

Abstract
Impairment of consciousness and other signs of cerebral dysfunction are common complications of severe Plasmodium falciparum malaria. Although the majority of patients make a complete recovery a significant minority, particularly children, have sequelae. The pathological process by which P. falciparum malaria induces severe but usually reversible neurological complications has not been elucidated. Impairment of transport within nerve fibers could induce neurological dysfunction and may have the potential either to resolve or to progress to irreversible damage. β-amyloid precursor protein (β-APP) immunocytochemistry, quantified using digital image analysis, was used to detect defects in axonal transport in brain sections from 54 Vietnamese cases with P. falciparum malaria. The frequency and extent of β-APP staining were more severe in patients with cerebral malaria than in those with no clinical cerebral involvement. β-APP staining was often associated with hemorrhages and areas of demyelination, suggesting that multiple processes may be involved in neuronal injury. The age of focal axonal damage, as determined by the extent of the associated microglial response, varied considerably within tissue sections from individual patients. These findings suggest that axons are vulnerable to a broad range of cerebral insults that occur during P. falciparum malaria infection. Disruption in axonal transport may represent a final common pathway leading to neurological dysfunction in cerebral malaria.
Cerebral malaria (CM) is a diffuse, potentially reversible, encephalopathy, caused by infection with the protozoan parasite Plasmodium falciparum. CM presents clinically with convulsions and coma, and carries 15 to 20% mortality. The malaria parasite invades and develops within erythrocytes that sequester in the cerebral microvasculature and other vital organs by adhesion to specific endothelial receptors. 1,2 The pathophysiological consequences of severe malaria infection have not been satisfactorily resolved. In particular it is not known how parasitized red blood cells, which remain within the vascular space, influence parenchymal brain function to induce coma and death. Histopathological studies reveal a variety of neurological abnormalities including chromatolysis, neuronophagia, and decreased glial cell numbers. 2-5 These changes may result from several coexisting pathological processes including hypoxia, hypoglycemia, cerebral swelling, hemorrhage, and inflammation. Despite these findings in patients who have died from CM, neurological recovery from CM is usually rapid and the majority of patients who survive make a complete recovery within several days.
In common with other neurological conditions such as encephalitis, stroke, or neurodegeneration, the response of the brain to an initial parasite-induced insult may be limited to a small number of common mechanisms that can continue to operate after eradication of parasitized red blood cells and contribute independently. If these can be identified they represent attractive targets for novel neuroprotective therapies in CM that could be used as adjuncts to anti-malarial chemotherapy. Axonal injury (AI) is thought to be a common pathway of cerebral injury that occurs in diseases such as multiple sclerosis 6,7 and stroke. 8 Axonal injury disrupts neural integrity, the distribution of neurosecretory granules, and the transport of enzymes and chemicals involved in the formation of neurotransmitters and substances associated with trophic activity. β-amyloid precursor protein (β-APP) is a protein that is normally transported along the axon, and accumulates at the sites of AI. 9 Positive β-APP immunostaining of axons may identify axons with reversible structural and biochemical changes. 10 Axonal damage may occur gradually, leaving a window for therapeutic intervention during the early stages. 11,12
In this article we have examined evidence for AI in postmortem brain tissue from adult Vietnamese patients who died from P. falciparum malaria. We established a method to quantify the extent of AI using digital image analysis of APP-staining patterns, and determined whether there was any regional susceptibility to AI and related the extent of AI to clinical disease parameters. To define further the causative mechanisms we have correlated AI with other neuropathological features seen in fatal malaria such as demyelination, hemorrhage, sequestration, and glial responses.
