

Abstract
Progesterone receptor (PR) action is linked to epidermal growth factor (EGF) initiated signaling pathways at multiple levels; mitogen-activated protein kinases (MAPKs) are key mediators of this important cross-talk. Herein, we probed the effects of EGF on PR function and regulation of breast cancer cell growth. EGF stimulated rapid and transient phosphorylation of PR-B Ser294 relative to persistent phosphorylation of this site induced by the synthetic progestin, R5020. EGF induced nuclear translocation and DNA binding of unliganded wild-type, but not mutant PRs containing an Ala at position 294 (S294A). However, EGF alone induced little to no PR-B transcriptional activity; S294A PR-B was transcriptionally impaired. In contrast, pretreatment of cells with EGF (30 min) significantly increased the potency and efficacy of wild-type, but not S294A PR transcriptional activity in response to progestin, and enhanced ligand-dependent downregulation of wild-type but not S294A PR. Replacement of Ser294 with aspartic acid (S294D) to mimic phosphorylation at this site decreased receptor stability and, as predicted, heightened progestin-induced transcription relative to wild-type PR-B. RT-PCR demonstrated the Ser294 phosphorylation-dependence of selected PR target genes (TGFα and HB-EGF). Surprisingly, PR-B expressing cells growing in soft agar were highly responsive to EGF or progestin, and this was further stimulated by the combination of both hormones. Cells expressing S294A PR exhibited reduced soft agar growth, and were also sensitive to R5020 alone, but failed to respond to EGF. These results suggest that PR Ser294 is an important “sensor” for growth factor inputs that affects PR function and breast cancer cell growth in the absence of progestin or in the presence of low or “sub-threshold” progestin concentrations. PR function likely contributes to breast cancer progression when EGFR family members or their ligands are overexpressed, a condition that predicts low abundance, but highly active and nuclear PR.
Keywords: progesterone receptor, mitogen activated protein kinase, phosphorylation, epidermal growth factor, breast cancer, transcription, steroid
Introduction
The ovarian steroid hormones, estradiol and progesterone, control the ductal growth (1) and alveolar development (2), respectively, of the normal mammary gland and are believed to contribute to breast cancer development and progression. Similarly, epidermal growth factor receptor (EGFR) family members are expressed in both normal and malignant breast epithelial cells and their overexpression in breast cancers is predictive of a poor prognosis. In the normal maturing mammary gland, EGF augments the proliferative effects of progesterone and estrogen, to induce ductal sidebranching and lobuloalveolar development (3). Cross-talk between EGF and ovarian steroid hormone receptors likely continues in the malignant breast. Indeed numerous points of cross-talk between estrogen and progesterone and growth factor signaling events have been described (4–7). Additionally, progesterone addition to estrogen in hormone replacement therapy increases breast cancer risk (8,9).
At the time of breast cancer diagnosis, two thirds of tumors are steroid hormone-dependent. As tumors progress, they are more likely to become steroid hormone resistant, yet often retain their nuclear steroid receptors. In fact, receptor loss or mutation accounts for only 10–20% of clinically observed steroid-hormone resistant breast tumors (10). Thus it has been postulated that in the majority of resistant tumors, control over growth is assumed by locally acting autocrine or paracrine peptide growth factors, and in fact, the invasive cancers with the worst prognosis are those that are growth factor receptor positive and steroid-hormone resistant (11) (12). Growth factors regulate cell growth via the initiation of mitogenic intracellular signal transduction pathways after binding to high-affinity tyrosine kinase receptors on the cell surface, while steroid hormone receptors are ligand-activated transcription factors that can also function as cell membrane-associated signaling molecules (13).
One consequence of the activation of rapid signaling events (i.e. kinase cascades) by peptide growth factors or liganded steroid hormone receptors stationed at the cell membrane is the direct phosphorylation of nuclear steroid receptors. We have previously defined functional roles for phosphorylation of human progesterone receptor (PR) Ser294 by mitogen-activated protein kinase (14–16) and PR Ser400 by cell-cycle-dependent protein kinase two (17). Phosphorylation of these sites is regulated in response to progestins or growth factor mitogens, including EGF and heregulin, and can potentiate ligand-dependent PR transcriptional activity, as well as induce ligand-independent PR actions (17,18). Thus, phosphorylation of liganded steroid hormone receptors allows for positive “feed-forward”, perhaps insuring that appropriate (i.e. moderate to strong) genomic responses may occur in hormonally regulated tissues. Clearly however, robust kinase activation during breast cancer progression may “usurp” hormone action by driving receptor activities in the presence of very low or absent steroid hormone ligands (19). Additionally, differentially phosphorylated steroid hormone receptors regulate different subsets of genes, and remarkably with little to no overlap (20), thereby allowing for the “switching” of genetic programs in normal and malignant states.
Herein, we examined the effects of EGF on PR action and cell growth in human breast cancer cells. We find that EGF mediates PR nuclear translocation and interaction with PRE-sequences in the absence of progestin, and induces PR transcriptional “hypersensitivity” coupled to rapid PR turnover in the presence of sub-stimulatory levels of progestin. We also identify PR target genes whose transcription is dependent on PR Ser294 phosphorylation. Furthermore, phosphorylation of PR Ser294 is required for increased anchorage-independent breast cancer cell growth in response to EGF, but does not affect the cellular response to progestins. These studies illustrate how growth factors may usurp PR action, a process relevant to understanding hormonal regulation of malignant cell growth in breast cancers.
Experimental
Cell lines and reagents
T47D cell lines stably expressing PR isoforms (21) and HeLa cells transiently expressing GFP-tagged PR-B (15) were cultured as previously described. Phospho- and total Erk1/2 antibodies were purchased from Cell Signaling; phospho- (Ab12) and total (Ab-8) PR antibodies were purchased from Lab Vision. Actin antibody was obtained from Sigma. Secondary antibodies were purchased from Bio-Rad Laboratories. R5020 (10nM) was purchased from NEN Boston, MA.
Cell Fractionation, Immunoblotting and Confocal Microscopy
Cell Fractionation experiments, immunoblotting and confocal microscopy were performed as previously described (15).
Transcription Assays
T47D cells in 6 well plates were transfected with PRE-2x-TATA-luc plasmid (0.5ug), pRL-tk (Renilla; 10ng) and, in PR-null, pSG5-hPR1 (wt or mutant receptor constructs (20ng) using FuGENE 6 reagent (Roche). Starved cells were treated for 24 hours as indicated and luciferase assays were preformed in triplicate using the Dual-LuciferaseTM Reporter Assay (Promega).
Electrophoretic Mobility Shift Assay
T47D cells stably expressing wt or mutant PR-B were serum-starved for 24 hr and then treated as indicated. PR-B preparations (4ug, unless indicated otherwise) were incubated in DNA-binding buffer with P32-labeled PRE oligonucleotides for 30 min at 4°C. Precooled 5% polyacrylamide gels were loaded and run at 4 °C in TGE buffer. In controls, PR-B/PRE complexes were incubated in the presence of PR-specific antibodies and were competed with unlabled PRE or SP1-site containing oligonucleotides (22).
Reverse Transcriptase-PCR
Total cellular RNA was isolated with Trizol (Invitrogen) according to the manufacturer protocol. RNA was DNAse treated and cDNA was synthesized from 2ug total RNA using MMLV-RT and random primers (BD Biosciences) in a total volume of 20ul. cDNA was diluted 1:5 and 2ul was used for each PCR reaction with EGF, TGFα and HB-EGF specific primers. cDNA was diluted 1:50 for β-Actin specific PCR.
Cell cycle Analysis and Soft Agar Assay
Flow cytometry experiments (23) and anchorage independent growth assays (24) were performed and analyzed as previously described.
Results
EGF Induces PR Nuclear Association and PRE Binding
How does EGF alter PR function? Western blotting using phospho-specific antibodies indicated that EGF stimulation of T47D cells induces phosphorylation of PR Ser294 at 5-10 min, similar to that induced by R5020 at early time points (15,16). Both agents are known to activate MAPKs within 5–15 minutes (25,26). However, EGF is a stronger activator of MAPK relative to progestins at this time point, and liganded PRs are relatively transient, most likely due to the rapid down-regulation of liganded and Ser294 phosphorylated species (14). We therefore compared the kinetics of PR Ser294 phosphorylation in response to treatment of T47D-YB breast cancer cells with either EGF or R5020 for 5–60 min (Fig. 1A). Total and activated p42/p44 MAPKs were measured in the same cell lysates. Both EGF and R5020 stimulated a similar increase in the level of Ser294 phosphorylated PR between 5–15 min, consistent with the time course of activation of MAPKs by either agent (25,26) (see Fig. 1A). However, EGF-induced PR Ser294 phosphorylation was transient, dropping off between 30 and 60 min. Interestingly, PR Ser294 phosphorylation induced by R5020 remained persistent in the absence of detectable MAPK activity; total PR levels remained constant over the 60 min time course. EGF treatment caused a slight upshift in PR mobility (30–60 min), but this did not approach that seen for the fully active liganded receptor. MAPK activity assays, as measured using phospho-specific antibodies, indicated that both EGF and R5020 activated p42/p44 MAPKs at 5 min; R5020-induced MAPK activity was weak and transient relative to that induced by EGF (Fig 1A). Ser294 to alanine (S294A) mutant PR-B is not recognized by the phospho-Ser294 PR antibody, demonstrating the specificity of the antibody (Fig 1B). These data indicate that EGF and progestins can independently regulate PR Ser294 phosphorylation. Differences in the strength and duration of PR Ser294 phosphorylation relative to MAPK activation by either agent suggests that separate kinase pathways regulate this site in response to these hormones(15), and/or that liganded PRs are not readily dephosphorylated on Ser294 in response to modest activation of MAPKs.
