

Abstract
Fgf8 and Tbx1 have been shown to interact in patterning the aortic arch, and both genes are required in formation and growth of the outflow tract of the heart. However, the nature of the interaction of the two genes is unclear. We have utilized a novel Tbx1Fgf8 allele which drives Fgf8 expression in Tbx1-positive cells and an inducible Cre-LoxP recombination system to address the role of Fgf8 in Tbx1 positive cells in modulating cardiovascular development. Results support a requirement of Fgf8 in Tbx1 expressing cells to finely control patterning of the aortic arch and great arteries specifically during the pharyngeal arch artery remodeling process and indicate that the endoderm is the most likely site of this interaction. Furthermore, our data suggest that Fgf8 and Tbx1 play independent roles in regulating outflow tract development. This finding is clinically relevant since TBX1 is the candidate for DGS/VCFS, characterized clinically by variable expressivity and reduced penetrance of cardiovascular defects; Fgf8 gene variants may provide molecular clues to this variability.
Keywords: Tbx1, Fgf8, 22q11 Deletion syndrome, Cardiovascular development, Genetic interaction, Pharyngeal apparatus, Pharyngeal endoderm
Introduction
The transcription factor Tbx1 is required for the formation and growth of the pharyngeal apparatus, a vertebrate-specific structure that gives rise to components of the face, neck and heart. Tbx1 is expressed in pharyngeal ectoderm, mesoderm, and endoderm during development, and mutations of TBX1 have been identified in patients with DiGeorge syndrome/VCFS (Yagi et al., 2003). The cardiovascular defects typically seen in patients include interrupted aortic arch type B (IAA-B), persistent truncus arteriosis (PTA), tetrology of Fallot, ventricular septal defects (VSD), retro-esophageal right subclavian artery (reRSA), right aortic arch (RAA), and pulmonary atresia. In mice, haploinsufficiency of Tbx1 causes cardiovascular defects such as IAA-B, RAA and reRSA, due to maldevelopment of the 4th pharyngeal arch arteries (PAAs), while homozygous loss of Tbx1 causes PTA and VSD (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). The Tbx1 requirement in 4th PAA development is non-cell autonomous since Tbx1 is not expressed in the vessel itself (Zhang et al., 2005). Interestingly, the 4th PAA defects in Tbx1+/− embryos are fully penetrant at E10.5, while in term embryos, the incidence of aortic arch defects is approximately 30%, suggesting the presence of a Tbx1-dependent signal that triggers the recovery of 4th PAA growth delay. We have recently demonstrated that loss of one copy of Tbx1 in pharyngeal epithelia (endoderm and ectoderm) is sufficient to cause 4th PAA defects, demonstrating a requirement of Tbx1 in one or both of these tissues (Zhang et al., 2005).
The proper formation and maturation of the pharyngeal apparatus require a finely orchestrated series of tissue interactions, such that perturbation of any one of a number of inputs may result in similar phenotypic consequences (reviewed in Graham and Smith, 2001). For example, loss/decreased function of genes required in neural crest cells, endoderm or ectoderm, can result in similar cardiovascular defects, since pharyngeal arch artery remodeling into the mature aortic arch and outflow tract of the heart requires coordinated communication between all tissue types. For example, studies have shown that the total or partial loss of function of several genes expressed in different tissues, such as Raldh2 (Niederreither et al., 1999, 2001, 2002), Vegf (Stalmans et al., 2003), Crkl (Guris et al., 2001), Fgf8 (Abu-Issa et al., 2002; Frank et al., 2002), and Tbx1 (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001) can cause similar aortic arch artery patterning and outflow tract defects. Furthermore, two recent studies have examined the effects of genetic interaction of Crkl with either Tbx1 and/or Fgf8 and suggest that the genes cooperate synergistically to pattern the aortic arch in a dose-dependent manner (Guris et al., 2006; Moon et al., 2006). Thus, several key players required in patterning the aortic arch have been identified, however, how these genes interact has yet to be understood.
We had previously suggested the existence of a genetic interaction in the endoderm between Tbx1 and Fgf8 based on the finding that endoderm-only Fgf8 expression is down-regulated in Tbx1-null embryos, and Tbx1+/−;Fgf8+/− compound mutants showed a significant increase in arch artery defects vs. Tbx1 haploinsufficient mice (Vitelli et al., 2002b). Based on these data and the similarity of the cardiovascular phenotype of Fgf8 and Tbx1 mutant mice, we speculated that Fgf8 and Tbx1 may function in the same genetic pathway. In order to examine this putative pathway in vivo, we took a dual approach to test the effects of (1) forced expression of Fgf8 in the Tbx1 expression domain and (2) deletion of Fgf8 from Tbx1-positive cells. To this end, we generated a novel Tbx1-null allele driving the expression of the Fgf8 cDNA in the Tbx1 domain. We have also carried out the converse experiment, i.e., conditional deletion of Fgf8 in Tbx1-expressing cells using an Fgf8flox allele (Brown et al., 2004). Overall, results indicate that FGF8 (in excess or defect) modifies of only one aspect of the Tbx1 cardiovascular phenotype, namely the aortic arch patterning defects. In addition, the use of timed deletion of Fgf8 allowed us to temporally map the interaction of the two genes. We propose that such interaction is mainly relevant during the process of embryonic recovery observed in Tbx1 heterozygous mutants.
Methods
Gene targeting and genotyping
To generate the F8KIT construct and the Tbx1Fgf8 allele, we inserted a 0.8-kb Fgf8 cDNA, encoding isoform 8b, into the XcmI site of exon 5 of the Tbx1 gene, which encodes for a portion of the functionally essential T-box domain. This is the same site of insertion as the previously described LacZ knock-in reporter allele, a functionally null allele (Lindsay et al., 2001). The Fgf8 cDNA is flanked by a 5′ IRES and a 3′ poly-adenylation signal derived from a 300-bp fragment of the bovine growth hormone gene, followed by a HPRT minigene for positive selection. The F8KIT construct was linearized with SalI and electroporated into Hprt-minus AB2.2 ES cells. HAT-resistant clones were tested for homologous recombination by Southern blotting of HindIII-digested DNA probed with a 550-bp XcmI fragment located immediately distal to the 3′homology arm (Fig. 1A). Approximately 8% (23/288) of the HAT-resistant colonies resulted in the appropriately targeted allele. Of these, two ES cell lines were injected into blastocysts by the Baylor College of Medicine Darwin Transgenic Facility and produced 3 high-grade chimera which transmitted the mutation to their offspring. Offspring was genotyped by PCR using the following primers: F8KIT-TAR-F: taccttctgaggcggaaaga and F8KIT-TAR-R: cctcatacaaaggccagcat.
Fig. 1.
