A single nucleotide change from C367 to T was identified in one of the two PJS-associated IPMNs (case 2), resulting in a premature stop codon at position 123 (CAG [gln] to TAG [stop]; Figure 4 ▶ ). Based on the sequence analysis of the corresponding normal tissue, this mutation was considered germline in origin. One bp deletion at C650 occurred in an IPMN without PJS (case 12), causing a frame-shift mutation at codon 217. Both of these mutations were within the catalytic kinase domain of the STK11/LKB1 gene (codons 37–314), 23 suggesting the inactivating nature of these mutations.
Figure 4.
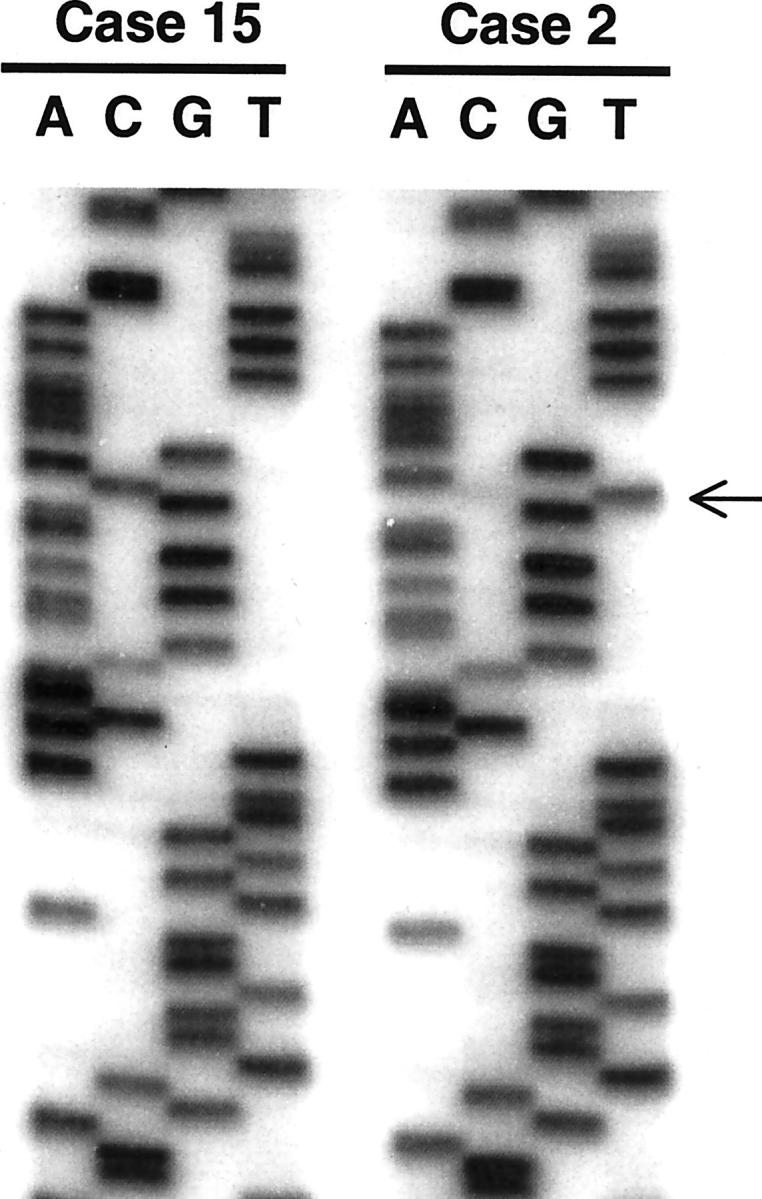
Non-sense mutation of the STK11/LKB1 gene (exon 2) in the PJS-associated IPMN (case 2). The arrow indicates a single nucleotide change from C to T, resulting in a premature stop codon at 123. The corresponding sequence in case 15 (sporadic IPMN) is wild type.
Methylation Analysis of the STK11/LKB1 Gene in IPMNs
Finally, we analyzed the 5′ CpG island of the STK11/LKB1 gene in 22 IPMNs using highly sensitive MSP technique. However, none of these neoplasms displayed hypermethylation of the STK11/LKB1 gene.
Discussion
In the present study, we investigated the role of STK11/LKB1 gene in 22 IPMNs associated with and without PJS. We found allelic loss at 19p13.3 in 7 of 22 (32%) IPMNs including both neoplasms from patients with PJS; that 3 of these 7 IPMNs showed LOH boundaries that localized to the STK11/LKB1 locus; and biallelic inactivation by germline/somatic mutation, coupled with LOH, in two (9%) IPMNs. Taken together, these findings suggest that STK11/LKB1 is one of the target genes involved in the development of IPMNs.