Figure 1.

EGF-induced phosphorylation of PR Ser294. A. Regulation of PR Ser294 by EGF. T47D cells stably expressing wt PR-B were treated with vehicle control (C), EGF (30ng/ml) (E), or R5020 (10nM) (R) for 5–60 min and PRs were visualized in cell lysates by Western blotting using phospho-Ser294- or total PR-specific antibodies. Total and active p42/p44 MAPKs were similarly measured in the same lysates using phospho- and total MAPK-specific antibodies. A darker exposure indicates the progestin-induced activation of MAPKs at 5 min. B. T47D cells expressing wt or S294A PR-B were treated with either EGF or R5020 for 15 or 60min, cells were harvested and Western blots performed using phospho-294 PR or PR specific antibodies.
Signals from growth factors, including EGF, may enhance the activity of liganded PRs by altering PR location (19) or association with co-regulatory molecules (27). EGF induces the rapid nuclear translocation of unliganded PRs that are suspected to regulate selected promoters (15). Translocation of PR is apparent in intact EGF-stimulated HeLa cells by as early as 5min, as measured using confocal microscopy (15). To examine the kinetics of PR nuclear translocation by EGF in breast cancer cells, a time course of EGF treatment was conducted in T47D cells stably expressing either wt or S294A mutant PR-B. Cell lysates were fractionated into cytoplasmic and nuclear fractions and subjected to Western blotting with PR-specific antibodies (Fig. 2A). Similar to our previous reports (15), the distribution of wt and S294A PR-B between the cytoplasmic and nuclear fractions is very similar in untreated T47D cells expressing either receptor (lanes 1–2). Interestingly, however, a short time course (0–60 min) of EGF treatment revealed that S294A PR-B is excluded from the nuclear fraction (or not stably associated with the nucleus) following at least 5min EGF treatment, and remains soluble or “cytoplasmic” throughout the 60min time course (compare lane 2 to lanes 4, 6, 8, and 10). Similarly, wt PR-B also appears to remain soluble or exit the nuclear fraction by at least 15min (lane 6). In contrast to S294A PR-B, wt PR-B subsequently undergoes nuclear translocation that is complete between 30 and 60min (lanes 8 and 10). At 60 min, the two receptors are differentially partitioned into the cytoplasmic or soluble (S294A) and nuclear (wt PR) fractions (lanes 9–10). We reported that both receptors undergo nuclear translocation in the presence of R5020 (Fig. 2A, lower panel), and with similar kinetics (between 30 to 60 min) (15), indicating that different mechanisms exist for nuclear association of phosphorylated unliganded PRs relative to liganded PRs.
Figure 2.

MAPK-dependent nuclear association of PR and EGF-induced PRE binding. A. Fractionation of nuclear and cytoplasmic PRs. T47D cells stably expressing wt or S294A PRs were placed in serum-free media for 24 hr and then treated without or with EGF (30ng/ml) for 0, 5, 15, 30, or 60 min. Cell lysates were then fractionated into soluble cytoplasmic (C) and nuclear (N) fractions. PRs were blotted using specific antibodies. In controls (lower panel), additional cultures of the same cell lines were treated with R5020 (10nM) for 60 min to demonstrate nuclear association of both wt and mutant PR-B using this method (15). B. Confocal microscopy showing MEK-1 induced nuclear association of wt, but not S294A PR-B in intact HeLa cells. HeLa cells expressing GFP-tagged wt or S294A PR-B were transiently transfected with empty vector (control), wt-Mek-1 or a constitutively active mutant form of Mek-1 (R4F-MEK-1). GFP-tagged PR-B was visualized using direct fluorescence confocal microscopy as in Methods. C. EMSA showing EGF-induced recruitment of unliganded wt PR-B to PRE sequences. T47D cells stably expressing wt PR-B were serum-starved for 24 hr and then treated with vehicle (EtOH) or R5020 (10nM; 60 min) to control for the specificity of PR/PRE interaction (upper panel). In the same experiment but on a separate gel, cells were treated with EGF for 15, 30, or 60 min (lower panel). Specific binding of wt PR-B to P32-labeled PRE oligonucleotides was measured in the absence and presence of unlabeled competitor PRE (c-PRE). Unless indicated, all reactions used 4μg nuclear extract (NEx) and a constant PRE concentration.
Studies using confocal microscopy of GFP-tagged receptors demonstrated that wt, but not S294A PR, appear exclusively nuclear following EGF treatment of T47D or HeLa cells (15). Thus, we do not observe the apparent “nuclear exclusion” of endogenous or tagged PRs by confocal microscopy (see below) (15). Rather, confocal studies consistently reveal that cytoplasmic S294A PR-Bs do not persistently associate with the nucleus in response to EGF, as do wt-PRs (15). Further, Ser294 phosphorylated receptors appear nuclear (15). These data suggest that cell fractionation may reflect the relative solubility of PRs that are associated with either fraction, rather than their true cellular location in intact cells; soluble but nuclear PRs may partition into the cytoplasmic fraction upon cell lysis. The very large nuclear to cytoplasmic ratio of T47D cells complicates the visualization of PR partitioning in intact cells (15). However, due to their large cytoplasm and cell body, HeLa cells are ideal for the clear visualization of GFP-tagged PRs using confocal fluorescence microscopy (15). GFP-PRs have been well-characterized, and behave as do endogenous PRs with regard to location and function in transcription assays (28). We therefore expressed GFP-tagged wt or S294A PR-B in HeLa cells. We showed previously that EGF-induced nuclear association of unliganded wt PRs was blocked by MEK inhibitors (15), suggesting that this event is MAPK-dependent. However, MEK inhibitors can indirectly alter processes important for nucleo-cytoplasmic shuttling (15). To confirm that nuclear association of unliganded PRs occurs by a MAPK and Ser294-dependent mechanism, we co-transfected HeLa cells growing on cover slips with GFP-tagged wt or S294A mutant PRs, and either vector control or a constitutively active mutant of MEK-1, which directly phosphorylates and activates p42/p44 MAPKs. Wt-MEK-1 does not appreciably activate MAPKs (29), and was included as a control for the overexpressed MEK-1 protein. Transfected HeLa cells were placed in serum-free steroid stripped media, fixed, and subjected to confocal fluorescence microscopy to visualize GFP-tagged PRs (Fig 2B). In cells expressing either control vector or wt MEK-1, both wt PR-B and the S294A mutant were similarly distributed between the cytoplasm and nucleus, consistent with our previous reports (15) and the data shown (Fig. 2B). Wt-MEK-1 expression induced the subtle “spreading” of cell cytoplasm or apparent “flattening” of the cell bodies relative to vector controls, but did not appreciably alter PR location. However, as reported with EGF (15), constitutively active MEK-1 induced the nuclear association of wt, but not S294A PR-B. These data support a mechanism for PR nuclear association that involves the action of the activated MAPK module and phosphorylation of PR Ser294. The expression and activity of each MEK-1 protein was confirmed by Western blotting and by transcription assays using a c-fos promoter-driven (MAPK-dependent) luciferase construct (not shown).
We wondered if nuclear, but unliganded PR is able to bind specifically to PRE sequences in response to EGF stimulation of cells. To address this question, we performed EMSA using T47D cells stably expressing PR-B (Fig. 2C). Cells growing in serum-free steroid-stripped media were treated with vehicle controls, R5020 or EGF, and nuclear extracts (4–12ug protein) were incubated with radio-labeled PRE-containing oligonucleotides, and subjected to non-denaturing gel electrophoresis. Unliganded wt PR-B weakly associated with an oligonucleotide sequence representing a canonical PRE (lanes 1 and 8). As expected, binding of PR to the PRE probe increased upon R5020 treatment of cells (lanes 1 and 2; lanes 7 and 8). The specificity of the PR-PRE association was confirmed by competition with 10–50X unlabeled PRE (lanes 5–6). A time course of EGF treatment (30ng/ml; 15–60min) revealed that EGF induced the binding of PR-B to the PRE in a time-dependent manner, beginning at 30min (lane 10), and ultimately approximating that induced following a 60min treatment with R5020 (compare lanes 7 and 9). These data suggest that unliganded PR can associate with PREs in response to EGF treatment of cells, consistent with the timing of EGF-induced PR nuclear association as measured by cell fractionation and confocal microscopy (Fig. 2A and B). S294A PR-B failed to associate with PRE sequences in response to EGF treatment of T47D cells (not shown), but clearly bound to PRE probe in response to R5020 (Fig 3C).