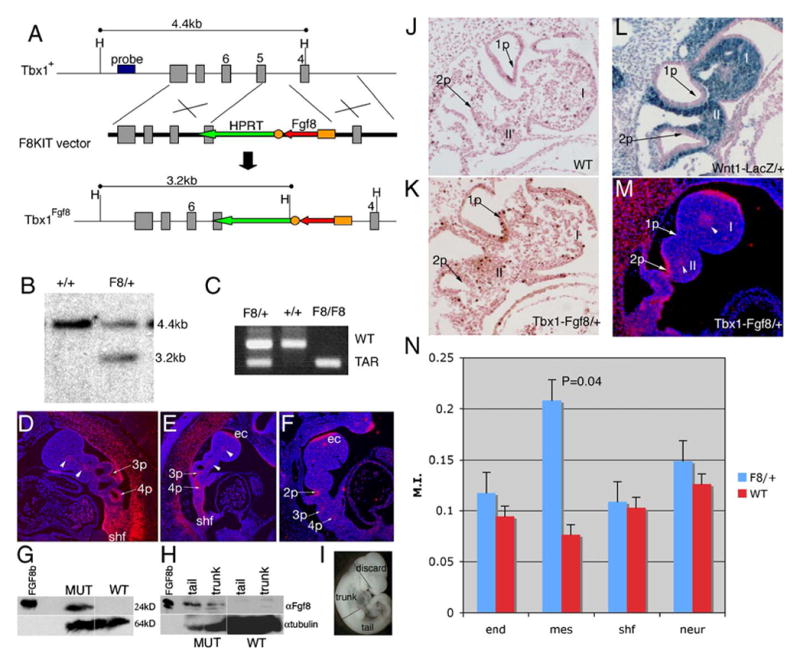
Generation and characterization of Tbx1Fgf8/+ knock-in mice. (A) Schematic of the F8KIT targeting construct and Tbx1Fgf8 allele, showing the disruption of a portion of the T-box domain encoded by exon 5. (B) Southern blot identifying a targeted (F8) vs. WT ES cell line. (C) PCR of yolk sac DNA. (D–F) RNA in situ hybridization with probes for Tbx1 on Tbx1+/− (D) or Fgf8 on Tbx1Fgf8/+ (E, M) or Tbx1+/− (F) embryos at E10 (D–F) and E9.5 (M). The embryo in panel E clearly shows expression of Fgf8 in all the endogenous Fgf8 domains seen in panel F such as the fronto-nasal process (fnp) and first arch ectoderm (ec), and in the same tissues normally expressing Tbx1 seen in panel D, including the core mesenchyme of the cranial arches (arrowheads), the secondary heart field (shf), and 3rd/4th pouches (3p, 4p). (G–I) Western blots of total protein from whole embryos (one per genotype) at E10.5 (G) or trunk and tail segments of embryos at E10.25 (H) show abundant FGF8 production in Tbx1Fgf8/+ vs. WT samples. Purified recombinant FGF8b (250 ng) protein was loaded as a control. Panel I shows how the embryos were dissected to generate panel H. (J–K) Sagittal sections through E9.5 WT (J) and Tbx1Fgf8/+ (K) embryos processed for phospho-histone H3 assay showing representative regions of 1st and 2nd arch mesenchyme in which positive cells were counted. (L) Xgal stained stage-matched Wnt1LacZ/+ embryo shows high NCC density in the 1st and 2nd pharyngeal arches (I, II). (N) Summary of phosphorylated histone H3 assay in Tbx1Fgf8/+ at E9.5 (23 somites). A significant increase in proliferation of the pharyngeal arch mesenchyme (P = 0.04) was seen as compared to other target regions of the embryo (endoderm and splanchnic mesenchyme/secondary heart field), or control regions in which Tbx1-driven Fgf8 is not expressed (neural tube).
The Fgf8+/− and Fgf8flox/flox mouse lines were supplied by Erik Meyers at Duke University, and the Hoxa3Cre/+ line from A.M. Moon at the University of Utah, Salt Lake City. Genotyping was performed as previously described (Brown et al., 2004; Macatee et al., 2003; Meyers et al., 1998). Wnt1LacZ/+ mice were obtained from A.P. McMahon.
RNA in situ hybridization and immunohistochemistry
Whole-mount and section in situ hybridization was performed as described in (Albrecht et al., 1997). Sense and antisense RNA probes were prepared by reverse transcription of DNA and labeled either with 35S-UTP (Perkin Elmer) or digoxigenin (Roche). Plasmids were obtained as follows: Tbx1 from V. Papaioannou, Fgf8 and Fgf4 from G. Martin, deletion-specific Fgf8-probe (PstI digested cDNA) and Gbx2 from E. Meyers, Raldh2 from K. Neiderreither, Spry4/2 from G. Minowada, Pax1 from R. Balling. At least three embryos per genotype per stage were hybridized with each probe.
Immunohistochemistry with phosphorylated Erk1/2 (Cell Signaling) was performed on whole-mount embryos according to the Rossant Lab Protocol (http://www.sickkids.ca/rossant/custom/protocols.asp), subsequently embedded in paraffin and sectioned at 20 μm, while immunohistochemistry with phosphorylated histone H3 (Upstate Biotechnology), and cleaved caspase 3 (Cell Signaling) antibodies were performed on ethanol-fixed and paraffin-embedded E9.5–11.5 embryos serially sectioned at 8 μm. The mitotic index was calculated by counting histone H3-positive cells/total cells (at least 1300) for each tissue and averaging the mitotic index of 4 embryos per genotype; P was calculated using Student's T test. LysoTracker-Red (Molecular Probes) was used on whole mount embryos at E10.5 as described in Vitelli et al. (2003).
Western blot analysis
Embryos at E10.5 were collected from Tbx1Fgf8/+ and wild-type crosses, staged by somite counting and either processed whole or dissected. Crude protein extracts were obtained from stage-matched whole embryos using iced RIPA buffer with protein inhibitor cocktail (Roche). For the dissected embryos, the head of each sample was discarded, and the remaining tissue was severed between somites 20 and 21, to separate the pharyngeal region from the rest of the embryo and the tail. These pieces were wrapped in foil, immediately frozen in liquid nitrogen and stored at −80°C until corresponding yolk-sac genotype was available. Three somite-matched (33 somites) samples per genotype per tissue were pooled, and proteins were extracted as above. About 20 μg of protein extract was run on a 12%/SDS/PAGE gel and transferred onto nylon filters. Filters were probed with monoclonal anti-mouse Fgf8 antibody (R&D Systems) at a 1:500 dilution in PBS/0.1% Tween overnight at 4°C, subsequently stripped and normalized with an anti-tubulin antibody (Sigma).
Skeletal preps
We collected E18.5 Tbx1+/+, Tbx1Fgf8/+, Tbx1+/− and TbxFgf8/− embryos and subjected them to skeletal preparations using Alcian Blue which stains cartilage and Alizarin Red for bone as described in Hogan et al. (1994).
Tamoxifen administration
In order to activate the Tbx1mcm allele and induce Cre recombination, we prepared a stock solution of 100 mg/ml Tamoxifen (Sigma) dissolved in ethanol and diluted 1:10 in autoclaved sesame oil (Sigma). Vortexing for 10 min produced an emulsion suitable for IP injection. Pregnant dams were exposed to a single dose of 75 mg/kg bodyweight of Tamoxifen at embryonic day (E) 7.5, E8.5 or E9.5, with E0.5 counted as noon of the day a vaginal plug was observed.
Results
Generation of the Fgf8 knock-in allele at the Tbx1 locus
In order to test the effect of FGF8 released by Tbx1-expressing cells, we generated a Tbx1-driven Fgf8 knock-in allele, Tbx1Fgf8 (Fig. 1A–C). We have previously utilized this same site of insertion to generate our Tbx1LacZ reporter allele, which reproduces endogenous Tbx1 expression and is a functionally null allele (Lindsay et al., 2001). Thus, by knocking in the F8KIT targeting construct, we expected to simultaneously inactivate Tbx1 and drive Fgf8-cDNA expression by Tbx1 regulatory elements. Using an appropriately targeted ES cell line, we were able to generate chimera which transmitted the Tbx1Fgf8 allele to their offspring. Tbx1Fgf8/+ mice were apparently healthy and fertile but exhibited an abnormally coiled tail, which required amputation (see below). To test whether the Fgf8 cDNA is expressed properly, we performed RNA in situ hybridization on embryos at E10 using Tbx1 and Fgf8 RNA probes on Tbx1Fgf8/+ embryos. Results showed robust expression of Fgf8 mRNA in all of the domains where Tbx1 is expressed, including the core mesenchyme of the arches, the pouch endoderm of arches 3–6 and the secondary heart field (Fig. 1D–F). In Tbx1Fgf8/+ embryos, the endogenous Fgf8 domains are also clearly visible (Fig. 1E). To test whether the Fgf8-transgene mRNA is translated, we extracted proteins from whole or segments of Tbx1Fgf8/+ embryos and subjected them to Western blot analysis using a monoclonal Fgf8 antibody. We found an abundant increase in levels of FGF8 protein in Tbx1Fgf8/+ vs. wild-type total protein extracts from embryos at E10.5 (38 somites, Fig. 1G). Since Fgf8 mRNA from the transgene is ectopically and abundantly expressed in the somites (see below), we wished to test whether the Tbx1Fgf8 allele produced detectable FGF8 in the pharyngeal region alone. We therefore separated trunk and tail regions of the embryos (shown in Fig. 1I) and extracted total protein from a pool of 3 stage matched trunks or tails for each genotype. The trunk region included the heart and the area of tissue surrounding the pharyngeal apparatus, while the tail region included all the structures caudal to the forelimb bud. We performed Western blots of trunk and tail segments and found abundant FGF8 protein in both fractions (Fig. 1H).