Materials and Methods
Case Selection
Specimens were taken at autopsy from adult Vietnamese patients who had died of severe P. falciparum malaria on the Malaria Research Ward, Center for Tropical Diseases, Ho Chi Minh City, Viet Nam. On admission, the presence of P. falciparum malaria parasites in the peripheral blood was detected by routine parasitological examination in all patients. The patients were part of a large double-blind trial of artemether versus quinine for the treatment of severe malaria in Viet Nam. 13 Two groups were defined prospectively, CM (n = 28) and non-CM (n = 26). CM was defined according to established World Health Organization guidelines as a Glasgow Coma score of 11 or less during the episode of severe malaria, 14 other causes of unconsciousness having been excluded, (eg, hypoglycemia, meningitis, or other encephalopathy) by clinical, biochemical, and cerebrospinal fluid (CSF) examination. Non-CM patients were those dying from severe malaria without coma, who had a range of clinical features typical of other vital organ system complications. A full autopsy was performed within 24 hours of death with consent from the family. All autopsy and specimen collection protocols were approved by the Ethical and Scientific Committee of the Center for Tropical Diseases.
Selection of Controls
Control cases were from a number of different causes of death collected at postmortem at the John Radcliffe Hospital, Oxford, UK, where specific written consent for retention of brain tissues for research purposes had been given. Sampling protocols and use of control UK tissues was approved by the Central Oxford Research Ethics Committee. The controls were cases in which standard neuropathological examination was normal. These included cases in which no history of neurological illness was recorded (acute and chronic deaths from other causes) and cases in which there was a past history of previous neurological illness that was not the direct cause of death. Ten control cases were age-matched to the malaria cases 15 and four controls were from elderly patients who died from a nonneurological illness. Autopsy delays varied but were predominantly conducted within 24 to 48 hours of death.
Specimen Collection
After brain fixation in 10% formalin for a minimum of 4 weeks, a formal brain cut was performed and samples taken systematically from various areas of the brain including cortex with deep white matter, internal capsule, pons, and cerebellum. These were embedded in paraffin and processed using standard methods. Hematoxylin and eosin slides of selected areas of the brainstem were examined to judge the pathological features of the disease, and unstained sections cut onto Snowcoat Extra slides (Surgipath, St. Neots, UK) for immunostaining.
β-APP Immunohistochemistry
β-APP immunohistochemistry was used to detect defects in fast axonal transport in brain sections. Sections 8 μm in thickness were dewaxed in Histoclear and rehydrated through graded alcohols to water. The sections were then microwaved in Tris-ethylenediaminetetraacetic acid, with the buffer being brought to boiling point throughout a 12-minute period. They were then treated with formic acid (Merck, Lutterworth, UK) for 5 minutes and rinsed thoroughly in Tris buffer. The sections were then incubated overnight at room temperature in 1:200 dilution of stock β-APP antibody clone 22c11 (Chemicon, Harrow, UK), diluted in Tris-buffered saline and 1% fetal calf serum. Bound antibody was visualized by incubation with biotinylated goat anti-mouse/rabbit immunoglobulin (DAKO, Cambridge, UK) followed by Streptavidin ABComplex alkaline phosphatase and visualized with the new fuchsin substrate system (DAKO, UK). Negative controls comprised sections immunostained as above apart from omission of the primary antibody. Positive controls included brain sections from patients with global hypoxic damage, infarction, and diabetic coma (data not shown). Appropriate concentrations of primary antibodies were determined using optimization on control and case tissues.
Quantitation of Axonal Injury and Image Analysis
AI was quantitated using a modified version of a semiautomated method described by Gentleman and colleagues. 16 Briefly, tissue sections were digitized using a Nikon LS-2000 slide scanner. Regions of focal axonal damage were selected by density thresholding in Adobe Photoshop 4.0 and the areas were calculated and summated using the public domain NIH image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) (Figure 1; A to C ▶ ). Neuronal cell bodies were excluded from the analysis by setting the minimum particle size to be detected as 5 pixels. The total area of the tissue section was then calculated. The amount of axonal damage or APP load was expressed as the area of tissue covered with β-APP staining divided by the total area of the section in square μm. One section was counted per site. Average β-APP load was calculated to provide an indication of the impact of AI on the brain as a whole and to adjust for the variability between different brain regions.