Abrogation of tumor-suppressor genes is a major mechanism that contributes to the development and progression of neoplasms. Inactivation of several tumor-suppressor genes including p16, DPC4, and p53 has been described in ductal adenocarcinoma of the pancreas. 10,36,37 Previous reports have shown that alterations in these tumor-suppressor genes are a relatively uncommon event in IPMNs, 15-17 though data on genetic analysis in IPMNs are limited so far. It has been hypothesized that retention of these tumor-suppressor functions in IPMNs may contribute to their better prognosis compared to conventional ductal adenocarcinomas. Alternatively, it is possible that other tumor-suppressor genes may play a role in the development of IPMNs.
In the present study, we demonstrate a relatively high frequency of LOH at 19p13.3 in IPMNs. LOH at STK11/LKB1 locus has been reported in sporadic cancers originating from the breast, colon, ovary, and pancreas. 38-43 Allelotype analysis using xenograft enrichment revealed loss of chromosome arm 19p in 29% of pancreatic adenocarcinoma. 44 Su et al 29 found LOH at STK11/LKB1 locus in 22 of 69 (32%) pancreatic cancers and, subsequently, somatic mutations in 4 of 100 (4%) cases. Interestingly, 1 of these 4 xenografted tumors was derived from a pancreatic adenocarcinoma that developed in association with an IPMN. This patient (case 12) was included in our present study, and this patient’s IPMN showed the same somatic nucleotide deletion (650delC), which was reported in their infiltrating adenocarcinoma. 29 These findings may provide the evidence for clonal progression from IPMNs to invasive carcinoma.
Sequencing analysis detected a germline mutation in one of the two PJS patients; however, another PJS-associated IPMN (case 1) did not show any sequence variants in the STK11/LKB1. The absence of mutations in this case may be due to large genomic deletions that are undetectable using PCR-based genetic analysis or non-coding region mutations that affect function. 45 Another possibility for the involvement of STK11/LKB1 is gene silencing by promoter methylation, as has been reported in hamartomatous polyps associated with PJS. 46 However, we were unable to find evidence for the 5′ CpG island methylation in our series of IPMNs. It is also possible that the allelic loss in the 19p13.3 region in IPMNs targets a yet-to-be-defined tumor-suppressor gene.
In summary, we demonstrate the LOH at 19p13.3 in 32% and biallelic inactivation of the STK11/LKB1 gene in 9% of IPMNs. Although further studies are warranted to determine the biological significance of this genetic change, our present results suggests a role of this gene in the progression of some IPMNs.
Footnotes
Address reprint requests to Michael Goggins, M.D., Departments of Pathology, Oncology, and Medicine, 632 Ross Building, 720 Rutland Avenue, The Johns Hopkins Medical Institutions, Baltimore, MD 21205. Email: mgoggins@jhmi.edu.
Supported by The National Institutes of Health SPORE grant in gastrointestinal malignancies (CA62924), the Lustgarten Foundation for pancreatic cancer research and the Michael Rolfe Foundation.
References
- 1.Fukushima N, Mukai K, Kanai Y, Hasebe T, Shimada K, Ozaki H, Kinoshita T, Kosuge T: Intraductal papillary tumors and mucinous cystic tumors of the pancreas: clinicopathologic study of 38 cases. Hum Pathol 1997, 28:1010-1017 [DOI] [PubMed] [Google Scholar]
- 2.Nagai E, Ueki T, Chijiiwa K, Tanaka M, Tsuneyoshi M: Intraductal papillary mucinous neoplasms of the pancreas associated with so-called “mucinous ductal ectasia”: histochemical and immunohistochemical analysis of 29 cases. Am J Surg Pathol 1995, 19:576-589 [DOI] [PubMed] [Google Scholar]
- 3.Loftus EV Jr, Olivares-Pakzad BA, Batts KP, Adkins MC, Stephens DH, Sarr MG, DiMagno EP: Intraductal papillary-mucinous tumors of the pancreas: clinicopathologic features, outcome, and nomenclature. Members of the Pancreas Clinic, and Pancreatic Surgeons of Mayo Clinic. Gastroenterology 1996, 110:1909–1918 [DOI] [PubMed]
- 4.Klöppel G: Clinicopathologic view of intraductal papillary-mucinous tumor of the pancreas. Hepatogastroenterology 1998, 45:1981-1985 [PubMed] [Google Scholar]
- 5.Paal E, Thompson LD, Przygodzki RM, Bratthauer GL, Heffess CS: A clinicopathologic and immunohistochemical study of 22 intraductal papillary mucinous neoplasms of the pancreas, with a review of the literature. Mod Pathol 1999, 12:518-528 [PubMed] [Google Scholar]