The Fgf8 knock-in modulates levels of cellular proliferation in the pharyngeal arch mesenchyme
Since partial loss of function of Fgf8 causes a decrease in cell survival (Abu-Issa et al., 2002; Frank et al., 2002) and Tbx1 conditional mutants show decreased levels of cellular proliferation (Xu et al., 2004, 2005), we tested cell survival and proliferation in the presence of the Fgf8 transgene. Immunohistochemistry with cleaved caspase 3 (E9.5) or LysoTracker analysis (E10.5) showed no significant differences in apoptosis between 3 pairs of stage matched Tbx1Fgf8/+ and WT embryos (not shown). Proliferation levels were assayed by counting phosphorylated histone H3-labeled cells in the pharyngeal endoderm, pharyngeal arch mesenchyme, splanchnic mesoderm of the secondary heart field region, and neural tube in 4 stage-matched Tbx1Fgf8/+ and WT embryos at E9.5. Although proliferation seemed slightly altered in all of the Tbx1Fgf8/+ tissues examined, we detected statistically significant increase in proliferation only in the pharyngeal arch mesenchyme of Tbx1Fgf8/+ embryos (P = 0.04, Figs. 1J, K, N). Neural crest-derived cells (NCC) should represent the majority of arch mesenchymal cells at this stage. To test this, we generated stage-matched Wnt1LacZ/+ embryos and stained with Xgal to reveal Wnt1-positive cells. Sections demonstrate that the cranial arch mesenchyme analyzed in the proliferation assay is densely populated by cells of NC origin (blue cells in Fig. 1L). We speculate that ectopic FGF8 produced by Tbx1-expressing cells from the Tbx1Fgf8 allele (Fig. 1M) may exert a non-cell autonomous effect on NCC proliferation.
Since it has been shown that Fgf4 and Fgf8 have redundant roles in limb development (Lu et al., 2006), we tested whether Fgf4 expression is changed in Tbx1Fgf8/+ embryos. Distribution of Fgf4 transcripts assayed by RNA in situ hybridization at E10 was apparently normal in Tbx1Fgf8/+ embryos (data not shown).
Tbx1Fgf8/+ mice exhibit axial skeletal abnormalities
All Tbx1Fgf8/+ animals (n = 55) presented with moderate to severe tail kinking or coiling (Fig. 2A–C). In the most severe cases, the abnormal tail required perinatal amputation as the coil formed a tight, ventrally located knot which interfered with mating and excretion of waste. Skeletal preparations at E18.5, revealed that the lumbar and sacral regions of the spine were affected in addition to the caudal region (Fig. 2D–I). The ventral aspect of each affected skeletal element appeared fused to the adjacent elements, forming a rigid, curved structure, which induced kyphosis and greatly reduced flexibility in the adult animal. The number of fused elements was proportional to the severity of the tail coiling. Normally, the caudal portion of the skeleton is not ossified at E18.5, and the anterior and posterior portions of adjacent vertebral units should be well separated and flank the nucleus polposus. In Tbx1Fgf8/+ mutants, the anterior and posterior portions of the original sclerotomal element failed to separate fully and appeared fused on the ventral aspect of the tail, resulting in a cup-like structure around the nucleus polposus (Fig. 2I). RNA in situ hybridization showed high Tbx1-driven Fgf8 mRNA levels in the ventral sclerotome at E11.5, where endogenous Fgf8 is not expressed (Fig. 2J–L).
Fig. 2.
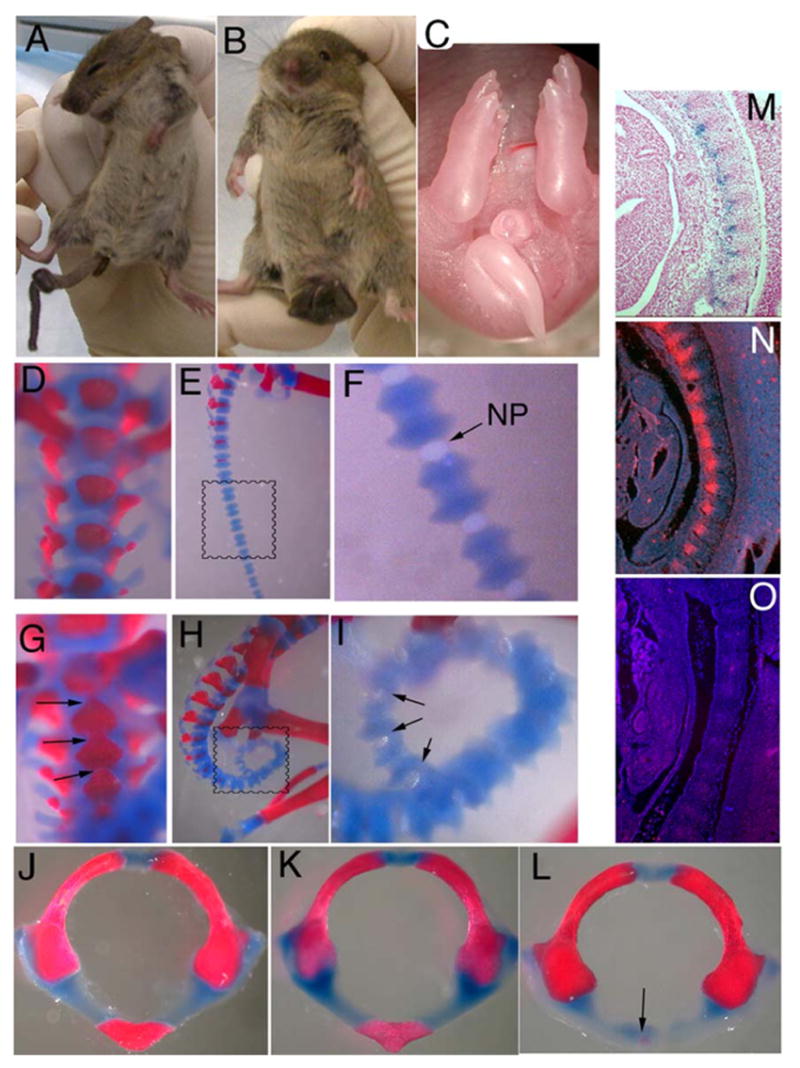
The skeletal phenotype of Tbx1Fgf8/mice. Mild (A) to severe (B) tail kinking was seen in all Tbx1Fgf8/+ mutants born and was evident at E18.5 (C). Skeletal preps at E18.5 showed fusion of caudal vertebral bodies (arrows in G and I) surrounding the nucleus pulposus (NP) in the mutants compared to WTcontrols (D–I) most likely caused by ectopic expression of Fgf8 in the ventral sclerotome, as seen by in situ hybridization with a Fgf8 RNA probe on Tbx1Fgf8/+ vs. WT embryos at E11.5 with 45 somite pairs (N, O, respectively; cranial is up, and dorsal is right). Tbx1 expression is detectable in this domain by X-gal staining of Tbx1LacZ/+ embryo at E11.5 (M). (J–L) The cranial portion of the axial skeleton was also abnormal in Tbx1Fgf8/+, manifesting as severe hypoplasia of the anterior arch of the atlas (arrow in L) compared to both WT (J) and Tbx1+/− (K) embryos at E18.5.