Figure 1.
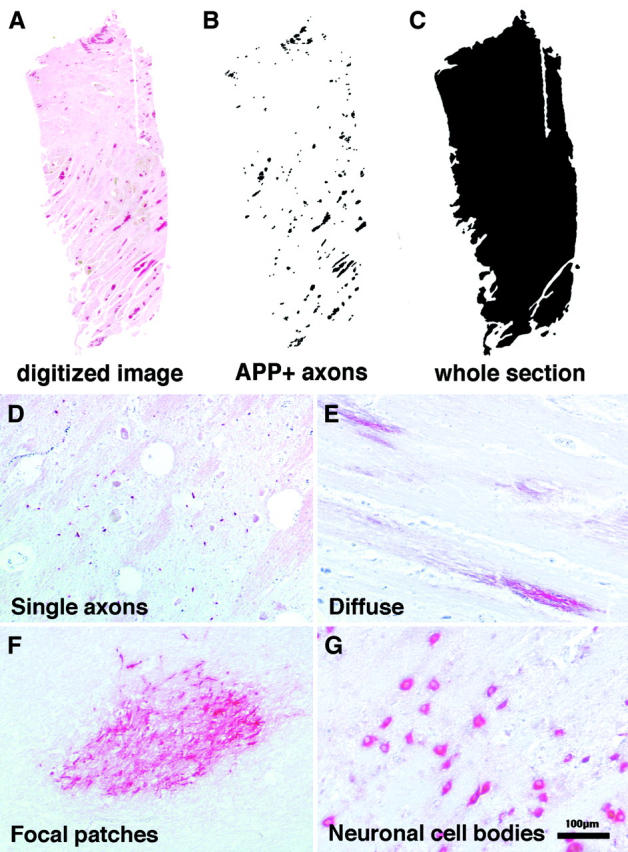
A–C: Quantitation of AI or APP load. A: Tissue sections were digitized using a Nikon LS-2000 slide scanner. B: Regions of focal axonal damage were selected by density thresholding and the areas were calculated and summated using NIH image. C: The total area of the tissue section was calculated. The amount of axonal damage or APP load was expressed as the area of tissue with APP staining divided by the total area of the section. D–G: Four different patterns of β-APP staining observed in the brains of fatal P. falciparum malaria cases: single axons (D), diffuse (E), focal patches (F), and neuronal cell bodies (G).
Statistics
Statistical analysis was performed using the Stata 6 (StataCorp, College Station, TX) program. Data were analyzed using nonparametric tests (Kruskal-Wallis test, Spearman rank correlation, and Fischer exact test). No adjustments for multiple comparisons were made, although for the purposes of interpretation and discussion P < 0.01 was regarded as significant.
Other Neuropathological Features
Serial sections were stained for other neuropathological features that have been reported in fatal malaria including: demyelination, visualized with Luxol Fast Blue Cresyl Violet; hemorrhage, visualized with an anti-glycophorin antibody (culture supernatant, JC159; DAKO); glial responses, visualized with an anti-glial fibrillary acidic protein (GFAP) antibody (culture supernatant, GF2; DAKO) for astrocytes, and an anti-CD68 (culture supernatant, clone KPI; DAKO) antibody for microglia. Those sections for GFAP and CD68 were microwaved in Tris-ethylenediaminetetraacetic acid and the rest of the antibody procedure was as described above.
Types of GFAP-staining patterns examined included: increased density of GFAP staining; increased process complexity or swelling of astrocytes around the ventricles, pial surface, and parenchymal vessels. Clasmatodendrosis, defined as fragmentation or beading of GFAP-labeled astrocyte processes, suggesting degenerative changes in astrocytes was also examined. Types of CD68-staining patterns included staining of intravascular leukocytes (predominantly monocytes but also polymorphonuclear leukocytes to a lesser degree); perivascular macrophages, defined as CD68+ cells found within the Virchow-Robin space; paravascular macrophages, CD68+ cells with a globoid morphology close to vessels but within the parenchyma proper; and finally hypertrophied or hyperplastic microglia with the classic ramified morphology. In addition, an approximate time scale of microglial responses to AI was used to estimate the timing of injury in individual lesions. 17,18 Slides were read blind to the clinical details of the patient by two independent observers (IM and GT).