Figure 3.

Simultaneous treatment of HeLa or T47D cells with EGF and R5020 does not alter PR transcription. Equal numbers of PR-null HeLa cells (A) or T47D cells (B) stably expressing either wt PR-B or mutant S294A PR-B were plated in growth media, and transfected the following day with PRE-luciferase and constitutive renilla plasmids using the FuGENE 6 reagent as described in Experimental. Following a 24-48 hr recovery period, cells were then placed in serum-free media for 24 hrs, and stimulated for an additional 24hr with either vehicle control (EtOH), R5020 (1.0nM), EGF (30ng/ml), or both agents, added simultaneously. Luciferase and renilla activities were measured in cell lysates. Bars represent the average and S.E.M. of triplicate measures; experiments were repeated a minimum of three times. C. Electrophoretic Mobility Shift Assay (EMSA) showing wt and mutant PR-B recruited to PRE-specific sites in DNA. T47D cells stably expressing either wt or S294A PR-B were treated with EtOH or R5020 (10nM) for 1 hr and nuclear extracts were subjected to EMSA using a P32-labeled PRE oligonucleotide. In controls, PR-B/PRE complexes shifted in the presence of PR-specific antibodies and were competed with cold PRE, but not SP1-site containing oligonucleotides.
Simultaneous Exposure to EGF and Progestin Does not Alter PR Transcription
We previously reported transcriptional synergy in progestin-treated cells transiently expressing activated MEKK1, a kinase pathway that mediates robust p42/p44 MAPK activation via the action of the classical Ras/Raf/MEK1–2 module in breast epithelial cells (16). Additionally, EGF and progestins synergistically activated transcription from the p21 and c-fos promoters that lack classical PRE sites, but presumably respond to liganded PR via the action of activated STATs (5,30) and/or PR interaction with SP-1 molecules (31). In contrast to studies with estrogen receptors, ligand-independent PR transcriptional activity in response to growth factors is seldom reported (17,18). To address whether EGF stimulation of breast cancer cells could similarly alter the effects of progestins on PRE-containing promoters, we transiently expressed a PRE-luciferase reporter gene in HeLa or T47D cells stably expressing the PR-B isoform (Fig. 3A–B). In these cell line models, progestins (10nM R5020) typically stimulated PRE-luciferase expression 10–20-fold above vehicle-treated controls, while EGF alone (30ng/ml) was without significant effect. In contrast to our previous reports using transiently expressed constitutively activated protein kinases (16,17), the combination of EGF and R5020 (simultaneously delivered) produced a modest increase in PR transcriptional activity relative to R5020 alone, but this did not reach statistical significance. The same treatments were without effect in control experiments using T47D-Y PR-null cells (not shown).
PR Ser294 is a hormone-inducible MAPK consensus site in the PR N-terminus that may serve as a direct “sensor” for MAPK-dependent input to PR transcriptional activity (6,13–16,19,32). Therefore, we included our well-characterized S294A PR mutant receptor in the above experiments (Fig. 3). S294A PR-B contains a single point mutation at Ser294 to Ala that renders it resistant to progestin-induced downregulation in multiple cell lines (14). S294A PR-B is transcriptionally impaired when stably expressed in T47D or HeLa cells (16). However, similar to wt PR-B, stably expressed S294A PR-B is able to stimulate MAPK activation, cyclin D1 expression, and S-phase entry (23) via the c-Src-dependent pathway induced upon progestin binding to cytoplasmic PRs (25). As expected, in HeLa or T47D cells, stably expressed S294A PR exhibited weakened transcriptional activity with R5020 alone, and this was not further enhanced when cells were treated with EGF (Fig 3A–B). S294A PR is capable of entering the nucleus in the presence of progestins (15) (Fig 2B). Electrophoretic gel mobility shift assays (EMSA) using a radio-labeled PRE oligonucleotide binding site demonstrated that both wt and mutant liganded PRs are able to bind DNA (Fig. 3C). These results indicate that although S294A PR is capable of ligand-binding and specific interaction at PRE-sites, Ser294 may be a key phosphorylation site required for PR transcription in this cell and promoter context.
EGF Pretreatment Induces PR Transcriptional Hypersensitivity to Progestin
The above data suggest that EGF may stimulate the formation of stable, but inactive transcription complexes that are perhaps “poised” for hormonal stimulation by progestins. In this case, phosphorylated but unliganded PRs may be capable of “sensing” low or sub-threshold steroid hormone concentrations. To test this concept, we again transiently transfected a PRE-luciferase reporter gene into T47D cells stably expressing either wt PR-B or S294A PR-B. Cells were then pretreated with EGF (30ng/ml) or vehicle control for 30min to allow for the formation of PR-PRE complexes (Fig. 2C), followed by treatment with increasing concentrations of R5020 (0.0 to 10nM). Surprisingly, EGF pretreatment significantly increased both the potency and efficacy of PR activation by progestin (Fig. 4A). “Hypersensitive” PR exhibited a dose-response curve that shifted to the left by two orders of magnitude; wt PR-B responded to 0.10 nM R5020 following EGF pretreatment relative to R5020 alone, in which PR required at least 10nM R5020 to reach a similar level of transcriptional activity. The total level or maximal transcription observed in the presence of 10nM R5020 was also significantly increased following EGF pretreatment. In contrast, a slight increase in S294A PR-B transcriptional activity was weakly apparent at 1.0 to 10.0 nM R5020 alone, but was not significantly altered by EGF pretreatment. EGF alone was without effect on the transcriptional activity of either receptor (Fig. 3). Additionally, simultaneous co-treatment with both EGF and R5020 did not alter the dose-response curve (not shown and see Fig. 3); a leftward shift was not observed until at least 15min EGF pretreatment (not shown).
Figure 4.
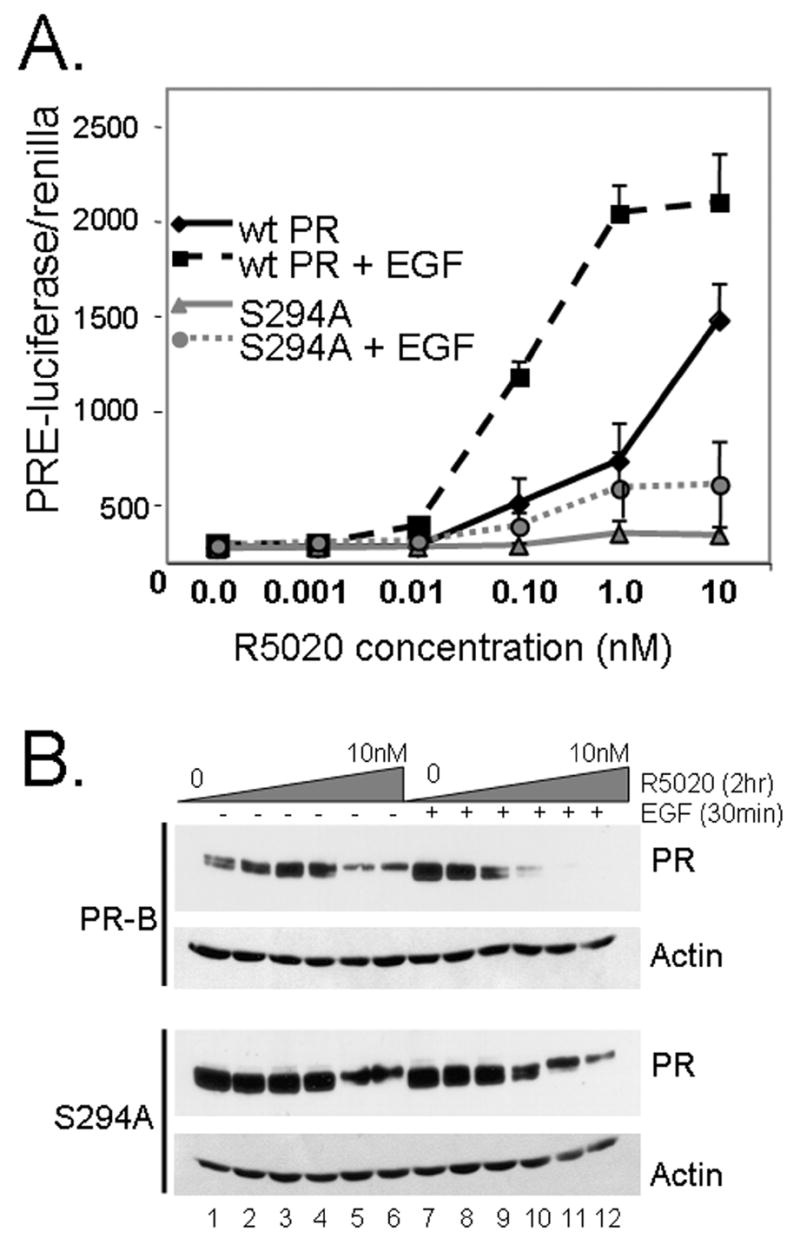
EGF pre-treatment confers hypersensitivity to liganded PR. A. Dose-response curve showing progestin-induced PR transcriptional activity following 30 min EGF pretreatment. T47D cells stably expressing either wt or S294A PR-B were transfected with PRE-luciferase and constitutive renilla reporters. Following a 24–48 hr recovery period, cells were placed in serum-free media for 24 hr, and then pre-treated without or with EGF for 30 min. Cells were then treated with increasing concentrations (0–10nM) of R5020 for 24 hr and luciferase/renilla activity was measured in cell lysates. Data points represent the mean and S.E.M. of triplicate measures; experiments were repeated a minimum of 4 times. Experiments in HeLa cells stably expressing PRs produced similar results (not shown). B. Western blotting showing increased turnover of liganded wt PR-B protein in T47D cells pretreated with EGF. Cells were pre-treated with EGF for 30 min as in part A except this was followed by a 2 hr R5020 treatment, and wt and S294A PRs or Actin proteins were measured in whole cell lysates by Western blotting using specific antibodies. Experiments were repeated twice in T47D cells and once in HeLa cells with similar results (not shown).