In addition, all Tbx1Fgf8/+ embryos showed abnormal anterior arches of the atlas and the axis, the first and second cervical vertebrae, respectively, compared to WT and Tbx1+/−embryos (Fig. 2M–O). The C2 vertebral body was missing in all mutants analyzed (n = 23), while the centrum of C1 was either absent (n = 9) or severely hypoplastic (n = 14). Surprisingly, similar defects have been described in homozygous Tbx1−/− and Chrd−/− mutants, which both show decreased Fgf8 expression in the pharyngeal region (Bachiller et al., 2003; Jerome and Papaioannou, 2001). However, all of the remaining skeletal elements that are defective in Tbx1−/− embryos formed normally in Tbx1Fgf8/+ mutants, including the middle ear ossicles and tympanic ring, the bones of the jaw and the palatine shelves (not shown).
Endogenous Fgf8 is first expressed in a caudal-to-rostral gradient peaking at the distal tail-bud where it is antagonized by an opposing Raldh2-dependent retinoic acid gradient, such that the interface of the two gradients defines the position of the determination front where the somites form (Dubrulle and Pourquie, 2004; Niederreither et al., 2003). Normal Raldh2 expression in the Tbx1Fgf8/+ mutants suggests that disruption of this gradient is not the mechanism of axial defects in these mice. Furthermore, the expression of Pax1, BMP4, Fgf4 and levels of phosphorylated Erk1/2 protein appeared normal in mutants at E10, suggesting that somite patterning and FGF signaling are initially normal (not shown).
The Fgf8 knock-in allele modifies the Tbx1 mutant phenotype
We examined the progeny derived from Tbx1Fgf8/+ and wild-type crosses at embryonic day E18.5 and at weaning (post-natal day (P) 21). The expected number of Tbx1Fgf8/+ fetuses was recovered at E18.5 (56% Tbx1Fgf8/+ vs. 44% wild type). In contrast, at weaning, we observed the loss of 10.5% of Tbx1Fgf8/+ (77 observed vs. 86 expected, n = 172). This loss is more than twice what we observed in similar mixed C57/129 background for Tbx1+/− mice at weaning (4.1%, 139 Tbx1+/− vs. 145 expected, n = 290), suggesting that forced expression of Fgf8 exacerbates the Tbx1 haploinsufficiency phenotype. Intercrosses between Tbx1Fgf8/+ mice yielded no homozygous Tbx1Fgf8/Fgf8 at weaning. When collected at E18.5, we recovered the appropriate proportion of Tbx1Fgf8/Fgf8 (10/41; 24%); all homozygous mutants show PTA, indistinguishable from that observed in Tbx1−/− embryos (Fig. 3C, F; see below).
Fig. 3.
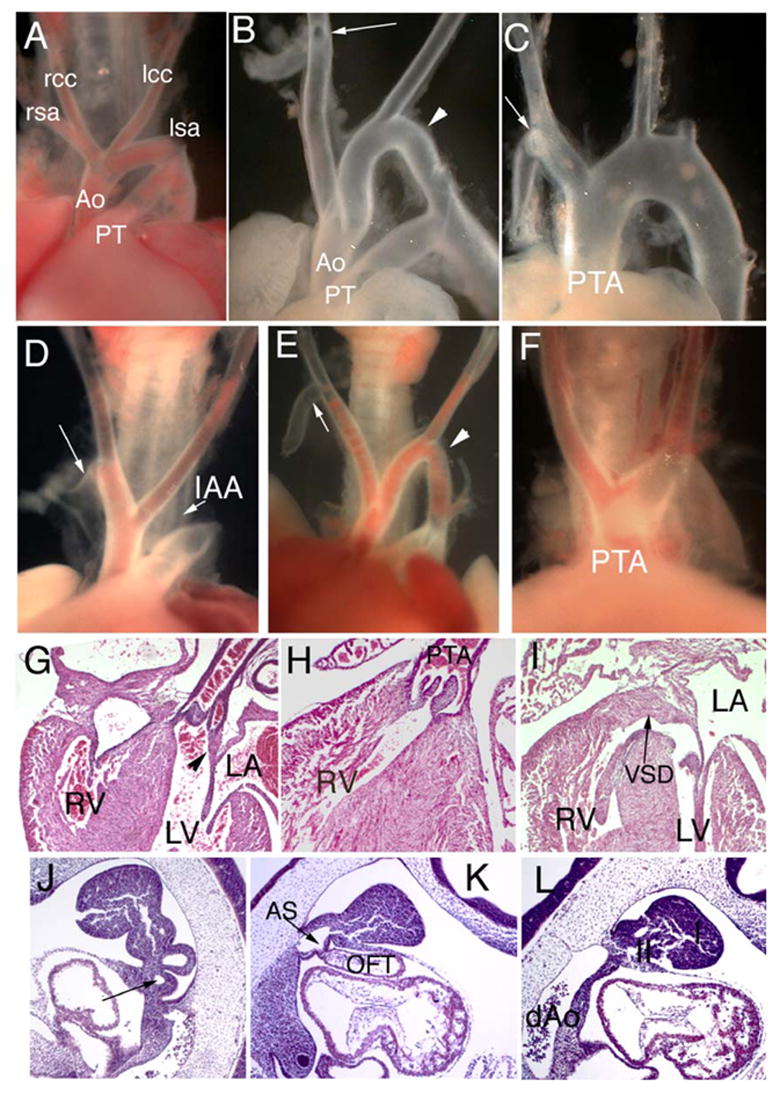
Normal cardiovascular development is not restored by the Tbx1Fgf8 allele. (A–I) Tbx1Fgf8/+ embryos at E18.5 (D-E) show interrupted aortic arch (IAA), aberrant branching of the right subclavian artery (arrows in D, E compared to RSA in A, WT) and cervical aortic arch (arrowhead in E) similar to the phenotype in Tbx1 haploinsufficient control embryos (B). Tbx1Fgf8/− (C) or Tbx1Fgf8/Fgf8 (F) show persistent truncus arteriosis (PTA) associated with VSD (I) and loss of continuity between mitral and semilunar valves (compare arrowhead in G, WT to H–I, Tbx1Fgf8/Fgf8). (J–L) At E10.5, sagittal sections of Tbx1Fgf8/+ show a hypoplastic 4th PAA (arrow in J) or abnormal pharynx lacking caudal arches and arch arteries in Tbx1Fgf8/− embryos (K–L); rcc, lcc, right and left common carotid arteries; rsa, lsa, right and left subclavian arteries, Ao, aorta; PT, pulmonary trunk; RV, LV, LA, right or left ventricle or atrium; AS, aortic sac; dAo, dorsal aorta; I, II, first and second pharyngeal arches.
In order to determine if the forced expression of Fgf8 can modulate the Tbx1-mutant phenotype, we examined Tbx1Fgf8/+ and Tbx1Fgf8/Fgf8 embryos at E10.5 and 18.5. At E18.5, the heart defects observed in Tbx1Fgf8/+, and Tbx1Fgf8/− or Tbx1Fgf8/Fgf8 were identical to those seen in Tbx1+/− and Tbx1−/−, respectively (Fig. 3A–L). Specifically, we observed retroflex right aortic arch, cervical aortic arch, cervical or retro-esophageal right subclavian artery in 75% of Tbx1Fgf8/+ mutants (Table 1). Interestingly, the incidence of cardiovascular defects in Tbx1Fgf8/+ was approximately twice the incidence of the defects caused solely by Tbx1 haploinsufficiency (75% vs. 35%, respectively; n = 114, P = 0.0008; Table 1) consistent with the increased lethality at weaning. In all the Tbx1Fgf8/Fgf8 or Tbx1Fgf8/− fetuses examined (n = 13), we observed persistent truncus arteriosis (PTA) associated with right (n = 6) or left (n = 16) aortic arch, and in only one case did we observe double aortic arch with aortic dilation (n = 1) (Fig. 5F). In all mutants, the PTA communicated solely with the right ventricle, the truncal valve had lost continuity with the mitral valve, and there was a ventricular septal defect (VSD) (Fig. 5F–H), as seen in Tbx1−/− embryos (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Vitelli et al., 2002a). We did not observe intracardiac defects in Tbx1Fgf8/+.
Table 1.