Results
Patterns of β-APP Staining in Severe Malaria
Four different patterns of β-APP staining were observed in the brains of fatal P. falciparum malaria cases. These included strong positive staining of single axons (Figure 1D) ▶ , diffuse patches of parenchymal staining (Figure 1E) ▶ , more focal parenchymal patches (Figure 1F ▶ and Figure 2; A, C, E, and G ▶ ), and staining in neuronal cell bodies (Figure 1G) ▶ . Combinations of the four different patterns could be found on the same section.
Figure 2.
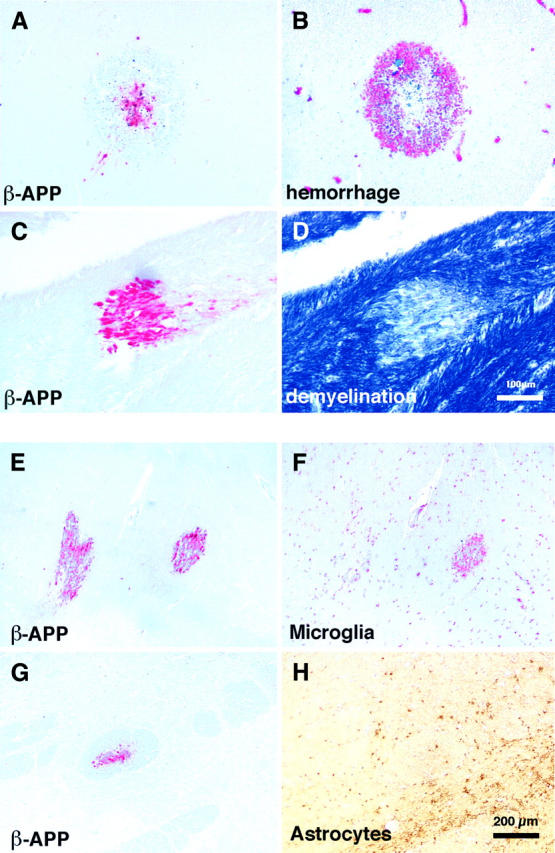
A and B: Serial sections stained for either β-APP to visualize areas of AI (A) or glycophorin to identify hemorrhages (B). In this region of the brain AI was associated with the center of ring hemorrhages. C and D: Serial sections stained for β-APP to visualize areas of AI (C) or Luxol Fast Blue Cresyl Violet to identify demyelination (D). In this case AI was associated with demyelination. E and F: Serial sections stained for either β-APP to visualize areas of AI (E) or CD68 to identify microglia (F). In this region of the brain one focal region of AI was associated with a microglial response whereas the other focus only showed a mild microglial response. This suggests different ages of the axonal lesions. G and H: Serial sections stained for β-APP to visualize areas of AI (G) or an anti-GFAP antibody to identify astrocytes (H). Invariably, astrocytes did not respond to axonal lesions.