In a similar set of experiments, protein was harvested for PR and actin Western blotting from parallel cultures of T47D cells stably expressing either wt or S294A PR, and pretreated with EGF or vehicle control (30min) prior to addition of increasing concentrations of R5020 (Fig. 4B). In the absence of EGF pretreatment, wt PR were activated, as indicated by gel up-shift, in the presence of 1.0 and 10.0nM R5020 (Fig. 4B; lanes 5–6). Following EGF pretreatment however, wt PR-B up-shifted in the presence of much lower concentrations of R5020 relative to that observed with R5020 alone (compare lanes 1–4 to lanes 7–10). Additionally, EGF pretreatment of cells expressing wt PR-B resulted in the rapid downregulation of liganded PR in the presence of 0.10 to 10nM R5020 (lanes 10–12). PR transcriptional activity is inversely related to PR stability (16,19); transcriptionally active PR turnover very rapidly and these events appear to be coupled via the phosphorylation of PR Ser294 (14–16,19). Consistent with these results, the ability of S294A PR-B to upshift in the presence of R5020 was unaltered by EGF pretreatment, and S294A PR were resistant to ligand-induced downregulation relative to wt PR-B (compare lanes 10–12 for each receptor). Although they can bind PRE sequences, treatment with EGF alone did not fully upshift wt PRs (Fig. 1A) or alter their rate of turnover in T47D cells (16).
Increased turnover and heightened transcriptional activity of phospho-mimic PR
To confirm that reversible phosphorylation of PR Ser294 mediates changes in PR hormone responsiveness coupled to altered rates of PR turnover, we created PR in which Ser294 was replaced with aspartic acid (S294D). The negatively charged Asp residue at PR-B amino acid position 294 is predicted to mimic phosphorylation at this site, creating a mutant that acts as a constitutively phosphorylated PR. Western blot analysis of HeLa cells transiently transfected with S294D PR showed that upon treatment with R5020, S294D PR-B exhibited a gel upshift (Fig. 5A), characteristic of ligand-binding and phosphorylation at multiple sites (33). As we predicted, the S294D mutant was destabilized relative to wt receptor. In time courses conducted using transient assays we saw complete loss of S294D PR at 6 hours of R5020 treatment, while wt PR persisted for at least 24 hrs. In T47D-Y cells stably expressing S294D PR-B the effect was more subtle; however an increase in S294D PR-B receptor turnover relative to wt PR-B was apparent in multiple experiments (Fig 5B; see 24 hrs).
Figure 5.

Phospho-mimic S294D PR-B exhibits decreased stability and increased transcriptional activity. A. Western blots showing ligand-induced downregulation of wt and S294D PR-B. HeLa cells were transiently transfected with either wt or S294D PR-B and treated with or without R5020 for 1–24 hours. Western blots for PR were performed using PR specific antibodies. B. T47D cells stably expressing either wt or S294D PR-B were treated for either 8 or 24 with R5020 and Western blots were performed using PR specific antibodies. C. HeLa cells were transiently transfected with wt, S294D or S294A PR-B, PRE-luciferase, and pRL-TK (Renilla). Cells were serum-starved for 24 hrs and then treated for 24 hours with R5020, EGF, or both agents (added simultaneously) and luciferase units in whole cell lysates were normalized to the signal from renilla as a transfection control.
We then tested the transcriptional activity of S294D PR-B in the presence of R5020, EGF, or both agents. HeLa cells were transiently transfected with wt PR-B or mutant S294D or S294A PR-B receptors and PRE-luciferase constructs (Fig 5C); because S294D PR-B is predicted to behave as a phosphorylated receptor cells were hormone treated simultaneously. In HeLa cells, the transcriptional activity of wt PR-B was augmented in the presence of EGF relative to R5020 alone. S294D PR-B exhibited robust transcriptional activity in the presence of R5020 alone, and this approached the activation of wt PR-B treated with both EGF and R5020. PR negative breast cancer cells, BT-549, transiently transfected with S294D PR-B also displayed heightened PRE-driven transcription relative to wt PR (data not shown). As expected (Fig. 3A), S294A PR-B was transcriptionally inactive (Fig. 5C). These data indicate that reversible phosphorylation of PR Ser294 is critical for regulation of liganded PR-B transcriptional responses in the presence of growth factors. However, regulation of this site does not appear to fully rescue PR hypersensitivity (Fig. 4A). Other phosphorylation sites on PR-B, such as MAPK consensus site Ser345, contribute to transcriptional synergy (14). In addition, kinase inputs to PR action also target co-activator recruitment (27).
Ser294 phosphorylated PR regulates a subset of endogenous genes
In addition to facilitating PR hypersensitivity on experimental PRE-driven promoter constructs, can EGF pretreatment of cells alter the expression of endogenous genes? To examine the expression of selected PR gene targets, we performed RT-PCR on cDNAs derived from T47D cells stably expressing either wt or S294A PR-B. In a first set of experiments, cells were treated for 12 or 18 hours with R5020 and total cell RNA was isolated with Trizol and DNAse treated. Purified RNA was then used to synthesize cDNA with MMLV-RT and random primers (see Experimental). PCR reactions were carried out with cDNA templates and primers specific for EGF, TGFα and Actin. R5020 induced increased levels of EGF transcript in both wt and S294A PR-B expressing cells. However, TGFα transcripts increased only in progestin-treated cells expressing wt PR-B (Fig 6A). These data predict that phosphorylation of PR Ser294 is important for progestin-induced upregulation of a specific subset of PR-responsive genes.
Figure 6.
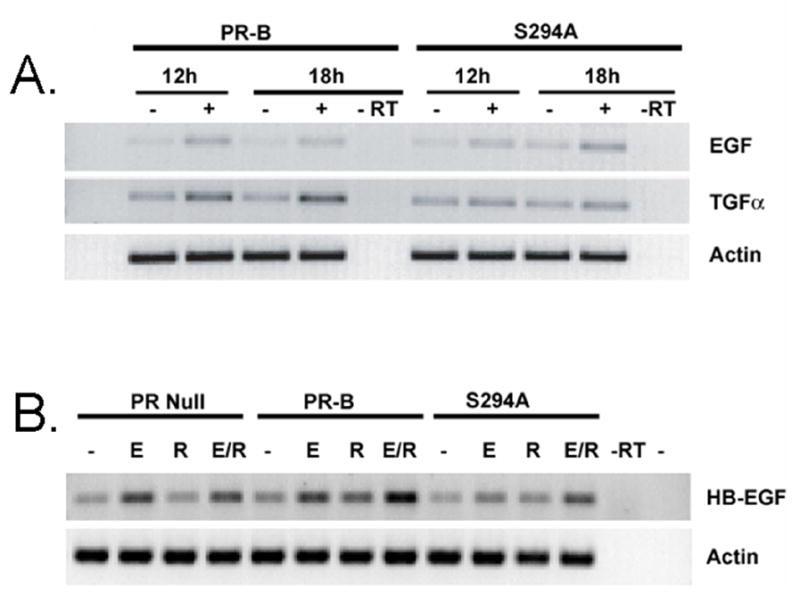
Phosphorylation of Ser294 PR-B is required for the regulation of a subset of endogenous PR target genes. A. RT-PCR showing differential upregulation of selected EGFR ligand transcript levels in response to phosphorylation of PR Ser294. T47D cells stably expressing either wt or S294A PR-B and treated with or without R5020 for 12 or 18 hours, and total cellular RNA was isolated with Trizol (Invitrogen) according to the manufacturer’s protocol. RNA was DNAse treated and cDNA was synthesized form 2ug of total RNA using MMLV-RT and random primers (BD Biosciences) in a total volume of 20ul. cDNA was used for PCR with EGF, TNFα and Actin specific primers and run on agarose gels. B. T47D-Y cells (PR null), and wt or S294A PR-B expressing cells were treated with vehicle or EGF for 30 min then vehicle or R5020 for 1 hour. cDNA was generated as above and PCR performed using HB-EGF or Actin specific primers.