Frequency of aortic arch defects in Tbx1Fgf8/+ at E18.5
| Genotype | Normal | Abnormal |
|---|---|---|
| Tbx1+/+ | 44 | 0 |
| Tbx1+/− | 19 | 7 (35%) |
| Tbx1Fgf8/+ | 11 | 33 (75%) |
Defects are twice more frequent in Tbx1Fgf8/+ compared to Tbx1+/− (P = 0.0008).
Fig. 5.
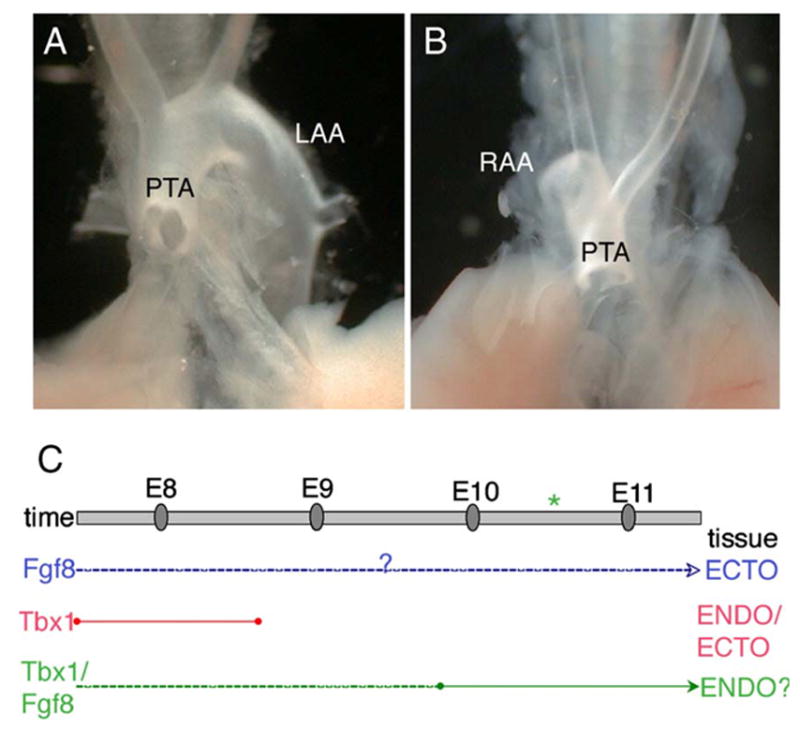
(A–B) Hoxa3-driven ablation of Tbx1 causes OFT defects. Comparison of Tbx1−/− (A) and Hoxa3Cre/+;Tbx1fl/− (B) embryos at E18.5 reveals an identical PTA that is associated either with right sided retroflex aortic arch (RAA) or left aortic arch (LAA). (C) Proposed tissue and time map of Tbx1-Fgf8 interaction. This diagram summarizes the data presented here and elsewhere regarding the requirements of Fgf8 (blue) and of Tbx1 (red) in arch artery development, and the potential timing and location of the Tbx1/Fgf8 interaction (green). Fgf8 is required in the ectoderm (Macatee et al., 2003), but the developmental time point has not been established (dashed line). Tbx1 is required after E7.5 and before E9 (Xu et al., 2005) in the pharyngeal ectoderm and/or endoderm (Zhang et al., 2005). Forced Fgf8 expression in the Tbx1 domain affected only the 4th PAA haploinsufficiency phenotype, and analysis of the timed conditional deletion mutants showed that the Tbx1/Fgf8 interaction is important at least after E9.5, yet it may also occur earlier (dashed line). Thus, we speculate that the Tbx1/Fgf8 interaction probably occurs in the endoderm and is required for embryonic recovery from 4th PAA hypoplasia (asterisk).
Consistent with the phenotype observed at E18.5, histological sections at E10.5 revealed hypoplasia of the 4th pharyngeal arch arteries in Tbx1Fgf8/+ and hypoplasia of the caudal pharyngeal apparatus in Tbx1Fgf8/Fgf8 embryos (Fig. 5I–J) identical to Tbx1+/− and Tbx1−/− embryos, respectively.
The thymic, palate and external ear abnormalities characteristic of Tbx1 mutants were not modified by the presence of the Fgf8 knock-in allele (data not shown) Thus, Fgf8 expression in the Tbx1 expression domain enhances the aortic arch phenotype of Tbx1 mutants but does not affect other associated abnormalities.
Forced Fgf8 expression in the Tbx1 domain is not sufficient to cause aortic arch abnormalities
To establish whether ectopic Fgf8 expression per se confers aortic arch abnormalities, we crossed Tbx1Fgf8/+ to Dp1/+ mice, which harbor a duplication carrying two functional copies of Tbx1 (Lindsay et al., 1999). All of the Dp1/Tbx1Fgf8 embryos at E18.5 examined (n = 18; see Table 2) had normal aortic arch patterning and thymus size indicating that the aortic arch phenotype observed in Tbx1Fgf8/+ animals is due to Tbx1 haploinsufficiency and that the expression of Fgf8 in the Tbx1 domain alone is not sufficient to cause those abnormalities. Conversely, skeletal abnormalities associated with the Tbx1Fgf8 allele were not rescued by the duplication, indicating that this is a gain-of-function phenotype.
Table 2.
The Tbx1Fgf8 allele does not per se cause aortic arch defects
| Genotype | Normal | Abnormal |
|---|---|---|
| Tbx1+/+ | 31 | 0 |
| Dp1/+ | 15 | 0 |
| Tbx1Fgf8/+ | 11 | 9 (45%) |
| Dp1/Tbx1Fgf8/+ | 18 | 0 |
Normal aortic arch patterning is restored in the presence of the Dp1 allele carrying two functional copies of Tbx1, suggesting that the phenotype of Tbx1Fgf8/+ mutants is attributable to an aggravating effect of excess Fgf8 in the context of Tbx1 haploinsufficiency.
Inactivation of Fgf8 in Tbx1-expressing cells
To address if Tbx1-expressing cells are an essential source of FGF8 during cardiovascular development and to establish when in embryogenesis such expression may be required, we used a Tamoxifen-inducible Tbx1-driven Cre allele, Tbx1mcm, to delete the Fgf8flox conditional allele from Tbx1-positive cells. If Tbx1 activates expression of Fgf8, we expect Tbx1mcm/+;Fgf8fl/− embryos to enhance the Tbx1+/−phenotype, since Tbx1mcm is a functionally null allele (Xu et al., 2004). Hence, we crossed Tbx1mcm/+;Fgf8+/− mice to Fgf8fl/+ or Fgf8fl/fl mice and subsequently exposed pregnant dams to a single dose of Tamoxifen (75 mg/kg body weight) at either E7.5, 8.5, or 9.5. Embryos were harvested at E9.5, E10.5, and E18.5. Once Tamoxifen is cleared, no further Cre-mediated deletion will occur, although the Tbx1 locus may remain transcriptionally active (the half-life of Tamoxifen is 11.9 h (Robinson et al., 1991).
In order to confirm tissue specific loss of Fgf8 in Tbx1-positive cells, we performed RNA in situ hybridization using an Fgf8 exon 4–5 specific probe (Brown et al., 2004). We tested E9.5 or E10.25 embryos exposed to Tamoxifen at E7.5, E8.5 or E9.5. In compound Tbx1mcm/+;Fgf8fl/−embryos injected at E7.5, expression of Fgf8 mRNA was irregular in the pharyngeal endoderm and persisted in the ectoderm, as compared to Tbx1mcm/+;Fgf8fl/+ control embryos (Fig. 4A–B), indicating that the deletion is inefficient at this stage of induction. In Tbx1mcm/+;Fgf8fl/− mutants exposed to Tamoxifen at E8.5, the endodermal expression of Fgf8 decreased to barely detectable levels, yet persisted in the caudal ectoderm covering the arches (Figs. 4C–D). Control embryos injected at E9.5 and collected at E10.25 showed low but evident levels of Fgf8 expression within the caudal endoderm, while signal persisted robustly in the ectoderm and cranial pouches (Fig. 4E). In Tbx1mcm/+; Fgf8fl/− littermates, although signal persisted in the cranial pouches, no Fgf8 expression was detectable in the caudal pouch endoderm (Fig. 4F), consistent with efficiency of Tbx1mcm-mediated recombination at this stage of Tamoxifen exposure (Xu et al., 2005). In all the embryos tested, the Fgf8 signal remained strong in the regions of the embryo that do not express Tbx1, such as the isthmus, first arch ectoderm (Figs. 4C–D) and the apical ectodermal ridge (not shown).