The single-axon staining pattern was often seen in axonal tracts that had been cut transversely (Figure 1D) ▶ . In longitudinal sections this pattern was often seen in mildly swollen axonal segments (Figure 1E) ▶ or as a punctuate profile along an axonal segment. The intensity of β-APP staining in the single axons and the diffuse parenchymal staining pattern were not as intense as that found in the focal patches and often lacked an associated microglial response, suggesting that these were fresher lesions. The diffuse staining pattern was characterized by the lack of clear demarcation of the lesion boundaries. Occasionally, large areas of tissue sections were covered with waves of diffuse axonal staining. Although the lesion borders were not clearly demarcated, the β-APP product was confined exclusively to the axon cylinder, with negligible background staining. It was often difficult to associate diffuse AI with other pathological features. The focal patches pattern of AI were invariably intensely immunoreactive for β-APP. The areas of injury were clearly demarcated and consisted of up to hundreds of linear arrays of damaged axonal segments that appeared as a spectrum of focally disrupted continuous axonal swellings to grossly swollen, club-shaped or, on rare occasions, disconnected axonal bulbs (Figure 1F ▶ and Figure 2; A, C, E, and G ▶ ). The number of patients with diffuse or focal AI somewhere in the brain was significantly higher in CM patients compared with non-CM patients (Table 1) ▶ .
Table 1.
The Number of Patients with Axonal Injury Was Significantly Higher in the CM Group Compared with Non-CM or Control Groups
| Patient group | No. of patients with AI | No. of patients without AI | Chi P value (versus control) | Chi P value (versus non-CM) |
|---|---|---|---|---|
| Control | 6 | 8 | — | 0.26 |
| Non-CM | 11 | 15 | 0.26 | — |
| CM | 23 | 5 | 0.01 | 0.002 |
Correlation between AI and Other Neuropathological Features
Focal patches were often associated with hemorrhage (14 of 49, 29% of patients; Figure 2, A and B ▶ ) or bald patches of secondary demyelination (19 of 47, 40% of patients; Figure 2, C and D ▶ ). However, in some cases, it was clear that AI was uniquely and directly the result of hemorrhage. In these cases, staining was found exclusively within the center of ring hemorrhages (Figure 2, A and B) ▶ . We did not observe an exclusively perivascular distribution of AI within a single section. However, there was a positive correlation between average AI and vascular sequestration (P = 0.0001).
There was no significant correlation between the presence or absence of intravascular leukocytes and CM or AI. Intravascular leukocytes were not an uncommon finding being found in 78% of malaria cases (Figure 3A) ▶ reflecting parasite infection. The degree of intravascular leukocyte sequestration varied, with some vessels showing individual cells, whereas others showed packing with large plugs of cells (Figure 3A) ▶ . The leukocytes were predominantly CD68+ monocytes but mixed populations were also found. In addition, no correlation was found between the presence or absence of peri/paravascular macrophages or perineuronal macrophages and CM or AI. Enhanced CD68+ perivascular (Figure 3B) ▶ , paravascular (Figure 3C) ▶ , and perineuronal macrophage (Figure 3D) ▶ responses were found in 78%, 48%, and 71% of the malaria patients, respectively.
Figure 3.
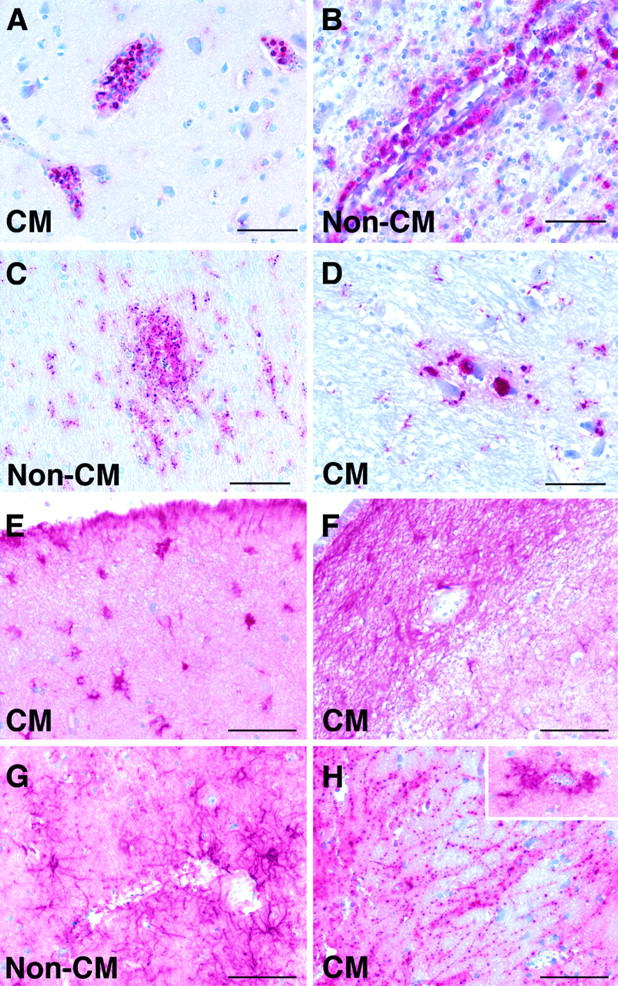