In similar experiments, we tested the effects of R5020 in combination with EGF on HB-EGF transcript levels, which are known to be progestin regulated (34). PR-null T47D cells (T47D-Y) or T47D cells stably expressing wt or S294A mutant PR-B were compared. In PR negative cells, EGF but not progestin treatment induced increased expression of HB-EGF transcripts (Fig 6B). However, in PR-B expressing cells, HB-EGF expression was weakly upregulated in response to either R5020 or EGF, but exhibited robust expression via the combination of both hormones. These effects were greatly diminished in cells expressing the phospho-mutant S294A PR-B. These results suggest that EGF-induced PR Ser294 phosphorylation influences a subset of PR-B target genes (Fig. 6A), and surprisingly, can augment the expression of EGF-responsive genes (Fig. 6B). Heavily phosphorylated and rapidly cycling PRs may mediate increased expression of EGFR family ligands in breast cancers. Tumor cell proliferation in response to locally acting growth factors, may in part be facilitated by PR-dependent autocrine signaling (35).
PR Ser294 acts as a Sensor for the Mitogenic Actions of EGF
Phosphorylation of PR Ser294 is critical for PR hypersensitivity (Fig. 4). Does PR Ser294 phosphorylation also act as a “sensor” for EGF signaling to cell proliferation? We wondered if PRs may confer increased sensitivity of breast cancer cells to the growth promoting properties of EGF or other growth factors present in serum, in part via the phosphorylation of PR Ser294. We therefore examined the ability of T47D cells stably expressing either wt or S294A mutant PR-B to proliferate in response to EGF and/or progestins (Fig. 7). During the creation of the S294A mutant PR-B cell lines, we noted that clones containing S294A PR-B exhibit reproducible differences in cell morphology relative to clones expressing wt PR (Fig. 7A). When plated on plastic tissue culture dishes into growth media supplemented with 5% serum, S294A PR-B containing cells are less adherent and appear as more rounded single cells or small cell clumps relative to cells containing wt PR-B, and although easily propagated, they generally fail to flatten over time into multi-cell colonies with well-defined cell-cell borders, as do cells containing wt PR-B. The plating efficiency of S294A PR-B cell clones is roughly 25–30% lower than that of wt PR-B clones. This may indicate that PR Ser294 is important for the targeting of PRs to a subset of genes involved in cell shape and/or adhesion (36,37). T47D cells stably expressing S294D phospho-mimic PR appear morphologically comparable to cells expressing wt PR-B, and exhibit an equivalent plating efficiency (data not show).
Figure 7.

Role of PR Ser294 in cell morphology and proliferation. A. Ser294-dependent changes in T47D cell morphology. T47D cells stably expressing either wt or S294A PR-B were plated in standard tissue culture dishes and maintained in 5% serum. Live cells were examined using phase-contrast microscopy (images are 10X power). B. Flow cytometry of T47D cells treated with EGF and progestins. T47D cells stably expressing either wt or S294A PR-B were placed in serum-free media for 48 hr, and then treated with vehicle (EtOH), R5020 (10nM), EGF (30ng/ml), or both agents for 18 hr. DNA content in fixed cells was determined by flow cytometry as described in Methods. Bars represent the mean and S.E.M. for triplicate determinations of the percentage of total cells in the G1, S, and G2 phases of the cell cycle; experiments were repeated a minimum of three times and in multiple clones of S294A cells. C. EGF induced T47D cell growth in soft-agar is Ser294-dependent. T47D cells stably expressing wt or S294A PR-B were plated in soft-agar supplemented with either vehicle (EtOH), EGF (30ng/ml), R5020 (10nM), or both agents. Once solidified, soft-agar cultures were overlayed with growth media containing the same concentrations of each agent; spent media was replaced with fresh media every 48 hrs. Colonies were counted following 16 days continuous treatment. Bars represent the average number of colonies per field (n=9) plus/minus the S.E.M. Experiments were repeated at least 5 times with similar results.
We then measured the ability of cells stably expressing wt or S294A mutant PR-B to proliferate in response to hormones using flow cytometry (Fig. 7B). Cell numbers were adjusted to account for plating efficiency, and equal numbers of plated cells expressing either wt or S294A PR-B were serum-starved (48hr) and then treated for 24hr with vehicle control (EtOH), R5020 (10nM), EGF (30ng/ml), or both agents. Equal numbers of cells were subjected to FACS analysis to measure DNA content using flow cytometry. In control conditions (starved cells treated with EtOH), fewer S294A PR containing cells were in S phase relative to wt PR containing cells, suggesting that these cells may be more easily synchronized by serum starvation relative to cells containing wt PR-B. However, R5020 induced robust S-phase entry in both cell lines, indicating that cells expressing transcriptionally impaired PRs are quite capable of responding to progestins. We recently reported that progestin-induced S-phase entry is MAPK-dependent and occurs via the PR-dependent activation of c-Src kinases (25), but independently of the classically defined transcriptional activity of PRs (23). T47D cells containing wt PR-B were not further stimulated to proliferate in response to EGF alone. This is not surprising, as breast cancer cell lines growing in vitro generally appear to be unresponsive to EGF alone (38), perhaps due to their relatively high basal growth rates (25–30% S phase cells) in control (starved) conditions. In support of this interpretation, EGF stimulated a weak response in S294A PR-containing cells. Interestingly, the combination of both EGF and R5020 further enhanced the proliferative response of cells expressing wt PR-B, but not S294A PR-B, above the level reached in response to R5020 alone. These data indicate that PR, via Ser294, confers increased responsiveness of breast cancer cells to proliferative signaling via the EGF pathway, specifically when PR are liganded (Fig. 7B).
Progestins have biphasic effects on breast cancer cell growth in vitro, with an initial peak of S phase entry (18 hrs) and proliferation that may last from one (38,39) to multiple rounds (40) of cell division followed by cell growth inhibition (38,39). The response to progestins is influenced by the presence of additional hormones, such as estrogens (40) or by growth factors, including EGF (38). Additionally, long-term exposure to progestins may increase cell survival (41). To address the responsiveness of T47D breast cancer cells expressing wt or S294A mutant PR-B under conditions of long-term hormone exposure that more closely mimic anchorage-independent tumor growth in vivo, cells were seeded into soft agar in the presence of vehicle, R5020, EGF, or both agents. Colonies growing in soft agar were counted following 16 days continuous hormone treatment (Fig. 7C). At two weeks (Fig. 7) following seeding of equal viable cell numbers in soft agar, cells expressing S294A PR exhibited fewer colonies relative to cells expressing wt PR, suggesting that PR Ser294 phosphorylation and/or PR-dependent transcriptional events contribute to the ability of these cells to survive and grow in soft agar, regardless of the presence of progestin. Indeed, we were surprised to find that in the absence of progestins, EGF induced increased soft agar growth of cells containing wt PR-B, but not S294A PR-B. These data suggest that unliganded but Ser294 phosphorylated PR mediates the tumor cell anchorage-independent growth promoting effects of EGF when cells are seeded in soft agar. R5020 also induced increased growth in soft agar, but in contrast to EGF, similar increases in the total number of colonies of each cell line were observed at the two-week time point (Fig. 6A), consistent with the ability of either receptor to activate the c-Src-dependent MAPK pathway (23). Notably, the combination of both R5020 plus EGF enhanced soft agar growth only in T47D cells expressing wt PR-B.
These data suggest that T47D anchorage-independent cell growth and survival in soft agar in the presence of EGF is in part mediated by unliganded, but phosphorylated PRs, and that PR Ser294 may be an important determinant of growth factor responsiveness of human breast cancer cells, independently of progestins. The actions of PR to induce colony formation and growth in the presence of EGF alone are most likely mediated by the transcriptional actions of the receptor rather than its ability to transiently activate c-Src and MAPKs (25), as the S294A mutant receptor is transcriptionally impaired in this setting (Fig. 1 and (16), but capable of MAPK activation (35). Further, the inability of EGF to transactivate PR as measured using a PRE-driven promoter (Fig. 3) supports the existence of growth regulatory genes whose expression is regulated primarily by unliganded but phosphorylated PRs, perhaps functioning at non-classical promoters lacking proximal PRE elements, as in the case of PR-B dependent upregulation of IRS-1 (19).
Discussion
Herein, we have focused on the regulation of human PR by EGF. Remarkably, although significant differences exist in the regulation of PRs by EGF relative to their progestin ligands, there appears to be considerable overlap. We find that EGF signaling can induce changes in PR phosphorylation (Fig. 1A), nuclear association (Fig. 2A and B), and DNA-binding (Fig. 2C) that can mimic the actions of progestins. Although EGF alone failed to induce appreciable PR transcriptional activity as measured on PRE promoter elements (Fig. 3), or downregulation of the unliganded receptor (19), these actions of liganded PRs are dramatically enhanced in the presence of EGF (Fig. 4). Notably, pretreatment of T47D breast cancer cells with EGF left-shifted the dose-response curve for PR activation by R5020 at least two orders of magnitude and significantly increased total PR transcriptional activity at saturating ligand concentrations (Fig. 4), while simultaneous treatment was only weakly effective in the same cell line (Fig. 3). Phosphorylation of PR (42) and/or its coactivators (43,44) by MAPKs may contribute to these effects (Fig. 5). Additional mechanisms of ligand-dependent and –independent PR action are discussed below.
