

Abstract
Cholestasis is associated with retention of potentially toxic bile acids and profound cytoskeletal alterations of hepatocytes. Given the well-established cytoprotective role of hepatocyte keratins this study aimed to determine the effects of cholestasis on the cytokeratin (CK) intermediate filament network in mouse liver. Mice were subjected to common bile duct ligation or sham operation. Mice were also fed a cholic acid or ursodeoxycholic acid (UDCA)-supplemented diet (0.1%, 0.5%, and 1%) or control diet for 7 days. CK 8 and CK 18 expression was studied by competitive reverse transcriptase-polymerase chain reaction, in situ hybridization, Western blot analysis, and immunofluorescence microscopy. Common bile duct ligation and cholic acid feeding significantly stimulated CK 8 and CK 18 mRNA and protein levels compared to controls, whereas UDCA had no effect. CK overexpression was accompanied by pronounced phosphorylation. Our results show that potentially toxic bile acids induce hepatocytic CK 8 and CK 18 expression and phosphorylation whereas nontoxic UDCA has no effect on CKs. Thus, increased hepatocellular CK expression and phosphorylation in cholestasis may be caused by retention of toxic bile acids and reflect a hepatocellular stress response with potential beneficial effects.
Cholestatic liver disorders were found to be associated with profound alterations of all three components of the hepatocyte cytoskeleton, namely microtubules, microfilaments, and cytokeratin (CK) intermediate filaments (IFs). 1-3 Hydrophobic bile acids accumulate during cholestasis and affect microtubular motors, such as kinesin and dynein, resulting in impaired vesicle movement to the canalicular membrane and reduced bile flow. 1-4 Disruption of microfilaments, which normally form a contractile web around the bile canaliculus and regulate tight junction permeability, results in dilated canaliculi, leaky tight junctions, and loss of microvilli. All these changes may not only be consequences of but also contribute to cholestasis. 1-4 Hepatocytic IFs consisting of CK 8 and CK 18 mechanically support the bile canaliculus (pericanalicular sheath) but their role in bile secretion remains unclear. 5,6 Increased density of IFs in experimental obstructive cholestasis in rats results in the thickening of the pericanalicular sheath and has mainly been attributed to increased canalicular pressure as a result of biliary obstruction. 7-9 However, primary biliary cirrhosis and disorders associated with nonmechanical intrahepatic cholestasis, such as alcoholic hepatitis, nonalcoholic steatohepatitis, and Wilson’s disease, are also associated with profound cytoskeletal alterations including the formation of Mallory bodies (MBs), which are cytoplasmic inclusions in hepatocytes consisting of CKs and non-CK components. 10,11 The CK-IF network has long been considered as being a rather static structure responsible for mechanical stability of cells. More recently, however, the importance of CKs for maintenance of functional integrity of hepatocytes has been demonstrated in several gene knockout mouse models. 11-16 Mutations of CKs in mice are associated with higher susceptibility to hepatotoxins, indicating that CKs may also have important nonmechanical functions, such as defense against toxic injury. 12,16 The importance of CK mutations for the pathogenesis of human liver diseases has been emphasized by the recent finding of mutations in the CK 8 gene in patients with cryptogenic cirrhosis. 17 In addition, hyperphosphorylation of CKs seems to be involved in cytoprotection against toxic stress. 6,11,15,18 Marked overexpression and hyperphosphorylation of CK 8 and CK 18 were also demonstrated in human alcoholic hepatitis, which is also frequently accompanied by cholestasis. 18,19 Therefore, the aim of this study was to investigate whether bile acids play a causal role in overexpression and phosphorylation of CK 8 and CK 18. For this purpose we studied expression and phosphorylation of CKs in different mouse models associated with elevated bile acid levels.
Materials and Methods
Animals and Materials
Male Swiss Albino mice (strain Him OF1 SPF) were obtained from the Institute of Laboratory Animal Research, University of Vienna School of Medicine, Himberg, Austria, housed with a 12-hour light-dark cycle and permitted ad libitum consumption of water and a standard mouse diet (Marek, Vienna, Austria). Experiments were performed with 2-month-old mice weighing 25 to 30 g. The experiments described in this article were approved by the local ethics committee and followed the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” (National Academy of Sciences) as published by the National Institutes of Health (NIH publication 86-23, revised 1985). Cholic acid (CA) was obtained from Aldrich (Steinheim, Germany), ursodeoxycholic acid (UDCA) was kindly provided by the Falk Foundation (Freiburg, Germany).
Animal Experiments
Common Bile Duct Ligation (CBDL)
All surgical procedures were performed under sterile conditions. To study the effects of obstructive cholestasis on hepatic CK expression, the common bile duct was ligated under general anesthesia (10 mg Avertin intraperitoneally) close to the liver hilum immediately below the bifurcation and dissected between the ligatures as described previously. 20 Cholecystectomy was performed after ligation of the cystic duct. Controls underwent a sham operation with exposure, but without ligation of the common bile duct and removal of the gallbladder. The livers were excised under general anesthesia 3 and 7 days after surgery, respectively. Aliquots of liver tissue were frozen in liquid nitrogen for molecular analysis and immunohistochemistry or fixed in 4% neutral buffered formaldehyde solution and paraffin-embedded for light microscopy and in situ hybridization. Serum samples from each mouse were stored at −70°C for analysis of AST/ALT, alkaline phosphatase, and total bile acid levels.