Patterns of CD68 (A–D) and GFAP (E–H) labeling in the brains of patients who died with CM or non-CM. A: Strong labeling of intravascular leukocytes, predominantly monocytes. B: Strong labeling of perivascular macrophages in the Virchow-Robin space. C: Ramified parenchymal microglia in the paravascular location. D: Foamy perineuronal microglia/macrophages. E: Hypertrophied astrocytes in the subpial region. F: Astrogliosis in the subventricular region. G: Perivascular astrocyte response. H: Clasmatodendrosis, suggesting degenerative changes in astrocytes. Inset: Strong perivascular astrocyte response in a different region of the same patient’s brain. Scale bar, 100 μm.
Age of Axonal Lesions and Relationship to Glial Responses
There was considerable heterogeneity in the estimated age of the AI within individual patients. Whether the AI occurred during the agonal stage or considerably before death could be estimated by the extent of the associated glial response, associated secondary pathological changes, and the extent of AI along the spectrum of changes described above. Twenty-nine of 34 (85%) patients showed microglial clusters around APP+ axons, suggesting that the lesions were several days old. This value also included those patients who had combinations of AI lesions with and without associated microglial responses on the same section (Figure 2, E and F) ▶ . It was clear that in some patients the AI in different regions of the same section developed simultaneously whereas in other cases they occurred at different times.
We saw no focal astrocyte responses to AI (Figure 2, G and H) ▶ and there was no significant association between astrocyte responses and CM. However, astrocyte responses were not uncommon in patients with severe malaria: 52% of severe malaria patients showed some degree of generalized astrogliosis (Figure 3) ▶ , 73% showed astrocyte responses in the subpial region (Figure 3E) ▶ , 76% in the parenchymal perivascular region (Figure 3, G and H ▶ , insert), and 33% in the subventricular regions (Figure 3F) ▶ . Clasmatodendrosis (Figure 3H) ▶ was found in 20 patients (36%) with severe malaria but was not associated with CM or AI. There was a correlation between the occurrence of clasmatodendrosis and hemorrhages in the same section (P = 0.03).
Distribution of Axonal Lesions
There also was heterogeneity within and between cases in the extent of AI in different brain regions. In some cases, it was evident that AI occurred within one brain region before another was affected. The APP load was calculated for each brain region analyzed: cortex including deep white matter, internal capsule, pons, and cerebellum (Table 2 ▶ and Figure 4 ▶ ). Further, an overall average APP load for each individual patient was calculated. There were significant differences in the regional β-APP staining in the malaria group as a whole compared with controls, with the exception of the cortex where no significant difference was found (P = 0.57). However, separate analyses comparing the amount of AI in patients dying of CM and those dying of non-CM showed a significantly higher degree of AI in CM in cortex (P = 0.01), internal capsule (P = 0.01), and pons (P = 0.0003) as well as the overall average over all brain regions (P = 0.002). However, there was no significant difference between the two groups in the amount of AI in the cerebellum (P = 0.25). There were highly significant differences between the average amount of AI and region distributions in CM patients compared with controls (P < 0.01) with the exception of the cortex (P = 0.13). There were no significant differences between non-CM and control patients in the amount of AI for any brain region (cortex, P = 0.58; internal capsule (IC), P = 0.08; pons, P = 0.98; cerebellum (CB), P = 0.05). These results are summarized in Figure 4 ▶ .