Functions of Liganded PR
In addition to its classical action as a ligand-activated transcription factor, PR-B has been shown to rapidly activate MAP kinase pathways by direct interaction with c-Src-family tyrosine kinases, an event that requires progesterone-binding to PR and occurs in the cytoplasm and/or associated with cell membranes (25). Although this has been described as a “non-genomic” action of liganded PR and appears to be modest relative to MAPK activation in response to EGF (Fig. 1A), PR-dependent kinase activation can mediate significant changes in gene expression independently of PR transcriptional activity. For example, cyclin E and D1 protein expression in response to progestins is mediated by PR-dependent activation of MAPKs, and MAPK-dependent regulation of the cyclin D1 proximal promoter by progestins occurs independently of PR transcriptional activity (23). MAPK activation in response to PR-ligand interaction may also provide a means of “feed-forward” regulation via direct phosphorylation of these receptors and/or their coactivators (43,44). Because the strength and duration of kinase activation differs greatly between growth factor and steroid hormone signals (Fig 1A), sites of hormonally regulated phosphorylation may allow for an additional growth-factor “sensor” input to liganded PR in the presence of EGF. This may in part explain enhanced transcription and proliferation in the presence of both EGF and R5020 (Figs. 3 and 7B). However, the requirement for 30 min EGF pretreatment in order to achieve transcriptional synergy (Figs. 2–4) suggests that additional mechanisms are at play. Notably, PR translocation into the nucleus (Fig. 2B) and DNA-binding (Fig. 2C) occur within the same time frame (30–60 min). Presumably, phosphorylation events trigger the formation of productive transcription complexes at the PRE that are then perhaps poised to respond to subphysiological concentrations of PR agonist. Linkage between PR hyperactivity and PR turnover (Fig. 4B) suggests that such complexes likely contain ubiquitin E3 ligases, as has been shown for activated ER (45). We reported that both PR downregulation and PR transcriptional synergy in the presence of MEKK1 and progestins were similarly blocked by inhibitors of p42/p44 MAPKs (PD98059 or UO126), the 26S proteasome (lactacystin), or nuclear export (leptomycinB) (14,15).
Much is known about the determinants of the total level of gene expression at a saturating hormone concentration. These include the actions of well-characterized coactivators, corepressors, kinases and chaperones, chromatin remodeling complexes, and components of RNA polymerase II. Most transcription studies are performed at a constant hormone concentration, and factors that increase the total level of transactivation of a given promoter-gene pair often do not alter the position of the dose-response curve (46). Thus, the mechanisms of transcriptional hypersensitivity as they relate to shifting the dose-response curve are poorly defined. However, it seems clear that reversible phosphorylation events that shift the position of the dose-response curve, or the setting of the EC50 (the concentration of hormone required for a half-maximal response), provide a simple means for adjusting the amount of response to a single concentration of steroid hormone within an organism and during regulated changes in physiological status (Fig. 8). Historically, the position of the dose-response curve was thought to be determined by the affinity of steroid hormone binding to cognate receptors under the assumption that this was the rate-determining step. Thus, equality between the EC50 and the dissociation constant of ligand-receptor interactions, or Kd, defined a receptor-mediated response. This view has changed as it has become clear that the EC50 can vary widely for different genes regulated by the same receptor within the same cell and that changing the growth conditions can dramatically alter the EC50 for a single gene (46), just as the inclusion of EGF (as pretreatment) sensitizes PR to low concentrations of progestins at PRE-driven promoters (Fig 4) and the phosphorylation of Ser294 is required for the regulation of some gene targets, but not others (Fig 6).
Figure 8.

Model for regulation of PR hypersensitivity via phosphorylation events. Several kinases regulate PR via direct phosphorylation of multiple serine residues, including Ser294 (16) and Ser400 (17). These growth factor inputs can create receptors with increased transcription in response to lower concentrations of progestins, or hypersensitive to ligand. MAPK phosphorylation of PR Ser294 in cells exposed to EGF primes PR for activity upon ligand binding by localizing it to the nucleus and inducing DNA binding. Phosphorylation of PR provides a mechanism for the cells to alter its regulation of a subset of genes when exposed to EGF and progestins, thus altering its hormone responsiveness. In tumors with upregulated kinase activity heavily phosphorylated PR are predicted to be rapidly turning over and highly active on a select set of promoters.
Recent examples of diverse factors or agents that produce a similar left-ward shift in the dose-response curve for steroid hormone receptors include the expression of Ubc9 for GR (47), the addition of short-chain fatty acids for ER and PR (48), and constitutively activated Ras signaling for AR (49). The latter two examples were associated with robust MAPK activation (48,49), as illustrated herein for the actions of EGF on PR (Fig. 4). How might kinases or other factors alter the EC50? It is now believed that the EC50 is largely influenced by the ratio of coactivators and corepressors, which bind to receptor agonist and antagonist complexes in a reversible equilibrium and with different affinities. Other factors that modify (i.e. kinases or SUMO or Ubi ligases) or bind these coregulators thus also directly or indirectly perturb the apparent EC50 of receptor-ligand interactions by altering the affinities of coregulator for liganded receptor (in the case of phosphorylation or sumoylation) or by competing for a limiting cofactor pool (in the case of direct binding of factor to coregulator). The concept that specificity in gene transcription is achieved by the assembly of the “enhanceosome”, a higher-order three-dimensional transcription factor/enhancer DNA complex, arose in the mid-1990s (50). The inherent cooperativity in enhanceosome complex assembly provides the sigmoidal shape of the dose-response curve (50). Embedded synergy in transcription ensures that a specific gene will be selected for activation only if all the components are present in the same nucleus. Enhanceosome components may include numerous kinases (NfkappaB) and other modifying enzymes (ubiquitin E3 ligases), steroid hormone or nuclear receptors (GR, VDR), and additional general and/or architectural transcription factors (AP1, ELK1, HMGA1), and activate transcription by recruiting chromatin-modifying activities and basal transcription factors to nearby promoters (50). Thus, EGF via the actions of downstream MAPKs and subsequent phosphorylation events, may act to recruit or stabilize favorable interactions between co-factors in PR-containing enhanceosomes at selected promoters. The requirement for EGF pretreatment (Fig. 4) may reflect the timing for nuclear accumulation of unliganded PR, modification or expression of additional required factors, and/or rate-limiting steps in the equilibrium binding of multiple factors within large dynamic transcription complexes. We are in the process of unraveling these possibilities using protein-protein interaction and CHIP assay approaches.
Ligand-independent PR Actions
PRs are capable of mediating transactivation of diverse genes by direct binding to PRE sequences, or indirectly by tethering to other transcription factors such as SP1 (31), AP1 (51,52), or STATs (5,30), or by stimulation of upstream cytoplasmic kinases such as c-Src and MAPKs (25,26) or JAK2 (30), that then serve as direct inputs to transcription factor function. Mechanistic overlap between these types of regulation perhaps ensures that the appropriate genes will be selectively activated under a given stimulus or combination of stimuli. In contrast to studies with ER (53) or AR (54,55), ligand-independent actions of PRs at specific promoters are seldom reported. Labriola et al (18) demonstrated a 2–4 fold activation of a PRE-reporter by heregulin in human breast cancer cells. CDK2 synergistically activated liganded PRs (17,27,56), and induced robust ligand-independent PR transcriptional activity in p27-null mouse embryonic fibroblast or breast cancer cells or during p27 knock-down of p27+ human breast cancer cells (17). We found that EGF could mimic the actions of CDK2 at PRE-driven promoters, but only when p27 levels remained low (17). Such studies have been limited by our incomplete knowledge of PR/progestin-regulated gene expression, especially for genes whose promoters lack consensus PRE sequences (19,36). A surprising diversity of genes whose promoters lack clear PRE sequences are differentially regulated by PR-A and PR-B isoforms (36), including a large number of genes regulated by unliganded PR-A or PR-B, but not their liganded derivatives (37). Notably, PR-A is not phosphorylated on Ser294 in vivo (57), suggesting that direct phosphorylation of PRs may contribute to differences in PR isoform function.