Bile Acid Feeding
To study the differential effects of bile acids on CK expression and phosphorylation, mice were fed a diet supplemented with toxic CA or hydrophilic nontoxic UDCA in different concentrations (0.1%, 0.5%, or 1%) for 7 days. 21
Determination of mRNA Copy Numbers by Competitive Reverse Transcriptase-Polymerase Chain Reaction
mRNA copy numbers for CK 8, CK 18, and glyceraldehyde-3-phosphate dehydrogenase were determined by competitive reverse transcriptase-polymerase chain reaction as previously described by Zatloukal and colleagues. 16
In Situ Hybridization (ISH)
Synthesis of 35S-Labeled Mouse CK 8 Probe
Labeled sense and antisense transcripts were synthesized in a 20-μl reaction mixture containing 1 μg of linearized plasmid, 10× transcription buffer (0.4 mol/L Tris-HCl, pH 8.0, 60 mmol/L MgCl2, 20 mmol/L spermidine, 100 mmol/L dithiothreitol), 20 mmol/L dithiothreitol, rNTP-labeling mixture (1 mmol/L each of ATP, CTP, GTP; Boehringer Mannheim, Mannheim, Germany), 40 U Inhibit-ACE (5′→3′ Inc, Boulder, CO), 120 μCi α-35S UTP (Amersham, Buckinghamshire, UK), and 20 U of either SP6 or T7 RNA polymerase (Boehringer Mannheim). After an incubation period of 2 hours at 37°C, DNA was digested with 2 U DNase (RNase-free, Boehringer Mannheim) for 10 minutes at 37°C and the reaction was stopped with 2 μl of 0.5 mol/L ethylenediaminetetraacetic acid (EDTA), pH 8.0. Unincorporated nucleotides were removed using a MicroSpin S-200 HR column (Pharmacia, Uppsala, Sweden). Fifty μl of hydrolysis buffer (80 mmol/L NaHCO3, 120 mmol/L Na2CO3, 120 mmol/L dithiothreitol) were added to 50 μl of the eluted sample and hydrolysis was performed at 60°C for 45 minutes to obtain an average sample size of 150 bp. After addition of 5 μl of stopping solution (0.2 mol/L Na acetate, 10 mmol/L dithiothreitol, 1% glacial acetic acid) the sample was precipitated with LiCl/isopropanol. The washed pellet was resuspended in 100 μl of 50% formamide containing 25 mmol/L dithiothreitol.
Pretreatment of Paraffin Sections
Four-μm paraffin sections mounted on silanized glass slides were deparaffinized and postfixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 minutes at room temperature. After rinsing with Tris-buffered saline (TBS) (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl) sections were treated with 0.2 mol/L of HCl for 10 minutes. After washing in TBS sections were incubated in 20 μg/ml of proteinase K (Sigma Chemical Company, St. Louis, MO) in TBS containing 2 mmol/L of CaCl2 for 15 minutes at 37°C. The reaction was stopped with TBS for 5 minutes at 4°C. After treatment with 0.5% acetic anhydride in 100 mmol/L Tris, pH 8.0, for 10 minutes, sections were rinsed with TBS, dehydrated in graded ethanol, and air-dried.
Hybridization and Autoradiography
For hybridization the labeled sample (1 × 106cpm per section) was diluted in 50 μl of hybridization buffer containing 12.5 mmol/L phosphate buffer, pH 6.8, 12.5 mmol/L Tris, 0.4 mol/L NaCl, 3 mmol/L EDTA, 1.25× Denhardt, 50% formamide, 12.5% dextran sulfate, 0.1 mol/L dithiothreitol, 100 nmol/L S-rATP (Boehringer Mannheim), 60 ng t-RNA, and 30 ng poly(A). The sections were hybridized with the diluted probe overnight at 50°C in a humid chamber containing 2× standard saline citrate (0.3 mol/L NaCl, 30 mmol/L Na citrate, pH 7.0) and 50% formamide. Thereafter, sections were washed with formamide washing buffer (10 mmol/L phosphate buffer, pH 6.8, 10 mmol/L Tris-HCl, pH 7.7, 0.3 mol/L NaCl, 5 mmol/L EDTA, 0.1× Denhardt, 0.07% β-mercaptoethanol, and 50% formamide) at 45°C once for 1 hour and once for 2 hours, followed by washing in 10 mmol/L Tris-HCl, pH 7.4, 0.5 mol/L NaCl, 2.5 mmol/L EDTA, and 0.07% β-mercaptoethanol two times for 15 minutes. After RNase A treatment (20 μg/ml, Boehringer Mannheim) in the same buffer for 30 minutes at 37°C, washing was continued overnight in the formamide washing buffer at 37°C. On the next day there was a high-stringent wash in 2× standard saline citrate and 0.07% β-mercaptoethanol for 30 minutes at 45°C and in 0.1× standard saline citrate and 0.07% β-mercaptoethanol for 30 minutes at 45°C. After dehydration in graded ethanol, air-dried sections were coated with Ilford K2 photoemulsion (Ilford Ltd., Mobberly, Cheshire, UK). After 1 to 3 weeks of exposure slides were developed with Kodak D19 developer (Eastman Kodak, Rochester, NY) and counterstained with hematoxylin and eosin.
Western Blotting of CK 8 and CK 18
Snap-frozen liver tissue was homogenized in buffer containing 10 mmol/L NaH2PO4, pH 7.4, 5% sodium dodecyl sulfate, and 10% β-mercaptoethanol. Protein concentration was determined using the Bradford method. 22 Samples (20 μg protein per lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. 23 Equal loading amounts of protein were confirmed by Coomassie blue staining. For immunoblotting proteins were electrotransferred to nitrocellulose membranes (BioRad, Hercules, CA). 24 After blocking with 3% nonfat milk in PBS, CKs were detected using a monoclonal mouse antibody against CK 8 (Ks 8.7; Progen, Heidelberg, Germany) in a dilution of 1:500 and a monoclonal mouse antibody to CK 18 (K18.04, Progen) in a dilution of 1:1000. In addition, β-actin was detected using a monoclonal mouse anti-β-actin antibody (Sigma) in a dilution of 1:5000. After washing in PBS, blots were incubated with horseradish peroxidase-conjugated rabbit anti-mouse immunoglobulins (DAKO, Glostrup, Denmark) in a dilution of 1:1000. Specific binding was detected using the enhanced chemiluminescence (ECL) Western blotting detection system (Amersham) and exposing the blots to Trimax XDA Plus films (3M Imation, White City, OR). Band intensities were determined with a Docu Gel V video densitometer (MWG-Biotech, Munich, Germany) and RFLP-Scan Software (Scanalytics, Billerica, MA). Accuracy of the ECL method for quantification of CK protein levels was determined by standard curves (not shown).