Table 2.
The Extent of Axonal Injury Was Significantly Higher in CM Patients Compared with Non-CM and Control Patients
| Patient group | Brain region APP load ± SD | Average APP load (average ± SD) | |||
|---|---|---|---|---|---|
| Cortex | Internal capsule | Pons | Cerebellum | ||
| Control | 0.2 ± 0.4 | 0.06 ± 0.2 | 0.005 ± 0.01 | 0 | 0.06 ± 0.2 |
| Non-CM | 0.5 ± 2.4 | 0.8 ± 2.1 | 0.04 ± 0.1 | 0.06 ± 0.1 | 0.3 ± 0.9 |
| CM | 0.4 ± 0.8 | 1.7 ± 3.2†§ | 0.9 ± 1.6*¶ | 0.3 ± 0.6† | 0.9 ± 1.3‡¶ |
*P < 0.01 compared with controls;
†, P < 0.001 compared with controls;
‡, P < 0.0001 compared with controls;
§P < 0.01 compared with non-CM;
¶P < 0.001 compared with non-CM. The amount of axonal damage or APP load was expressed as the area of tissue covered with APP staining divided by the total area of the section.
Figure 4.

Graphs showing APP load in various areas of the brain for control, non-CM, and CM patient groups. Areas analyzed included cortex with deep white matter, internal capsule, pons, and cerebellum. Each dot represents an individual patient.
Clinicopathological Correlations of Axonal Injury
To investigate the significance of these patterns of AI, we performed a detailed clinicopathological correlation between the average AI and APP load within individual brain regions and the presence of a number of clinical and biochemical parameters using all malaria patients. These included the time to death from admission; incidence and duration of convulsions; coma duration, admission and lowest Glasgow coma score; hematocrit; plasma levels of glucose, creatinine, and lactate; CSF pressure; CSF protein and white cell count. Clinical data analyzed included the number of World Health Organization criteria confirming the diagnosis of severe malaria in an individual patient as a measure of the severity of multiorgan disease, and the presence of individual criteria including shock, pulmonary edema, hyperparasitemia, jaundice, anemia, and acute renal failure. Of all these parameters only Glasgow coma score, CSF protein, plasma lactate, and time to death showed significant correlation with AI staining or load, although not in all brain regions examined (results are summarized in Figures 5 and 6 ▶ ▶ ).
Figure 5.

Graphs showing APP load versus clinical findings in individual patients, including CSF pressure, hypoglycemia, acute renal failure, number of severe malaria World Health Organization criteria, diagnosis of CM, and CM alone. Each dot represents an individual patient.
Figure 6.

Graphs showing APP load versus plasma lactate on admission, CSF protein, lowest Glasgow coma score, and time until death. Each dot represents an individual patient. APP load (%) = area of tissue covered with injured axons divided by the total area of the section. Average APP load = average of APP load found in cortex, internal capsule, pons, and cerebellum of an individual patients.
The strongest correlation with AI was the Glasgow coma score (Figure 6) ▶ . There were positive associations (negative correlation) between depth of coma as judged by Glasgow coma score and the degree of APP load, ie, the worse the coma the more APP, in all brain regions with the exception of the cerebellum. Significant correlations existed between average AI or AI in the internal capsule and CSF protein (Figure 6) ▶ . There were also significant correlations between average AI or AI in the pons and plasma lactate levels on admission and time until death (Figure 6) ▶ . Negative correlations were found between admission levels of creatinine as a measure of renal failure (Figure 5) ▶ and AI in the cortex, pons, and cerebellum but not in the internal capsule or when AI was expressed as a whole brain average. There was no augmentation of AI with increasing numbers of severe criteria (Figure 5) ▶ . There were five patients with CM alone, without other organ complications. The average AI load varied considerably within this group (0.0009, 0.03, 0.4, 0.82, 4.51; Figure 5 ▶ ).