Little information exists on the role of PR target genes in cell growth control. However, nearly half of all genes identified using gene-array approaches encode cell adhesion and membrane-bound proteins or proteins involved in membrane-initiated signaling (36,37). PR is known to upregulate c-myc, STAT5A, cyclin D1, TGF-α, and EGFR mRNAs (39), and progestins synergize with EGF to induce increased expression of c-myc, c-fos, p21 and cyclins D1 and E (58). Recently, the growth promoting effects of PR (i.e. S-phase entry) have been shown to require signaling to c-Src and MAPK, and occur independently of the transcriptional activity of PRs (23). Clearly however, rapid activation of intracellular kinases requires progestin binding to cytosolic or membrane associated PRs (25). Recall that both wt and mutant S294A PR-B receptors are equally capable of activating c-Src and MAPK, and inducing cyclin D1 expression (23), consistent with the ability of R5020 to induce either cell line to undergo S-phase entry (Fig. 5) and colony growth in soft-agar (Fig. 6). In combination, EGF and progestins can augment S-phase entry by mechanisms that involve both membrane and nuclear actions of liganded PRs (56) (Fig. 5B). However, breast cancer cells are often insensitive to mitogenic stimulation by EGF alone (38). Thus, we were surprised to find significant ligand-independent actions of PR that also map to its transcriptional activity with regard to EGF-induced colony growth in soft agar (Fig. 6). EGF stimulated colony formation and growth of cells expressing wt PR-B. The growth promoting effects were completely dependent on PR Ser294, suggesting a requirement for PR transcriptional activity. Progestin-induced upregulation of Bcl-xL and protection of human breast cancer cells from apoptosis has been reported (40). This property of liganded PR may in part explain the increased risk of breast cancer development observed among women taking estrogen plus a progestin during hormone replacement therapy (8,9).
Genes that are regulated by EGF via the actions of unliganded PRs are largely unknown. However, Jacobsen et. al (37) reported at least 51 genes regulated by unliganded PRs, only 9 of which were also regulated by liganded PRs. Further, a large subset of the genes regulated by unliganded PRs were involved in membrane-initiated signaling events or coordination of extracellular signaling, and known to influence tumor cell biology (36,37). For example, basal expression of IRS-1 in T47D cells is insensitive to progestins, but requires Ser294 phosphorylation of unliganded PR-B, but not PR-A (19). In the absence of progesterone, PR isoforms influence the biology and treatment response of ER+ tumors, and can block Taxol-induced apoptosis (37); PR-A rich tumors are especially aggressive (59,60). In mouse xenograft models, the presence of PR isoforms reduced the overall growth of estrogen-dependent ER+ tumors. However, B-rich tumors were twice the size of A-rich tumors, and tamoxifen inhibited the growth of A-rich, but not B-rich tumors (60).
PRs are independent markers of breast cancer prognosis, irrespective of progestational status (60–62). Further, ER+/PR+ tumors clearly metastasize (63). Progression of breast cancer is often correlated with overexpression of growth factors and receptors capable of establishing autocrine and/or paracrine growth-stimulatory loops. The tumor microenvironment is thus predicted to exhibit increased concentrations of locally acting growth factors, including EGF and IGF-1. Our studies suggest that these agents (i.e activators of MAPKs) can usurp hormone-dependent actions (Fig. 2–4), and are predicted to drive breast cancer cell growth in part via the actions of unliganded PRs (Fig. 6). Further analysis of genes regulated by growth factors in a PR-dependent manner (independently of PR ligands) is required in order to more effectively target these pathways for breast cancer prevention and treatment. In addition to the activation of PRs via ligand-independent pathways (17,18), MAPK-dependent events may dramatically lower the EC50 for gene activation by liganded PR (Fig. 4), ER (64), or AR (65). As all three receptors are commonly expressed in advanced breast cancers (66), inclusion of antiprogestins, antiandrogens, and the relevant kinase inhibitors to existing antiestrogen therapies may offer a more powerful treatment alternative as combination therapy.
Acknowledgments
We thank Kate Horwitz (University of Colorado Health Sciences Center, Aurora, CO) for the gift of the T47D cell lines (PR-null, PR-A and PR-B expressing variants). We thank Natalie Ahn (University of Colorado) for the MEK-1 constructs. This work was supported by NIH grants R01 CA123763 (formerly DK053825) and R21 CA116790 (to C.A.L.), and Department of Defense Predoctoral Fellowship Grants, number W81XWH-05-1-0257 (to A.R.D.) and number DAMD17-03-1-0390 (to E.J.F).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conneely OM, Jericevic BM, Lydon JP. Progesterone receptors in mammary gland development and tumorigenesis. J Mammary Gland Biol Neoplasia. 2003;8(2):205–214. doi: 10.1023/a:1025952924864. [DOI] [PubMed] [Google Scholar]
- 2.Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr, Shyamala G, Conneely OM, O’Malley BW. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 3.Haslam SZ, Counterman LJ, Nummy KA. Effects of epidermal growth factor, estrogen, and progestin on DNA synthesis in mammary cells in vivo are determined by the developmental state of the gland. J Cell Physiol. 1993;155(1):72–78. doi: 10.1002/jcp.1041550110. [DOI] [PubMed] [Google Scholar]
- 4.Thorne C, Lee AV. Cross talk between estrogen receptor and IGF signaling in normal mammary gland development and breast cancer. Breast Dis. 2003;17:105–114. doi: 10.3233/bd-2003-17110. [DOI] [PubMed] [Google Scholar]
- 5.Richer JK, Lange CA, Manning NG, Owen G, Powell R, Horwitz KB. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J Biol Chem. 1998;273(47):31317–31326. doi: 10.1074/jbc.273.47.31317. [DOI] [PubMed] [Google Scholar]
- 6.Lange CA, Richer JK, Shen T, Horwitz KB. Convergence of progesterone and epidermal growth factor signaling in breast cancer. Potentiation of mitogen-activated protein kinase pathways. J Biol Chem. 1998;273(47):31308–31316. doi: 10.1074/jbc.273.47.31308. [DOI] [PubMed] [Google Scholar]
- 7.Woodward TL, Xie JW, Haslam SZ. The role of mammary stroma in modulating the proliferative response to ovarian hormones in the normal mammary gland. J Mammary Gland Biol Neoplasia. 1998;3(2):117–131. doi: 10.1023/a:1018738721656. [DOI] [PubMed] [Google Scholar]
- 8.Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M, Lane D, Rodabough RJ, Gilligan MA, Cyr MG, Thomson CA, Khandekar J, Petrovitch H, McTiernan A. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative Randomized Trial. Jama. 2003;289(24):3243–3253. doi: 10.1001/jama.289.24.3243. [DOI] [PubMed] [Google Scholar]
- 9.Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362(9382):419–427. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- 10.Ali S, Coombes RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. 2000;5(3):271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- 11.Nicholson RI, Johnston SR. Endocrine therapy--current benefits and limitations. Breast Cancer Res Treat. 2005;93 (Suppl 1):S3–10. doi: 10.1007/s10549-005-9036-4. [DOI] [PubMed] [Google Scholar]
- 12.Putti TC, Pinder SE, Elston CW, Lee AH, Ellis IO. Breast pathology practice: most common problems in a consultation service. Histopathology. 2005;47(5):445–457. doi: 10.1111/j.1365-2559.2005.02246.x. [DOI] [PubMed] [Google Scholar]
- 13.Lange CA. Making sense of cross-talk between steroid hormone receptors and intracellular signaling pathways: who will have the last word? Mol Endocrinol. 2004;18(2):269–278. doi: 10.1210/me.2003-0331. [DOI] [PubMed] [Google Scholar]
- 14.Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97(3):1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol. 2003;17(4):628–642. doi: 10.1210/me.2002-0378. [DOI] [PubMed] [Google Scholar]
- 16.Shen T, Horwitz KB, Lange CA. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol. 2001;21(18):6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24(24):10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labriola L, Salatino M, Proietti CJ, Pecci A, Coso OA, Kornblihtt AR, Charreau EH, Elizalde PV. Heregulin induces transcriptional activation of the progesterone receptor by a mechanism that requires functional ErbB-2 and mitogen-activated protein kinase activation in breast cancer cells. Mol Cell Biol. 2003;23(3):1095–1111. doi: 10.1128/MCB.23.3.1095-1111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu M, Lange CA. MAP kinases couple multiple functions of human progesterone receptors: degradation, transcriptional synergy, and nuclear association. J Steroid Biochem Mol Biol. 2003;85(2–5):147–157. doi: 10.1016/s0960-0760(03)00221-8. [DOI] [PubMed] [Google Scholar]
- 20.Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006;16(6):301–307. doi: 10.1016/j.tcb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, Horwitz KB. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994;54(14):3868–3877. [PubMed] [Google Scholar]
- 22.Richer JK, Lange CA, Wierman AM, Brooks KM, Tung L, Takimoto GS, Horwitz KB. Progesterone receptor variants found in breast cells repress transcription by wild-type receptors. Breast Cancer Res Treat. 1998;48(3):231–241. doi: 10.1023/a:1005941117247. [DOI] [PubMed] [Google Scholar]