Immunofluorescence Microscopy
To study alterations of the IF network, immunofluorescence microscopy was performed using the polyclonal rabbit CK antibody 50K160 recognizing CK 8 and CK 18 25,26 as well as the monoclonal mouse anti-K7 (Monosan, Uden, Netherlands) and anti-K19 (Amersham) recognizing CK 7 and CK 19, respectively. In addition, the monoclonal antibody LJ4 (kindly provided by Bishr Omary, Palo Alto, CA) directed against CK 8 phosphorylated at Ser 79 was used. 16 Immunofluorescent specimens were analyzed with a MRC 600 (BioRad) laser-scanning confocal device attached to a Zeiss Axiophot microscope. The fluorescent images were collected using the confocal photomultiplier tube as full frame (768 × 512 pixels). For dual labeling, separate excitation wavelengths (488 nm for fluorescein isothiocyanate; 568 nm for tetramethylrhodamine B isothiocyanate) from a krypton/argon ion laser were used.
Histology
At the time of harvesting, mouse livers were fixed in 4% neutral buffered formaldehyde solution and embedded in paraffin. Hematoxylin and eosin-stained sections were coded and examined by two pathologists (HD, KZ), who were blinded in regard to the treatment groups.
Routine Serum Biochemistry and Bile Acid Measurements
Serum biochemistry (ALT and AST) was performed by routine clinical chemistry testing on a Hitachi 717 analyzer (Boehringer Mannheim). Alkaline phosphatase and total serum bile acid levels were determined for assessment of the degree of cholestasis. For determination of total serum bile acid levels a commercially available 3α-hydroxysteroid dehydrogenase assay (Merck, Darmstadt, Germany) was used. Tests were performed in duplicate (Tables 1 and 2) ▶ ▶ .
Table 1.
Serum Liver Enzymes after CBDL
| Group | AST | ALT | AP | BA |
|---|---|---|---|---|
| Control | 202 ± 60 | 38 ± 7 | 40 ± 20 | 5 ± 1 |
| CBDL, 3 day | 672 ± 290* | 414 ± 200* | 1196 ± 270* | 1131 ± 413* |
| CBDL, 7 day | 581 ± 42* | 399 ± 57* | 1360 ± 608* | 288 ± 61* |
Values are means ± SD. AST, aspartate aminotransferase (U/L); ALT, alanine aminotransferase (U/L); AP, alkaline phosphatase (U/L); BA, bile acids (μMol/L).
*P < 0.05 mice challenged with CBDL versus sham-operated mice (control).
Table 2.
Serum Liver Enzymes in Bile Acid-Fed Mice
| Group | AST | ALT | AP | BA |
|---|---|---|---|---|
| Control | 203 ± 53 | 32 ± 7 | 43 ± 15 | 4 ± 0.8 |
| Cholate 0.1% | 226 ± 80 | 254 ± 76* | 233 ± 31* | 11 ± 6* |
| Cholate 0.5% | 268 ± 83 | 287 ± 87* | 387 ± 2* | 37 ± 19* |
| Cholate 1% | 360 ± 168* | 313 ± 9* | 402 ± 61* | 58 ± 36* |
| UDCA 1% | 148 ± 15 | 35 ± 5 | 101 ± 53 | 91 ± 35* |
Values are means ± SD. AST, aspartate aminotransferase (U/L); ALT, alanine aminotransferase (U/L); AP, alkaline phosphatase (U/L); BA, bile acids (μMol/L).
*P < 0.05 bile acid fed mice versus standard diet-fed mice (control); animals have also been used for a separate study and their biochemical data have been reported previously. 39
Statistical Analysis
In each group five animals were studied at each time point. Data are reported as arithmetic means ± SEM. Statistical analysis was performed using Student’s t-test as appropriate, or analysis of variance with posttesting when three or more groups were compared. A P value <0.05 was considered significant.
Results
CBDL Induces Hepatic CK 8 and CK 18 Expression
Obstructive cholestasis (CBDL) for 3 days led to biliary type of hepatocytic necroses (resembling bile infarcts), predominantly in acinar zones 1 and 2 that were infiltrated by variable numbers of neutrophils. Interlobular bile ducts were elongated with dilated lumina and irregular epithelium. The surrounding portal tissue was edematous and infiltrated by neutrophils. No pronounced ductular reaction was observed. Necroses were more prominent in 7-day-ligated than in 3-day-ligated mouse livers. In agreement with the histological findings serum transaminases, alkaline phosphatase, and serum bile acid levels were significantly elevated in comparison to controls (for details see Table 1 ▶ ).
CBDL resulted in a significant increase of CK 8 and CK 18 mRNA levels after 3 and 7 days (Figure 1A) ▶ . CK 8 and CK 18 proteins were also increased after 3 and 7 days of CBDL (Figure 1B) ▶ compared to sham-operated animals, whereas β-actin expression remained constant (Figure 1B) ▶ . After CBDL, increased CK 8 mRNA was observed particularly in hepatocytes surrounding bile infarcts (Figure 1C) ▶ , in hepatocytes in acinar zone 1, and also to a minor degree in bile duct epithelial cells (Figure 1D) ▶ as revealed by in situ hybridization. Increased CK 8 and CK 18 protein expression after CBDL was also detected by immunofluorescence microscopy (Figure 1, E and F) ▶ . CBDL resulted in an increased density of the cytoplasmic IF network predominantly around bile canaliculi (Figure 1F ▶ , arrowheads). Because neoexpression of CK 7 and CK 19 has previously been described in hepatocytes in human cholestatic liver disease, 27 additional immunostainings using antibodies against CK 7 and CK 19 were performed on CBDL livers showing expression only in the apical submembraneous cytoplasm of cholangiocytes but negative staining of hepatocytes for these CKs (not shown).