- 6.Sohn TA, Yeo CJ, Cameron JL, Iacobuzio-Donahue CA, Hruban RH, Lillemoe KD: Intraductal papillary mucinous neoplasms of the pancreas: an increasingly recognized clinicopathologic entity. Ann Surg 2001, 234:313-322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Traverso LW, Peralta EA, Ryan JA, Jr, Kozarek RA: Intraductal neoplasms of the pancreas. Am J Surg 1998, 175:426-432 [DOI] [PubMed] [Google Scholar]
- 8.Sho M, Nakajima Y, Kanehiro H, Hisanaga M, Nishio K, Nagao M, Ikeda N, Kanokogi H, Yamada T, Nakano H: Pattern of recurrence after resection for intraductal papillary mucinous tumors of the pancreas. World J Surg 1998, 22:874-878 [DOI] [PubMed] [Google Scholar]
- 9.Yamao K, Ohashi K, Nakamura T, Suzuki T, Shimizu Y, Nakamura Y, Horibe Y, Yanagisawa A, Nakao A, Nimuara Y, Naito Y, Hayakawa T: The prognosis of intraductal papillary mucinous tumors of the pancreas. Hepatogastroenterology 2000, 47:1129-1134 [PubMed] [Google Scholar]
- 10.Goggins M, Kern SE, Offerhaus JA, Hruban RH: Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol 1999, 10:4-8 [PubMed] [Google Scholar]
- 11.Satoh K, Sasano H, Shimosegawa T, Koizumi M, Yamazaki T, Mochizuki F, Kobayashi N, Okano T, Toyota T, Sawai T: An immunohistochemical study of the c-erbB-2 oncogene product in intraductal mucin-hypersecreting neoplasms and in ductal cell carcinomas of the pancreas. Cancer 1993, 72:51-56 [DOI] [PubMed] [Google Scholar]
- 12.Tada M, Omata M, Ohto M: Ras gene mutations in intraductal papillary neoplasms of the pancreas: analysis in five cases. Cancer 1991, 67:634-637 [DOI] [PubMed] [Google Scholar]
- 13.Z’Graggen K, Rivera JA, Compton CC, Pins M, Werner J, Fernandez-del Castillo C, Rattner DW, Lewandrowski KB, Rustgi AK, Warshaw AL: Prevalence of activating K-ras mutations in the evolutionary stages of neoplasia in intraductal papillary mucinous tumors of the pancreas. Ann Surg 1997, 226:491-498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujii H, Inagaki M, Kasai S, Miyokawa N, Tokusashi Y, Gabrielson E, Hruban RH: Genetic progression and heterogeneity in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 1997, 151:1447-1454 [PMC free article] [PubMed] [Google Scholar]
- 15.Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, Bassi C, Lemoine NR, Scarpa A: Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001, 84:253-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sessa F, Solcia E, Capella C, Bonato M, Scarpa A, Zamboni G, Pellegata NS, Ranzani GN, Rickaert F, Klöppel G: Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53, and c-erbB-2 abnormalities in 26 patients. Virchows Arch 1994, 425:357-367 [DOI] [PubMed] [Google Scholar]
- 17.Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, Wilentz RE, Argani P, Sohn TA, Yeo CJ, Cameron JL, Kern SE, Hruban RH: Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol 2000, 157:755-761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas : a new marker of DPC4 inactivation. Am J Pathol 2000, 156:37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue H, Furukawa T, Sunamura M, Takeda K, Matsuno S, Horii A: Exclusion of SMAD4 mutation as an early genetic change in human pancreatic ductal tumorigenesis. Genes Chromosomes Cancer 2001, 31:295-299 [DOI] [PubMed] [Google Scholar]
- 20.Jeghers H, McKusick VA, Katz KH: Generalized intestinal polyposis and melanin spots of the oral mucosa, lip, and digitis: a syndrome of diagnostic significance. N Engl J Med 1949, 241:1031-1036 [DOI] [PubMed] [Google Scholar]
- 21.Hemminki A, Tomlinson I, Markie D, Jarvinen H, Sistonen P, Bjorkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, de la Chapelle A, Aaltonen LA: Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet 1997, 15:87-90 [DOI] [PubMed] [Google Scholar]
- 22.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391:184-187 [DOI] [PubMed] [Google Scholar]
- 23.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M: Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998, 18:38-43 [DOI] [PubMed] [Google Scholar]