Fig. 4.
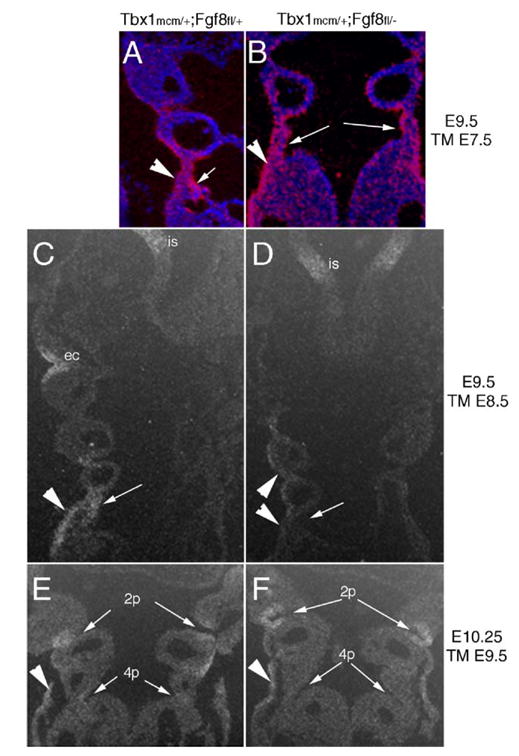
Timed, tissue-specific loss of Fgf8. RNA in situ hybridization with a deletion-specific probe that does not detect the recombined Fgf8flox allele reveals differential loss of Fgf8 in the pharyngeal epithelia of Tbx1mcm/+;Fgf8fl/− (B, D, F) as compared to control Tbx1mcm/+;Fgf8fl/+ (A, C, E) depending on the timing of deletion induction. Embryo stage and stage of Tamoxifen (TM) exposure is shown adjacent to each set of panels. (A–B) Injection at E7.5 does not cause reduced expression of Fgf8 in the pharyngeal ectoderm (arrows) while the endoderm expression appeared patchy in Tbx1mcm/+;Fgf8fl/− embryos at E9.5. (C–D) When TM is injected at E8.5, expression is reduced in the caudal ectoderm and lost in the endoderm, while other regions continue to express Fgf8 robustly (i.e., isthmus, is). (E–F) In embryos at E10.25 exposed to TM at E9.5 caudal endoderm expression of Fgf8 is weak in control embryos (E) and absent in Tbx1mcm/+;Fgf8fl/− mutants, while the ectoderm expression is strong in both mutant and control embryos.
Fgf8 dosage reduction in Tbx1-expressing cells is sufficient to enhance the Tbx1 haploinsufficiency phenotype
We next sought to address the effect of timed ablation of Fgf8 in the Tbx1 domain. The conditional deletion of a single copy of Fgf8 in Tbx1mcm/+; Fgf8fl/+ compound mutants at E18.5 exposed to Tamoxifen at E9.5 caused a significant increase in the penetrance of aortic arch defects compared to Tbx1mcm/+; Fgf8+/+ embryos (Table 3; 63% vs. 37%, P = 0.04). The penetrance of these defects in E18.5 Tbx1mcm/+;Fgf8fl/+ (TM E9.5) embryos is statistically not different from that observed in Tbx1mcm/+;Fgf8+/− animals (63% vs. 78%, P = 0.2). Thus, conditional deletion of one Fgf8 copy in Tbx1 expressing cells at E9.5 has similar phenotypic consequences as the germ line knockout of one Fgf8 allele. On the other hand, deletion of Fgf8 with TM at E7.5 and E8.5 did not result in phenotypic enhancement. This may be due to inefficient deletion of Fgf8 in the critical domains at these time points or to a non-requirement for Fgf8 in Tbx1-positive cells at these stages.
Table 3.
Frequency of aortic arch defects at E18.5 after timed ablation of Fgf8 in the Tbx1 domain
| Genotype | E7.5 | E8.5 | E9.5 |
|---|---|---|---|
| Abnormal/Total | |||
| Tbx1mcm/+; Fgf8+/+ | 16/41 (39%)*,^ | ||
| Tbx1mcm/+; Fgf8+/− | 29/37 (78%)* | ||
| Tbx1mcm/+; Fgf8flox/+ | 16/31 (52%) | 10/31 (32%) | 19/31 (63%)^ |
| Tbx1mcm/+; Fgf8flox/− | 21/26 (81%) | 28/32 (88%) | 33/37 (89%) |
Comparison of Tbx1mcm/+; Fgf8+/+ and Tbx1mcm/+; Fgf8+/− reconfirmed that Tbx1 and Fgf8 interact in vivo as the frequency of defects increases significantly in Tbx1mcm/+; Fgf8+/− embryos (*P = 0.006). Tbx1-driven deletion of one copy of Fgf8 in Tbx1mcm/+; Fgf8flox/+ embryos exposed to TM at E9.5 is sufficient to recapitulate the effect of germ-line ablation of Fgf8, since the frequency of defects in E9.5 induced Tbx1mcm/+; Fgf8flox/+ embryos is significantly greater that in Tbx1mcm/+; Fgf8+/+ (^P = 0.04) and closely approached the frequency seen in Tbx1mcm/+; Fgf8+/− (78% vs. 63%; P = 0.2).
Further reduction of Fgf8 dosage in Tbx1mcm/+;Fgf8fl/−animals exposed to Tamoxifen at E9.5 increased the percentage of abnormal animals to 89% (n = 37), but the change was not statistically significant when compared to the penetrance in Tbx1mcm/+;Fgf8fl/+ animals (89% vs. 63%, P = 0.2). Furthermore, we did not identify any additional phenotypic abnormality in Tbx1mcm/+;Fgf8fl/− compared to Tbx1mcm/+;Fgf8+/+ animals. In particular, we did not find outflow tract defects in these mutants (Table 2).
These data, while indicating that Tbx1-expressing cells are a critical source of FGF8 at or after E9.5 for the Tbx1-Fgf8 interaction, also suggest that under the conditions tested, the phenotypic significance of this interaction is limited to the development of the 4th PAAs.
Fgf8 and Tbx1 are required in different tissues or stages in OFT morphogenesis
Fgf8 is crucial for outflow tract development (Abu-Issa et al., 2002; Frank et al., 2002; Hu et al., 2004; Macatee et al., 2003), and the results shown above suggest that Tbx1-expressing cells are not a critical source of FGF8 for that developmental role. Thus, it is yet to be determined at what developmental stage and in which tissue Fgf8 is required for outflow tract development. It has been previously shown that Hoxa3Cre-driven ablation of Fgf8 causes aortic arch defects but not outflow tract defects (Macatee et al., 2003). Deletion of one copy of Tbx1 using the same Cre-driver, which is active in the pharyngeal ectoderm from E8.25 (10 somites) and endoderm from E8.5 (12 somites), is sufficient to cause aortic arch defects (Zhang et al., 2005), but the homozygous deletion of Tbx1 in the Hoxa3 domain has not been tested. Therefore, we obtained Hoxa3Cre/+;Tbx1flox/−embryos at E18.5 and examined their outflow tract phenotype. Embryos exhibited PTA, which is also seen in Tbx1−/− animals (Figs. 5A–B). Thus, expression of Tbx1, but not of Fgf8, in the Hoxa3 domain is crucial for outflow tract development. These data, together with the finding that outflow tract development is unaffected in Tbx1mcm/+; Fgf8flox/− vs. Tbx1mcm/+;Fgf8flox/− and Tbx1Fgf8/Fgf8 vs. Tbx1−/− mutants, suggest that Tbx1 and Fgf8 are required at different developmental times or in different tissues for outflow tract development.