Figure 1.
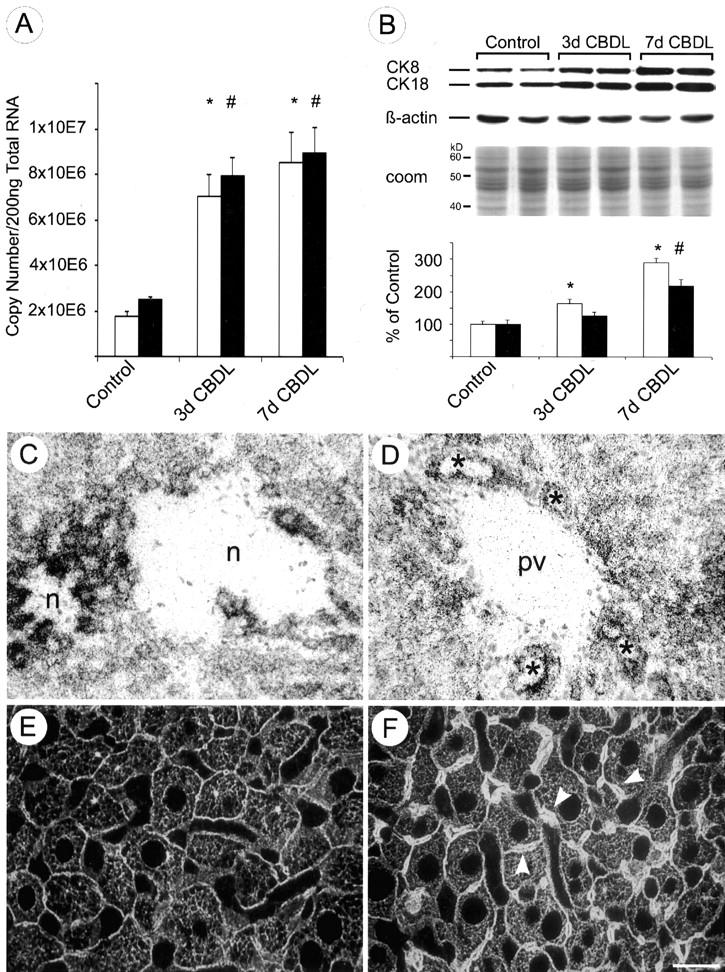
Effects of CBDL on hepatic CK 8 and CK18 expression and the intermediate filament cytoskeleton. A: Competitive reverse transcriptase-polymerase chain reaction revealed an approximate threefold increase of CK 8 (open bars) and an approximate fourfold increase of CK 18 (filled bars) mRNA after CBDL for 3 and 7 days, respectively. Data (means + SEM) are expressed as copy numbers per 200 ng of total RNA (n = 5 animals in each group; * and #, P < 0.05, CBDL versus sham-operated controls). B: CBDL resulted in a twofold and threefold increase of CK 8 protein (open bars) and a 1.5-fold and twofold increase of CK 18 protein (filled bars) after 3 and 7 days, respectively (n = 5 animals in each group; * and #, P < 0.05, CBDL versus sham-operated controls), whereas β-actin expression remained unaffected. Loading of equal amounts of proteins was confirmed by Coomassie blue staining (coom; to avoid redundancy only shown for Figure 1 ▶ ). In situ hybridization for CK 8 mRNA revealed a marked increase in CK 8 mRNA expression in hepatocytes surrounding bile infarcts (n, necrosis) (C) and in hepatocytes in zone 1 (periportal parenchyma) and in bile duct epithelial cells 7 days after CBDL (D) (asterisks indicate bile ducts; pv, portal vein). Immunofluorescence staining using a polyclonal antibody against hepatic CKs (50K160) of control mouse liver (E) and of mouse liver after CBDL (7 days) (F). Note the increased density of the IF network after CBDL, particularly around the dilated bile canaliculi (F, arrowheads). Scale bar, 20 μm. Original magnifications, ×20 (C and D).
CA Induces Hepatic CK 8 and CK 18 Expression
To further discriminate between mechanical (eg, increased canalicular pressure as present in CBDL) and toxic effects (eg, changes in bile acid pool composition) of cholestasis, mice were either fed a diet supplemented with potentially toxic CA or hydrophilic nontoxic UDCA. Feeding of CA (0.1%, 0.5%, 1%) for 7 days led to disseminated liver cell necroses and a significant increase in the number of mitotic hepatocytes. Feeding of 1% CA-supplemented diet was, in addition, associated with dilatation of interlobular bile ducts. Serum transaminase, alkaline phosphatase, and serum bile acid levels were also significantly elevated in CA-fed mice in comparison to controls (for details see Table 2 ▶ ). Feeding of UDCA did not result in liver cell necroses, increased mitotic figures, or elevated serum enzyme levels, but led to a dilatation of medium-sized and larger interlobular bile ducts.
Feeding of CA stimulated CK 8 and CK 18 mRNA and protein expression in a dose-dependent manner (Figure 2, A and B) ▶ . After 7 days of CA (0.1%, 0.5%, and 1%, respectively) feeding CK 8 and CK 18 mRNA levels were significantly increased. To determine whether the increase in CK 8 and CK 18 steady-state mRNA levels resulted in a comparable increase in protein, immunoblotting was performed with liver homogenates from CA-fed and control animals (Figure 2B) ▶ . CA feeding (1%) for 7 days resulted in a significant increase of CK 8 and CK 18 proteins compared to control animals. Interestingly, the increase of CK 8 always exceeded that of CK 18 and the dissociation between CK 8 and CK 18 was particularly prominent in mice fed 1% CA-containing diet. In situ hybridization revealed a marked increase of the CK-8 mRNA-related signal in hepatocytes, and, to a lesser degree, also in bile duct epithelial cells from CA-fed mice (Figure 2D) ▶ . CK 8 and CK 18 overexpression in CA-fed animals was also detected by immunofluorescence microscopy in tissue sections. Staining with a polyclonal antibody against CK 8 and CK 18 revealed in CA-fed mice a cytoplasmic IF skeletal meshwork with increased density, particularly at the periphery of enlarged hepatocytes and around bile canaliculi (Figure 2F) ▶ . Hepatocytes of CA-fed mice did not express CK 7 and CK 19 (not shown). Feeding the nontoxic UDCA had no significant effect on CK 8 and CK 18 mRNA and protein levels (data not shown) and on IF structure at any dose studied (Figure 3C) ▶ .