- 23.Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19(2):327–339. doi: 10.1210/me.2004-0306. [DOI] [PubMed] [Google Scholar]
- 24.Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003;63(3):627–635. [PubMed] [Google Scholar]
- 25.Boonyaratanakornkit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JL, Miller WT, Edwards DP. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell. 2001;8(2):269–280. doi: 10.1016/s1097-2765(01)00304-5. [DOI] [PubMed] [Google Scholar]
- 26.Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. Embo J. 1998;17(7):2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narayanan R, Adigun AA, Edwards DP, Weigel NL. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol Cell Biol. 2005;25(1):264–277. doi: 10.1128/MCB.25.1.264-277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hager GL. Studying nuclear receptors with green fluorescent protein fusions. Methods Enzymol. 1999;302:73–84. doi: 10.1016/s0076-6879(99)02011-x. [DOI] [PubMed] [Google Scholar]
- 29.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265(5174):966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 30.Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, Molinolo AA, Frahm I, Charreau EH, Schillaci R, Elizalde PV. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol. 2005;25(12):4826–4840. doi: 10.1128/MCB.25.12.4826-4840.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 32.Lange CA, Richer JK, Horwitz KB. Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol. 1999;13(6):829–836. doi: 10.1210/mend.13.6.0290. [DOI] [PubMed] [Google Scholar]
- 33.Takimoto GS, Hovland AR, Tasset DM, Melville MY, Tung L, Horwitz KB. Role of phosphorylation on DNA binding and transcriptional functions of human progesterone receptors. J Biol Chem. 1996;271(23):13308–13316. doi: 10.1074/jbc.271.23.13308. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z, Funk C, Roy D, Glasser S, Mulholland J. Heparin-binding epidermal growth factor-like growth factor is differentially regulated by progesterone and estradiol in rat uterine epithelial and stromal cells. Endocrinology. 1994;134(3):1089–1094. doi: 10.1210/endo.134.3.8119147. [DOI] [PubMed] [Google Scholar]
- 35.Faivre EJ, Lange CA. Progesterone Receptors Upregulate Wnt-1 to Induce EGFR transactivation and c-Src dependent sustained activation of Erk1/2 MAP Kinase in Breast Cancer Cells. Mol Biol Cell. 2006 doi: 10.1128/MCB.01539-06. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 37.Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol. 2005;19(3):574–587. doi: 10.1210/me.2004-0287. [DOI] [PubMed] [Google Scholar]
- 38.Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1) Mol Endocrinol. 1997;11(11):1593–1607. doi: 10.1210/mend.11.11.0006. [DOI] [PubMed] [Google Scholar]
- 39.Musgrove EA, Lee CS, Sutherland RL. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor alpha, epidermal growth factor receptor, c-fos, and c-myc genes. Mol Cell Biol. 1991;11(10):5032–5043. doi: 10.1128/mcb.11.10.5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore MR, Conover JL, Franks KM. Progestin effects on long-term growth, death, and Bcl-xL in breast cancer cells. Biochem Biophys Res Commun. 2000;277(3):650–654. doi: 10.1006/bbrc.2000.3728. [DOI] [PubMed] [Google Scholar]
- 41.Moore MR, Spence JB, Kiningham KK, Dillon JL. Progestin inhibition of cell death in human breast cancer cell lines. J Steroid Biochem Mol Biol. 2006;98(4–5):218–227. doi: 10.1016/j.jsbmb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Beck CA, Poletti A, Edwards DP, Weigel NL. Identification of a group of Ser-Pro motif hormone-inducible phosphorylation sites in the human progesterone receptor. Mol Endocrinol. 1995;9(8):1029–1040. doi: 10.1210/mend.9.8.7476977. [DOI] [PubMed] [Google Scholar]
- 43.Rowan BG, Weigel NL, O’Malley BW. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem. 2000;275(6):4475–4483. doi: 10.1074/jbc.275.6.4475. [DOI] [PubMed] [Google Scholar]
- 44.Lopez GN, Turck CW, Schaufele F, Stallcup MR, Kushner PJ. Growth factors signal to steroid receptors through mitogen-activated protein kinase regulation of p160 coactivator activity. J Biol Chem. 2001;276(25):22177–22182. doi: 10.1074/jbc.M010718200. [DOI] [PubMed] [Google Scholar]
- 45.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11(3):695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 46.Simons SS., Jr How much is enough? Modulation of dose-response curve for steroid receptor-regulated gene expression by changing concentrations of transcription factor. Curr Top Med Chem. 2006;6(3):271–285. doi: 10.2174/156802606776173465. [DOI] [PubMed] [Google Scholar]
- 47.Cho S, Kagan BL, Blackford JA, Jr, Szapary D, Simons SS., Jr Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol Endocrinol. 2005;19(2):290–311. doi: 10.1210/me.2004-0134. [DOI] [PubMed] [Google Scholar]
- 48.Jansen MS, Nagel SC, Miranda PJ, Lobenhofer EK, Afshari CA, McDonnell DP. Short-chain fatty acids enhance nuclear receptor activity through mitogen-activated protein kinase activation and histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2004;101(18):7199–7204. doi: 10.1073/pnas.0402014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakin RE, Gioeli D, Sikes RA, Bissonette EA, Weber MJ. Constitutive activation of the Ras/mitogen-activated protein kinase signaling pathway promotes androgen hypersensitivity in LNCaP prostate cancer cells. Cancer Res. 2003;63(8):1981–1989. [PubMed] [Google Scholar]
- 50.Merika M, Thanos D. Enhanceosomes. Curr Opin Genet Dev. 2001;11(2):205–208. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- 51.Tseng L, Tang M, Wang Z, Mazella J. Progesterone receptor (hPR) upregulates the fibronectin promoter activity in human decidual fibroblasts. DNA Cell Biol. 2003;22(10):633–640. doi: 10.1089/104454903770238102. [DOI] [PubMed] [Google Scholar]
- 52.Bamberger AM, Bamberger CM, Gellersen B, Schulte HM. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Natl Acad Sci U S A. 1996;93(12):6169–6174. doi: 10.1073/pnas.93.12.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dutertre M, Smith CL. Ligand-independent interactions of p160/steroid receptor coactivators and CREB-binding protein (CBP) with estrogen receptor-alpha: regulation by phosphorylation sites in the A/B region depends on other receptor domains. Mol Endocrinol. 2003;17(7):1296–1314. doi: 10.1210/me.2001-0316. [DOI] [PubMed] [Google Scholar]
- 54.Huang ZQ, Li J, Wong J. AR possesses an intrinsic hormone-independent transcriptional activity. Mol Endocrinol. 2002;16(5):924–937. doi: 10.1210/mend.16.5.0829. [DOI] [PubMed] [Google Scholar]
- 55.Cenni B, Picard D. Ligand-independent Activation of Steroid Receptors: New Roles for Old Players. Trends Endocrinol Metab. 1999;10(2):41–46. doi: 10.1016/s1043-2760(98)00121-0. [DOI] [PubMed] [Google Scholar]
- 56.Faivre E, Skildum A, Pierson-Mullany L, Lange CA. Integration of progesterone receptor mediated rapid signaling and nuclear actions in breast cancer cell models: role of mitogen-activated protein kinases and cell cycle regulators. Steroids. 2005;70(5–7):418–426. doi: 10.1016/j.steroids.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 57.Clemm DL, Sherman L, Boonyaratanakornkit V, Schrader WT, Weigel NL, Edwards DP. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol. 2000;14(1):52–65. doi: 10.1210/mend.14.1.0413. [DOI] [PubMed] [Google Scholar]
- 58.Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK, Sutherland RL. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol. 1993;13(6):3577–3587. doi: 10.1128/mcb.13.6.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72(2):163–172. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- 60.Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004;10(8):2751–2760. doi: 10.1158/1078-0432.ccr-03-0141. [DOI] [PubMed] [Google Scholar]
- 61.Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, Elledge RM. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005;97(17):1254–1261. doi: 10.1093/jnci/dji249. [DOI] [PubMed] [Google Scholar]
- 62.Punglia RS, Kuntz KM, Winer EP, Weeks JC, Burstein HJ. The impact of tumor progesterone receptor status on optimal adjuvant endocrine therapy for postmenopausal patients with early-stage breast cancer: a decision analysis. Cancer. 2006;106(12):2576–2582. doi: 10.1002/cncr.21919. [DOI] [PubMed] [Google Scholar]
- 63.Alanko A, Heinonen E, Scheinin T, Tolppanen EM, Vihko R. Significance of estrogen and progesterone receptors, disease-free interval, and site of first metastasis on survival of breast cancer patients. Cancer. 1985;56(7):1696–1700. doi: 10.1002/1097-0142(19851001)56:7<1696::aid-cncr2820560738>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 64.Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273(21):13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- 65.Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, Wilson EM. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279(8):7119–7130. doi: 10.1074/jbc.M307649200. [DOI] [PubMed] [Google Scholar]
- 66.Buchanan G, Birrell SN, Peters AA, Bianco-Miotto T, Ramsay K, Cops EJ, Yang M, Harris JM, Simila HA, Moore NL, Bentel JM, Ricciardelli C, Horsfall DJ, Butler LM, Tilley WD. Decreased androgen receptor levels and receptor function in breast cancer contribute to the failure of response to medroxyprogesterone acetate. Cancer Res. 2005;65(18):8487–8496. doi: 10.1158/0008-5472.CAN-04-3077. [DOI] [PubMed] [Google Scholar]