Discussion
Axial skeletal defects in Tbx1Fgf8/+ mutants
Wild-type expression levels of Raldh2, Pax1, BMP4, Fgf4 and levels of phosphorylated Erk1/2 protein in Tbx1Fgf8/+ mutants at E10 suggests that somite patterning and FGF signaling is initiates normally in these mice. However, the hypoplasia of the anterior arches of the first two cervical vertebrae and the fusion of lumbar, sacral, and caudal vertebral bodies suggest that Fgf8 expression from the Tbx1Fgf8 allele perturbs a later developmental process. Furthermore, the different phenotypes seen in the cranial and caudal vertebral elements suggest that skeletal precursors may respond differently to misexpression of Fgf8 depending on their cranial-to-caudal position. Further studies are required to understand which regulatory network is disrupted in the axial skeleton of these mice.
The Tbx1Fgf8 allele enhances the Tbx1 haploinsufficiency phenotype
Expression analysis at the RNA and protein levels demonstrated that the Fgf8 cDNA knocked into the Tbx1 locus is robustly transcribed and translated. Proliferation levels of the pharyngeal arch mesenchyme increased significantly in response to Tbx1-driven Fgf8 expression. However, in contrast to our hypothesis, Fgf8 expressed by the Tbx1-positive cells did not rescue any of the phenotypic abnormalities of Tbx1 mutants. Conversely, we observed an increase in aortic arch defects in Tbx1Fgf8/+ mice, indicating that additional Fgf8 expression in these cells, in the context of Tbx1 haploinsufficiency, can worsen the aortic arch phenotype caused by loss of one copy of Tbx1. Conditional ablation of Fgf8 in Tbx1 expressing cells also enhanced the haploinsufficiency phenotype. Thus, although our data support that at least one critical source of FGF8 for aortic arch patterning resides within Tbx1-expressing cells, the effect of forced Fgf8 expression in these cells is also detrimental. The latter finding may result from an Fgf8 dosage effect. The Tbx1Fgf8 allele may produce an excess of Fgf8, which may overcompensate the loss of Tbx1 function, thus phenocopying the 4th PAA defects see in TBX1 overexpressing animals (Liao et al., 2004). However, we found that per se, overexpression of Fgf8 does not cause aortic arch defects since increasing the Tbx1 dosage by introducing the Dp1 allele suppresses the Tbx1Fgf8 aortic arch phenotype rather than enhancing it. Thus, it appears that Tbx1-driven Fgf8 expression can exert its phenotypic effect only on a faulty 4th PAA, and that haploinsufficiency of Tbx1 is a prerequisite to uncovering sensitivity of the 4th PAA to increased dosage of Fgf8 in the Tbx1 domain.
Dosage sensitivity to Fgf8 has been well documented in the development of several organs including the cardiovascular system (Abu-Issa et al., 2002; Frank et al., 2002), salivary glands (Jaskoll et al., 2004), and forebrain (Storm et al., 2003). Recently, Waldo et al. (2005) speculated that an observed increase in proliferation of splanchnic mesenchymal cells in the cardiac neural crest-ablated chick is due to excessive FGF8 and/ or FGF4 signaling in the pharyngeal arches, which contributed to arch artery and outflow tract defects in this model (Waldo et al., 2005). These data provide further support for the hypothesis that either too much or too little FGF can be detrimental, and that Fgf8 levels must be finely titrated for normal cardiovascular development to progress.
Time and spatial resolution of the Fgf8-Tbx1 interaction in vivo
In vitro studies can test the transactivation of a target gene by a transcription factor. However, this is an oversimplification of in vivo interactions that occur in a developmental process. Our data show that in the context of Tbx1 haploinsufficiency, the timed conditional deletion of one Fgf8 copy in Tbx1 expressing cells at E9.5 has similar phenotypic consequences on cardiovascular development as the germ line knockout of one Fgf8 allele. However, homozygous loss of Fgf8 in these cells at E9.5 does not significantly aggravate the phenotype, although the incidence does rise to 89%. The lack of statistical significance may be attributable to near saturation of the experimental system, since the frequency of defects in double germ line heterozygous Tbx1+/−;Fgf8+/− embryos is already 78%. Alternatively, the 4th PAA may have a threshold of tolerance to decreased Fgf8 to which it responds. Indeed, a dose-dependent but non-linear epistatic response to variations of Fgf8 levels has been reported during embryonic submandibular salivary gland development, providing the precedent for a threshold-like response to Fgf8 signaling (Jaskoll et al., 2004). In addition, since Fgf8 encodes a diffusable molecule and Tbx1mcm-induced recombination may not be highly efficient, adjacent non-deleted cells may supply compensatory FGF8 to their neighbors.
Published data suggest that the primary role of Fgf8 in 4th PAA development occurs in the ectoderm (Macatee et al., 2003). Data from the timed deletion of Tbx1 using an ubiquitous inducible Cre driver suggest that the critical time point for Tbx1 in 4th PAA development occurs around E7.5–8.5 (Xu et al., 2005). In this study, our data suggest that the Tbx1-Fgf8 interaction in 4th PAA growth occurs after E9.5. Thus, Fgf8 and Tbx1 do act to modify PAA development, yet this interaction does not occur at the time point at which Tbx1 is essential (E7.5–8.5), nor in the tissues where Fgf8 is required (ectoderm) since Tbx1mcm does not induce recombination in the ectoderm at this stage (Xu et al., 2005). Thus, Tbx1 and Fgf8 most likely have separate primary roles in supporting 4th PAA formation. We speculate that, instead of interacting with Tbx1 during its primary role in artery formation, Fgf8 modifies the 4th PAA recovery from arterial growth delay, a phenomenon observed in Tbx1 haploinsufficient mutants after E10.5 (Lindsay and Baldini, 2001). Our data suggest that the endoderm is the most probable source of Fgf8 exerting this later, "secondary" function on 4th PAA development, and is therefore the likely site of the Tbx1-Fgf8 interaction. These hypotheses are summarized in Fig. 5C.
Our hypotheses are consistent with recent evidence of genetic interaction between Tbx1 and Crkl (Guris et al., 2006). Crkl encodes an adaptor protein required in the FGF signal transduction pathway (Larsson et al., 1999) and is highly abundant in the neural crest derivatives and in the pharyngeal apparatus (Guris et al., 2001). While Crkl haploinsufficiency has no consequences for cardiovascular development, double heterozygous Crkl+/−;Tbx1+/− embryos between E16.5-P2 showed increased expressivity and penetrance of congenital heart defects compared to Tbx1+/− embryos (Guris et al., 2006). Over 97% of Crkl+/−;Tbx1+/− compound heterozygotes had aortic arch abnormalities compared to 33% for Tbx1+/−, while at E10.5 the frequency of defective 4th PAAs was similar in both genotypes (Guris et al., 2006). Although it is not clear whether the 4th PAAs fail to form altogether in Crkl+/−;Tbx1+/− mutants, the data suggest that Crkl haploinsufficiency severely impairs the Tbx1-dependent embryonic recovery from 4th PAA hypoplasia, implicating a role for FGF signaling in this process.
The Tbx1-Fgf8 interaction is relevant to aortic arch but not outflow tract development
Overall, our data support a strong genetic interaction in aortic arch development but do not provide evidence that this interaction is relevant to OFT development. The OFT phenotype due to complete Tbx1 loss of function was unchanged in Tbx1Fgf8/Fgf8 vs. Tbx1−/− mutants suggesting that the function of Tbx1 in OFT development is not mediated by Fgf8, and that the Fgf8 role in OFT development is not Tbx1-dependent. These data are also consistent with the fact that we do not observe a change in cell proliferation levels in the secondary heart field of Tbx1Fgf8/+ mutants, while Tbx1 loss of function mutants show reduced cell proliferation in this region (Xu et al., 2004). In addition, data from the Hoxa3Cre/+;Tbx1flox/− mutants suggest that the critical sources for Tbx1 and Fgf8 are regionally and/or temporally distinct.