Figure 2.
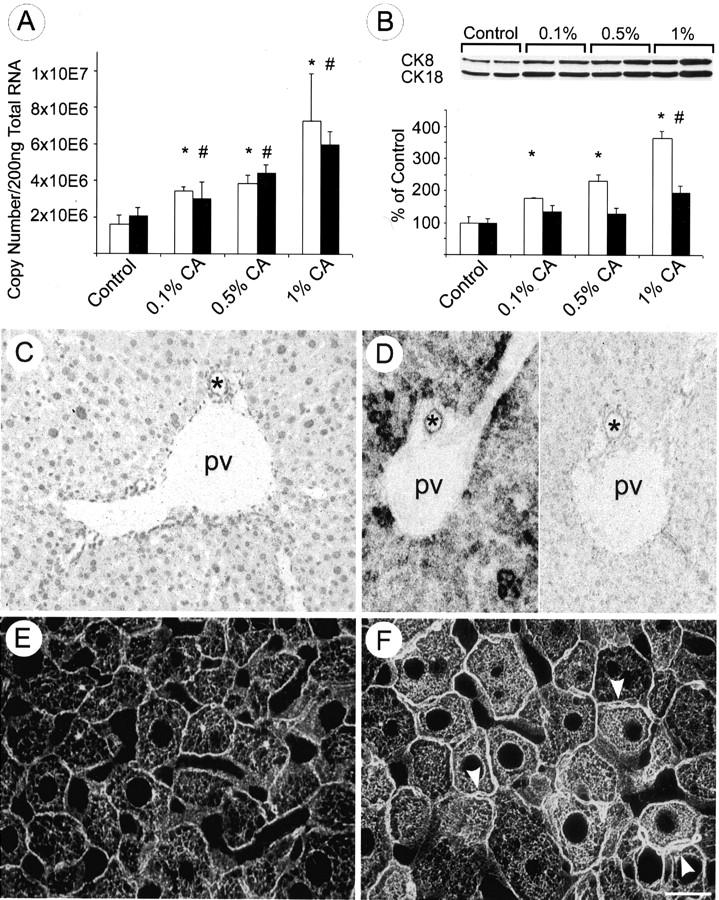
Effects of bile acid feeding on hepatic CK 8 and CK 18 expression and the intermediate filament cytoskeleton. A: Competitive reverse transcriptase-polymerase chain reaction revealed an increase of CK 8 (open bars) and CK 18 (black bars) mRNA after CA (0.1%, 0.5%, 1%) feeding for 7 days. Feeding a diet containing 1% CA led to an approximate threefold increase. Data (means + SEM) are expressed as copy numbers per 200 ng total RNA (n = 5 animals in each group; * and #, P < 0.05, CA-fed animals versus controls). B: CA feeding resulted in a dose-dependent increase of CK 8 protein (open bars) and CK 18 protein (black bars) levels. CK 8 protein levels increased twofold and 2.5-fold in 0.1 and 0.5% CA-fed animals, respectively, whereas CK 18 protein levels only slightly increased. However, 1% CA-fed mice showed 3.5-fold CK 8 and twofold CK18 elevation in comparison to controls. It is noteworthy that the increase of CK 8 clearly exceeded that of CK 18 (n = 5 animals in each group; * and #, P < 0.05, CA-fed animals versus controls). In situ hybridization for CK 8 mRNA in control liver (C) and in CA (1%, for 7 days)-fed mouse liver (D) (left side, anti-sense probe; right side, sense probe) showing that CA feeding resulted in a marked increase in CK 8 mRNA expression particularly in the periportal parenchyma. Immunofluorescence staining of IF cytoskeleton in normal liver (E) and increased density of the CK-IF network in CA-fed mouse liver particularly around the bile canaliculi (F, arrowheads; pv, portal vein; *, bile duct). Scale bar, 20 μm. Original magnifications, ×20 (C and D).
Figure 3.
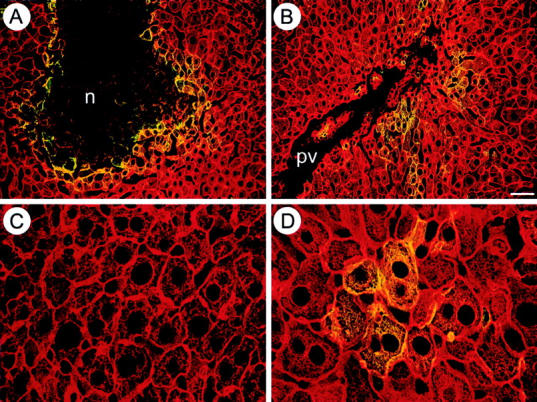
Cholestasis and CA induce phosphorylation of the CK intermediate filament cytoskeleton. Double-immunofluorescence microscopy was performed using the polyclonal CK antibody 50K160 against CK 8 and CK 18 and the monoclonal antibody LJ4 recognizing a phosphorylated epitope of CK 8. Overexpression of CKs after CBDL was accompanied by phosphorylation in cells surrounding bile infarcts (n) (A) and in hepatocytes in acinar zone 1 (B) (pv, portal vein). C: UDCA-fed mice showed normal architecture of the CK-IF cytoskeleton without phosphorylation. D: In contrast, CA-fed mice showed an increased density of the CK-IF network (red), particularly at the cell periphery and around bile canaliculi, accompanied by marked phosphorylation (yellow) in clusters of hepatocytes. Scale bar, 20 μm.