- 24.Tiainen M, Ylikorkala A, Makela TP: Growth suppression by Lkb1 is mediated by a G1 cell cycle arrest. Proc Natl Acad Sci USA 1999, 96:9248-9251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, Brien TP, Bozzuto CD, Ooi D, Cantley LC, Yuan J: The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell 2001, 7:1307-1319 [DOI] [PubMed] [Google Scholar]
- 26.Hizawa K, Iida M, Matsumoto T, Kohrogi N, Kinoshita H, Yao T, Fujishima M: Cancer in Peutz-Jeghers syndrome. Cancer 1993, 72:2777-2781 [DOI] [PubMed] [Google Scholar]
- 27.Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, Ahlquist DA, Podratz KC, Pittelkow M, Hartmann LC: Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998, 128:896-899 [DOI] [PubMed] [Google Scholar]
- 28.Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, Krush AJ, Yardley JH, Luk GD: Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987, 316:1511-1514 [DOI] [PubMed] [Google Scholar]
- 29.Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, Westerman AM, Entius MM, Goggins M, Yeo CJ, Kern SE: Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999, 154:1835-1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solcia E, Capella C, Klöppel G: Tumors of the pancreas. Atlas of Tumor Pathology, 3rd series. Edited by Rosai J, Sobin LH. Washington, DC, Armed Forces Institute of Pathology, 1997, pp 31–144
- 31.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 32.Goggins M, Hruban RH, Kern SE: BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol 2000, 156:1767-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Connolly DC, Katabuchi H, Cliby WA, Cho KR: Somatic mutations in the STK11/LKB1 gene are uncommon in rare gynecological tumor types associated with Peutz-Jegher’s syndrome. Am J Pathol 2000, 156:339-345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hruban RH, Canto MI, Yeo CJ: Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig Dis 2001, 19:79-84 [DOI] [PubMed] [Google Scholar]
- 36.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE: Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997, 57:1731-1734 [PubMed] [Google Scholar]
- 37.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG: Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997, 57:3126-3130 [PubMed] [Google Scholar]
- 38.Resta N, Simone C, Mareni C, Montera M, Gentile M, Susca F, Gristina R, Pozzi S, Bertario L, Bufo P, Carlomagno N, Ingrosso M, Rossini FP, Tenconi R, Guanti G: STK11 mutations in Peutz-Jeghers syndrome and sporadic colon cancer. Cancer Res 1998, 58:4799-4801 [PubMed] [Google Scholar]
- 39.Avizienyte E, Roth S, Loukola A, Hemminki A, Lothe RA, Stenwig AE, Fossa SD, Salovaara R, Aaltonen LA: Somatic mutations in LKB1 are rare in sporadic colorectal and testicular tumors. Cancer Res 1998, 58:2087-2090 [PubMed] [Google Scholar]
- 40.Bignell GR, Barfoot R, Seal S, Collins N, Warren W, Stratton MR: Low frequency of somatic mutations in the LKB1/Peutz-Jeghers syndrome gene in sporadic breast cancer. Cancer Res 1998, 58:1384-1386 [PubMed] [Google Scholar]
- 41.Forster LF, Defres S, Goudie DR, Baty DU, Carey FA: An investigation of the Peutz-Jeghers gene (LKB1) in sporadic breast and colon cancers. J Clin Pathol 2000, 53:791-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang ZJ, Ellis I, Zauber P, Iwama T, Marchese C, Talbot I, Xue WH, Yan ZY, Tomlinson I: Allelic imbalance at the LKB1 (STK11) locus in tumours from patients with Peutz-Jeghers’ syndrome provides evidence for a hamartoma-(adenoma)-carcinoma sequence. J Pathol 1999, 188:9-13 [DOI] [PubMed] [Google Scholar]
- 43.Wang ZJ, Churchman M, Campbell IG, Xu WH, Yan ZY, McCluggage WG, Foulkes WD, Tomlinson IPM: Allele loss and mutation screen at the Peutz-Jeghers (LKB1) locus (19p13.3) in sporadic ovarian tumours. Br J Cancer 1999, 80:70-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seymour AB, Hruban RH, Redston M, Caldas C, Powell SM, Kinzler KW, Yeo CJ, Kern SE: Allelotype of pancreatic adenocarcinoma. Cancer Res 1994, 54:2761-2764 [PubMed] [Google Scholar]
- 45.Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, Boland CR, Lynch HT, Chadwick RB, de la Chapelle A, Berg K, Eshleman JR, Yuan W, Markowitz S, Laken SJ, Lengauer C, Kinzler KW, Vogelstein B: Conversion of diploidy to haploidy. Nature 2000, 403:723-724 [DOI] [PubMed] [Google Scholar]
- 46.Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA, Herman JG: Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene 2000, 19:164-168 [DOI] [PubMed] [Google Scholar]