Fgf8 deletion with a transgenic Tbx1-Cre driver in Tbx1-Cre::Fgf8flox/− mice was shown to be associated with OFT defects, including PTA, double outlet right ventricle, transposition of the great arteries and Tetrology of Fallot (Brown et al., 2004). This discrepancy with the present study may be attributed to a more robust expression of the Cre in the transgenic line as compared to Tbx1mcm/+. Alternately, the transgenic Tbx1-Cre may show ectopic expression domains, for example at the level of the cardiac crescent and myotome (Brown et al., 2004), where endogenous Tbx1 is not expressed (Jerome and Papaioannou, 2001). The generation of a constitutively active Cre driver under the control of endogenous regulatory elements may provide further clarification of the observed phenotypic differences. Hu et al. have proposed that the essential Tbx1-Fgf8 interaction occurs in the OFT, based on the OFT-specific suppression of a transgene harboring a Tbx1-dependent Fgf8 enhancer in a Tbx1-deficient background (Hu et al., 2004). However, since the disruption of the 5 putative TBEs singly did not affect transgene expression in vivo, nor did mutation of up to 3 consensus sites in tandem, conclusive in vivo evidence of the significance of this Fgf8 enhancer has not been provided. In addition, Hoxa3Cre-driven deletion of Fgf8 does not cause OFT defects (Macatee et al., 2003), whereas we have shown in this study that ablation of Tbx1 by the same driver causes PTA. Although a mounting body of evidence has made it clear that both Tbx1 and Fgf8 are required for OFT development (Abu-Issa et al., 2002; Farrell et al., 2001; Frank et al., 2002; Jerome and Papaioannou, 2001; Lindsay et al., 2001; Macatee et al., 2003; Vitelli et al., 2002a; Waldo et al., 2005; Xu et al., 2004, 2005), our data imply that the Fgf8-Tbx1 interaction is not essential for OFT development.
Although it is still possible that Tbx1 has direct transcriptional activity on the Fgf8 gene in the endoderm, our in vivo data are more supportive of a complex picture in which the two genes do not interact directly, neither at the time point nor in the location of their primary individual roles in 4th PAA development. On the other hand, it is possible that the interaction concerns the signal transduction pathway rather than the expression of the ligand. Alternatively (or in addition), each gene functions in pathways that independently affect the same structure.
Overall, our genetic data confirm that the Tbx1-Fgf8 interaction is critical for 4th arch artery development, suggesting that such interaction is not relevant for other aspects of the Tbx1 mutant phenotype, and that it occurs mostly in the pharyngeal endoderm at mid-gestation. We propose that the interaction is essential for the embryonic recovery from arterial growth delay previously described in Tbx1 haploinsufficient mice. This finding is clinically relevant to 22q11 Deletion Syndrome since the Fgf8 dosage sensitivity of the 4th PAA in the context of Tbx1 haploinsufficiency may provide possible grounds for the variable penetrance of aortic arch defects seen in del22q11 patients. Further understanding the mechanism of the Tbx1-Fgf8 interaction in the endoderm may uncover an entry point for altering clinical severity of del22q11-associated aortic arch defects.
Acknowledgments
This work was supported by AHA0464133Y to FVand NIH, HL051524 to AB. We wish to thank A.M. Moon for Hoxa3Cre/+ mice, E. Meyers for Fgf8+/− and Fgf8flox/flox mice, K. Niederreither for assistance with whole mount in situ, the Darwin Transgenic Core at Baylor College of Medicine for blastocyst injections and E.A. Lindsay for critical reading of the manuscript.
References
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development. 2002;129:4613–4625. doi: 10.1242/dev.129.19.4613. [DOI] [PubMed] [Google Scholar]
- Albrecht U, Eichele G, Helms JA, Lu HC. Visualization of gene expression patterns by in situ hybridization. In: Daston GP, editor. Molecular and Cellular Methods in Developmental Toxicology. CRC Press, Inc.; New York: 1997. pp. 23–48. [Google Scholar]
- Bachiller D, Klingensmith J, Shneyder N, Tran U, Anderson R, Rossant J, De Robertis EM. The role of chordin/Bmp signals in mammalian pharyngeal development and DiGeorge syndrome. Development. 2003;130:3567–3578. doi: 10.1242/dev.00581. [DOI] [PubMed] [Google Scholar]
- Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol. 2004;267:190–202. doi: 10.1016/j.ydbio.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Dubrulle J, Pourquie O. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature. 2004;427:419–422. doi: 10.1038/nature02216. [DOI] [PubMed] [Google Scholar]
- Farrell MJ, Burch JL, Wallis K, Rowley L, Kumiski D, Stadt H, Godt RE, Creazzo TL, Kirby ML. FGF-8 in the ventral pharynx alters development of myocardial calcium transients after neural crest ablation. J Clin Invest. 2001;107:1509–1517. doi: 10.1172/JCI9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A, Smith A. Patterning the pharyngeal arches. BioEssays. 2001;23:54–61. doi: 10.1002/1521-1878(200101)23:1<54::AID-BIES1007>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–298. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Beddington R, Constantini F, Lacy E, editors. A laboratory manual. Cold Spring Harbor Press; Cold Spring Harbor: 1994. Manipulating the Mouse Embryo. [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–5502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- Jaskoll T, Witcher D, Toreno L, Bringas P, Moon AM, Melnick M. FGF8 dose-dependent regulation of embryonic submandibular salivary gland morphogenesis. Dev Biol. 2004;268:457–469. doi: 10.1016/j.ydbio.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Larsson H, Klint P, Landgren E, Claesson-Welsh L. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J Biol Chem. 1999;274:25726–25734. doi: 10.1074/jbc.274.36.25726. [DOI] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Baldini A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Hum Mol Genet. 2001;10:997–1002. doi: 10.1093/hmg/10.9.997. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Lu P, Minowada G, Martin GR. Increasing Fgf4 expression in the mouse limb bud causes polysyndactyly and rescues the skeletal defects that result from loss of Fgf8 function. Development. 2006;133:33–42. doi: 10.1242/dev.02172. [DOI] [PubMed] [Google Scholar]
- Macatee TL, Hammond BP, Arenkiel BR, Francis L, Frank DU, Moon AM. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development. 2003;130:6361–6374. doi: 10.1242/dev.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Min Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, St Jore B, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler PJ, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is Responsible for cardiovascular defects in Velo-Cardio-Facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. Crkl deficiency disrupts fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell. 2006;10:71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21:444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Messaddeq N, Schuhbaur B, Chambon P, Dolle P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development. 2001;128:1019–1031. doi: 10.1242/dev.128.7.1019. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Fraulob V, Chambon P, Dolle P. Retinaldehyde dehydrogenase 2 (RALDH2)-independent patterns of retinoic acid synthesis in the mouse embryo. Proc Nat Acad Sci U S A. 2002;99:16111–16116. doi: 10.1073/pnas.252626599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Roux IL, Schuhbaur B, Chambon P, Dolle P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. 2003;130:2525–2534. doi: 10.1242/dev.00463. [DOI] [PubMed] [Google Scholar]
- Robinson SP, Langan-Fahey SM, Johnson DA, Jordan VC. Metabolites, pharmacodynamics, and pharmacokinetics of tamoxifen in rats and mice compared to the breast cancer patient. Drug Metab Dispos. 1991;19:36–43. [PubMed] [Google Scholar]
- Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Gewillig M, Molin DG, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Plaisance S, Vlietinck R, Emanuel B, Gittenberger-de Groot AC, Scambler P, Morrow B, Driscol DA, Moons L, Esguerra CV, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P. VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med. 2003;9:173–182. doi: 10.1038/nm819. [DOI] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR. Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci U S A. 2003;100:1757–1762. doi: 10.1073/pnas.0337736100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002a;11:915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002b;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Hum Mol Genet. 2003;12:2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. 2005;281:66–77. doi: 10.1016/j.ydbio.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Xu H, Cerrato F, Baldini A. Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development. 2005;132:4387–4395. doi: 10.1242/dev.02018. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cerrato F, Xu H, Vitelli F, Morishima M, Vincentz J, Furuta Y, Ma L, Martin JF, Baldini A, Lindsay E. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]