Cholestasis and CA Feeding Induce CK 8 Phosphorylation
Because mutations of CK phosphorylation sites predispose mice to toxic liver injury, and cell stress induces CK phosphorylation 14,15 we studied the influence of obstructive cholestasis and bile acid feeding on the phosphorylation status of CK 8. Overexpression of CKs after CBDL was accompanied by CK phosphorylation in cells surrounding bile infarcts and hepatocytes in acinar zone 1 as revealed by a CK 8 phosphoepitope antibody (Figure 3, A and B) ▶ . Marked IF phosphorylation was also observed in clusters of hepatocytes in CA-fed animals (Figure 3D) ▶ . In contrast, UDCA-fed animals did not show phosphorylation of the CK-IF network (Figure 3C) ▶ .
Discussion
Cholestasis, defined as a decrease or even cessation of bile flow with intrahepatic and systemic retention of bile acids and other biliary constituents, is associated with profound changes of the hepatocyte cytoskeleton. 1-4,27 The pericanalicular CK-IF sheath is altered in various forms of experimental cholestasis. 1,7,9 Mechanical cholestasis, eg, bile duct ligation, leads to an increased density of the pericanalicular CK-IF network. 8,9 This is thought to reflect a compensatory mechanism of the hepatocyte to resist increased canalicular pressure associated with mechanical obstruction of bile flow. 9 Chronic cholestatic liver diseases in humans, such as primary biliary cirrhosis, are also associated with alterations of the CK-IF network including formation of MBs that consist of CK and non-CK components. 10,11 Increased reactivity of cholestatic hepatocytes with polyclonal CK antibodies and changes in the hepatocellular CK expression pattern in various cholestatic liver diseases further argue for a specific role of CK-IF alterations in cholestasis. 27 In addition, overexpression and phosphorylation of CKs have recently been demonstrated in human alcoholic hepatitis and in a mouse model of MB formation induced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) feeding, conditions that are also associated with cholestasis. 18,19,28 In the current study CBDL but also feeding of CA (but not nontoxic UDCA) resulted in marked overexpression of CK 8 and CK 18 and the phosphorylation of CK 8. These findings provide strong evidence for a key role of potentially toxic bile acids in the regulation of CKs, particularly in cholestatic conditions. Consequently, retention of toxic bile acids associated with cholestasis could also be causally involved in cytoskeletal alterations in human liver diseases, such as alcoholic hepatitis and nonalcoholic steatohepatitis, primary biliary cirrhosis, or Wilson’s disease.
The CK-IF network is important for the structural integrity of hepatocytes, 6 but CK proteins in addition seem to have important nonmechanical functions, including protection from toxic stress, as demonstrated by increased sensitivity of CK-8 knockout (CK 8(−/−)) mice to DDC toxicity and of mice overexpressing a dominant-negative human CK 18 serine 52-alanine mutant to griseofulvin toxicity. 6,14,16 Comparative studies with CK 8(−/−)- and CK-18 knockout (CK 18(−/−))-mice demonstrated the overall importance of CK 8 in MB formation and in protection from toxic stress as indicated by enhanced mortality and lack of MB formation in DDC-intoxicated CK 8(−/−) mice. 11,16 Although hepatocytes of both CK 8(−/−) and CK 18(−/−) mice lacked a cytoplasmic IF network, CK 18(−/−) animals did not show an increased mortality after DDC feeding but developed MBs even earlier compared to intoxicated wild-type mice. 16 Interestingly, feeding of potentially toxic CA (but not of the nontoxic hydrophilic UDCA) not only induced hepatic CK expression but resulted in a more pronounced increase of CK 8 in comparison to CK 18 and thus disturbance of the 1:1 CK 8:CK 18 ratio that is essential for correct IF formation. In addition, animals of the experimental groups with the highest bile acid levels showed the most pronounced increase of CK 8 expression, which further underlines the significance of CK 8 in toxic liver injury. Isolated CK 8 could either be degraded or aggregated and thus be the substrate of MB formation. 29
Increased CK synthesis and phosphorylation occurred in hepatocytes in acinar zone 1 and in the vicinity of bile infarcts that are areas with probably the highest bile acid concentrations and resulting toxic stress. Phosphorylation of CKs could represent a cellular defense mechanism and may support hepatocytes in handling bile acid-induced toxic stress, because CK phosphorylation decreases intracellular adenosine triphosphate levels and may enable the hepatocyte to maintain a phosphate reserve. 6 The lack of effects of nontoxic UDCA on CK 8 and CK 18 expression and phosphorylation further supports the concept of a toxic stress response. The detailed mechanisms of how stress-induced CK phosphorylation may protect against certain types of liver injury, however, remains an open question. 6
An alternative, although less attractive, explanation for hepatic up-regulation of CK-IFs in CA-fed mice could be a role of the CK-IF network in transcytosis and targeting of bile acid transporters to the canalicular membrane, similar to the role of other components of the cytoskeleton. 30-34 IFs are closely associated with the canalicular membrane and provide a structural scaffold of the canaliculus, which is believed to be important for its formation and maintenance. 1,2,7,9 However, evidence for the involvement of CK-IF in the regulation of bile flow is only fragmentary and the detailed role of CK-IF in bile formation has not yet been established. 1,6 CK 8(−/−) and transgenic mice with disrupted CK 8/18 network because of ectopic CK 14 expression showed markedly decreased bile flow and bile acid secretion. 12,35 Because canalicular secretion is the rate-limiting step in bile formation, 1,3 it is likely that this defect is located at the canalicular membrane. Interestingly, the livers of these mice are unable to handle higher bile acid concentrations, as shown by a decreased Tmax in response to increased bile acid load, 12,35 which is consistent with defective vesicular targeting of bile acid transporters to the bile canalicular membrane. In addition, a possible role for CKs in transcellular vesicle transport has recently been discussed based on the finding of abnormally dispersed zymogen granules in pancreatic acinar cells in mice expressing human CK 8 or a dominant-negative mutant transforming growth factor-βII receptor. 36 The authors speculated that the observed pancreatic insufficiency in these mice could be because of a defect in the processing and/or secretion of zymogen granules, suggesting that CKs may also be involved in vesicular targeting. In addition, recent studies demonstrated binding affinity of CK 18 for bilirubin, and the authors speculated that CK 18 may play a role as a membrane reservoir in the transport and secretion of bile pigments. 37 Together with our finding of CK 8 and CK 18 overexpression leading to an increased density of the CK-IF network and a thickened pericanalicular sheath in livers exposed to increased bile acid levels, a direct or indirect role of the CK-IFs in vesicular transport and targeting or bile secretion could be possible. However, the lack of effect of UDCA (known to stimulate bile secretion) on CK expression in the present study argues against a direct role of CKs in bile secretion. 38,39
This study provides the first evidence that bile acids lead to marked alterations of the CK-IF network. We could demonstrate that up-regulation of CK 8 and CK 18 in cholestatic conditions is not merely because of mechanical stress but mediated by potentially toxic bile acids. Furthermore, toxic bile acids induce CK phosphorylation, which could reflect a stress response against the toxic effects of bile acids.
Acknowledgments
We thank Dr. Bishr M. Omary (Department of Medicine, Palo Alto VA Medical Center and Stanford University, Palo Alto, CA) for providing antibodies against hyperphosphorylated CK epitopes, Dr. W. Erwa (Graz) and colleagues for performing liver function tests, Dr. R. Aigner (Graz) and colleagues for measurement of serum bile acid levels, and Dr. G. J. Krejs (Graz) for critical reading of the manuscript.
Footnotes
Address reprint requests to Helmut Denk, M.D., FRCPath., Department of Pathology, University of Graz, Auenbruggerplatz 25, A-8036 Graz, Austria. E-mail: helmut.denk@kfunigraz.ac.at.
Supported by grants 7401-MOB from the Austrian Science Foundation (to K. Z.), 8522 and 7171 from the Jubilee Funds of the Austrian National Bank (to M. T.), and the Joseph Skoda Prize from the Austrian Society of Internal Medicine (to M. T.).
Presented in part at the annual meeting of the American Association for the Study of the Liver Diseases, November 1999, Dallas, Texas, and published in abstract form (Hepatology 1999, 30:464A).
References
- 1.Trauner M, Meier PJ, Boyer JL: Molecular pathogenesis of cholestasis. N Engl J Med 1998, 17:1217-1227 [DOI] [PubMed] [Google Scholar]
- 2.Phillips MJ, Poucell S, Oda M: Mechanisms of cholestasis. Lab Invest 1986, 54:593-608 [PubMed] [Google Scholar]
- 3.Trauner M, Fickert P, Zollner G, Stauber RE, Zatloukal K, Denk H, Krejs GJ: Cellular and molecular mechanisms of cholestasis. Blum HE Maier KP Sauerbruch T Stadler GA eds. Liver Cirrhosis and Its Development. 2000, :pp 3-20 Kluwer Academic Publishers, Dodrecht [Google Scholar]
- 4.Reichen J, Krähenbühl, Zimmermann H: Impact of cholestasis on hepatic function: retention of cholephiles and their potential targets. Gentilini P eds. Cholestasis 1994, :167-175 Elsevier Science [Google Scholar]
- 5.Katsuma Y, Marceau N, Ohla M, French SW: Cytokeratin intermediate filaments of rat hepatocytes: different cytoskeletal domains and their three-dimensional structure. Hepatology 1988, 8:559-568 [DOI] [PubMed] [Google Scholar]
- 6.Omary MB, Ku N: Intermediate filament protein of the liver. Emerging disease association and functions. Hepatology 1997, 25:1043-1048 [DOI] [PubMed] [Google Scholar]
- 7.Kawahara H, Cadrin M, Perry G, Autilio-Gambetti L, Swierenga SHH, Metuzals J, Marceau N, French SW: Role of cytokeratin intermediate filaments in transhepatic transport and canalicular secretion. Hepatology 1990, 11:434-448 [DOI] [PubMed] [Google Scholar]
- 8.Ohta M, Marceau N, French SW: Pathologic changes in the cytokeratin pericanalicular sheath in experimental cholestasis and alcoholic fatty liver. Lab Invest 1988, 59:60-74 [PubMed] [Google Scholar]
- 9.Song JY, Van Noorden CJF, Frederiks WM: Alterations of hepatocellular intermediate filaments during extrahepatic cholestasis in rat liver. Virchows Arch 1997, 430:253-260 [DOI] [PubMed] [Google Scholar]
- 10.Gerber MA, Orr W, Denk H, Schaffner F, Popper H: Hepatocellular hyalin in cholestasis and cirrhosis: its diagnostic significance. Gastroenterology 1973, 64:89-98 [PubMed] [Google Scholar]
- 11.Denk H, Stumptner C, Zatloukal K: Mallory body revisited. J Hepatol 2000, 32:689-702 [DOI] [PubMed] [Google Scholar]
- 12.Nam-On K, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Oshima RG, Omary MB: Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J Clin Invest 1996, 98:1034-1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ku N, Michie S, Oshima RG, Omary MB: Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J Cell Biol 1995, 131:1303-1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ku N, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Omary MB: Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J Cell Biol 1998, 143:2023-2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao J, Ku N, Omary MB: Stress, apoptosis, and mitosis induce phosphorylation of human keratin 8 at ser73 in tissues and cultured cells. J Biol Chem 1995, 272:17565-17573 [DOI] [PubMed] [Google Scholar]
- 16.Zatloukal K, Stumptner C, Lehner M, Denk H, Baribault H, Eshkind LG, Franke WW: Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am J Pathol 2000, 156:1263-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ku N, Gish R, Wright TL, Omary MB: Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med 2001, 344:1580-1587 [DOI] [PubMed] [Google Scholar]
- 18.Stumptner C, Omary MB, Fickert P, Denk H, Zatloukal K: Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in Mallory bodies. Am J Pathol 2000, 156:77-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenner L, Trauner M, Fickert P, Stauber RE, Eferl R, Denk H, Zatloukal K: Overexpression of keratin-8 and -18 mRNA associated with Mallory body formation in patients with alcoholic hepatitis. Hepatology 1998, 28:A564 [Google Scholar]
- 20.Trauner M, Arrese M, Soroka CJ, Anathanarayanan M, Koeppel TA, Schlosser SF, Suchy FJ, Keppler D, Boyer JL: The rat canalicular conjugate export pump (Mrp2) is downregulated in intrahepatic and obstructive cholestasis. Gastroenterology 1997, 113:255-264 [DOI] [PubMed] [Google Scholar]
- 21.Van Nieuwkerk CM, Oude Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, Van Den Bergh Weerman MA, Offerhaus GJ: Effects of ursodeoxycholate and cholate feeding on liver diseases in FVB mice with disrupted mdr2 P-glycoprotein gene. Gastroenterology 1996, 111:165-171 [DOI] [PubMed] [Google Scholar]
- 22.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227:680-685 [DOI] [PubMed] [Google Scholar]
- 24.Towbin H, Staehelin T, Gordon J: Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci 1979, 76:1350-1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutter H, Zatloukal K, Winter G, Stumptner C, Denk H: Disturbance of keratin homeostasis in griseofulvin-intoxicated mouse liver. Lab Invest 1993, 69:576-582 [PubMed] [Google Scholar]
- 26.Zatloukal K, Denk H, Spurej G, Lackinger E, Preisegger KH, Franke WW: High molecular weight component of Mallory bodies detected by monoclonal antibody. Lab Invest 1990, 62:427-434 [PubMed] [Google Scholar]
- 27.Van Eyken P, Sciot R, Desmet VJ: A cytokeratin immunohistochemical study of cholestatic liver disease: evidence that hepatocytes can express ‘bile duct-type’ cytokeratins. Histopathology 1989, 15:125-135 [DOI] [PubMed] [Google Scholar]
- 28.Fickert P, Trauner M, Fuchsbichler A, Riegelnegg D, Eferl R, Kenner L, Zatloukal K, Denk H: The role of Kupffer cells on cytokeratin 8/18 expression in lipopolysaccharide or 3,5-diethoxycarbonyl-1,4-dihydrocollidine induced hepatic injury. Hepatology 1999, 30:A1567 [Google Scholar]
- 29.Stumptner C, Fuchsbichler A, Lehner M, Zatloukal K, Denk H: Sequence of events in the assembly of Mallory body components in mouse liver: clues to the pathogenesis and significance of Mallory body formation. J Hepatol 2001, 34:665-675 [DOI] [PubMed] [Google Scholar]
- 30.Boyer JL, Soroka CJ: Vesicle targeting to the apical domain regulates bile excretory function in isolated rat hepatocytes couplets. Gastroenterology 1999, 109:1600-1611 [DOI] [PubMed] [Google Scholar]
- 31.Dranoff JA, McClure M, Burgstahler AD, Denson LA, Crawford AR, Crawford JM, Karpen SJ, Nathanson MH: Short-term regulation of bile acid uptake by microfilament-dependent translocation of rat ntcp to the plasma membrane. Hepatology 1999, 30:223-229 [DOI] [PubMed] [Google Scholar]
- 32.Reichen J, Bermann MD, Berk PD: The role of microfilaments and microtubules in taurocholate uptake by isolated rat liver cells. Biochim Biophys Acta 1981, 643:126-133 [DOI] [PubMed] [Google Scholar]
- 33.Zegers MM, Zaal KJ, Van Ijzendoorn SC, Klappe K, Hoekstra D: Actin filaments and microtubules are involved in different membrane traffic pathways that transport sphingolipids to the apical surface of polarized HepG2 cells. Mol Biol Cell 1998, 9:1939-1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Nieuwerk CMJ, Elferink ORPJ, Groen AK, Ottenhoff R, Tytgast GNJ, Dingemans KP, van den Bergh Weerman MA, Offerhaus GJ: Effects of ursodeoxycholate and cholate feeding on liver disease in fvb mice with a disrupted mdr2 p-glycoprotein gene. Gastroenterology 1996, 111:165-171 [DOI] [PubMed] [Google Scholar]
- 35.Loranger A, Duclos S, Grenier A, Price J, Wilson-Heiner M, Baribault H, Marceau N: Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am J Pathol 1997, 151:1673-1683 [PMC free article] [PubMed] [Google Scholar]
- 36.Casanova ML, Bravo A, Ramirez A, Morreale de Esobar G, Were F, Merlino G, Vidal M, Iorcono JL: Exocrine pancreatic disorders in transgenic mice expressing human keratin 8. J Clin Invest 1999, 103:1587-1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohishi K, Mita T, Odani S, Takahashi T, Aoyagi Y, Munukata H, Tada K, Isemura S, Hsu CC, Isemura M: Bilirubin binding activity of cytokeratin 18 isolated from the porcine liver. Cell Struct Funct 1998, 23:325-331 [DOI] [PubMed] [Google Scholar]
- 38.Trauner M, Graziadei IW: Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver disease. Aliment Pharm Ther 1999, 13:979-996 [DOI] [PubMed] [Google Scholar]
- 39.Fickert P, Zollner G, Fuchsbichler A, Pojer C, Zenz R, Stumptner C, Pojer C, Zenz R, Lammert F, Stieger B, Meier PJ, Zatloukal K, Denk H, Trauner M: Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transport expression in mouse liver. Gastroenterology 2001, 120:170-183 [DOI] [PubMed] [Google Scholar]