Discussion
This is the first detailed clinicopathological study of P. falciparum malaria to provide a quantitative association between the clinical manifestations of central nervous system infection and the mechanisms of neurological injury during disease. It suggests a unifying mechanism that could explain the induction of neurological dysfunction with the potential to resolve or progress to irreversible damage during severe malaria infection. In contrast to our previous analyses of cerebral cellular stress and injury patterns, 15 the degree and extent of AI distinguishes between the groups of patients infected with P. falciparum with and without neurological complications during life. The only other pathological correlate that in our series, and in previous studies, quantitatively distinguishes CM from non-CM in this group of patients was the intensity of sequestration of parasitized erythrocytes within cerebral microvessels. In this study hemorrhage, myelin disruption, glial reaction, and the presence of intravascular leukocytes did not distinguish between the two groups. This does not necessarily exclude a contribution of these elements to the neuropathology of CM, as these responses are more dynamic and thus likely to change dramatically within short periods of time whereas the footprint of AI remains stable for several weeks.
Axons respond differently to changes in central nervous system metabolism compared with neurons or glial cells. Axons often extend for great distances from their cell bodies of origin, and may possibly therefore depend on local production of ATP to maintain ion gradients and sustain energy-consuming functions. They are therefore susceptible to ischemic or toxic damage in several different vascular territories. In P. falciparum malaria sequestration is highly variable between microvessels. The metabolic isolation means that axons may suffer energy deprivation that is independent of neuron cell bodies. 19 This may explain why we found no correlation with specific patterns of neuronal or glial injury in a subset of these patients 15 compared to the finding of significantly increased levels of AI in CM cases.
Although AI was a significant finding in the CM patients, the age and distribution of the lesions, as well as the association of AI with other pathological features, were heterogeneous. Pathological features differ widely between individual patients with CM. This implies that the cerebral insults to which axons are submitted/vulnerable during fatal P. falciparum malaria also differ. Disruption in axonal transport may represent a common pathway leading to potentially reversible neurological dysfunction. It is likely that AI occurs in patients who survive and recover from CM. These responses might be measured prospectively in living patients using proton magnetic resonance spectroscopic imaging for N-acetylaspartate, an index of axonal integrity and CSF from these patients for soluble markers of AI. However, magnetic resonance spectroscopic imaging facilities are currently not available in most malaria-endemic countries. This finding also makes AI a potential target for neuroprotective adjuvant therapy in CM.
By studying disruption of fast axonal flow by β-APP immunohistochemistry we have detected more subtle changes to neuronal function allowing a better understanding of the clinicopathological correlations. Although β-APP is a normal constituent of axons these levels are not detected by standard immunohistochemical techniques. β-APP rapidly accumulates at sites of injury, stains damaged axons within 2 hours after injury, and remains detectable in axons and bulbs for more than 2 weeks. 9,17,20 In this study, all fatal malaria cases died within 2 weeks of admission to hospital. Therefore all damaged axons should remain visibly labeled using this method. β-APP staining has been reported to occur in activated macrophages/microglia 21 but we observed clearly different staining patterns with CD68 and β-APP (Figure 2, E and F) ▶ .
To define further the possible mechanisms of AI we have analyzed multiple regions of the brain, stained serial sections for other pathological features, and correlated AI with other clinical and biochemical parameters. Two major distributional patterns of AI emerged from this study. The first pattern showed a comparable amount of APP load throughout all brain regions examined. The second pattern showed a predominance of AI in one brain region, typically the internal capsule or pons. This may reflect two different axonopathological mechanisms. The first potentially reflects an insult simultaneously affecting multiple brain regions, such as that which could be expected after a multisystem organ failure or vascular sequestration. In contrast the pattern affecting predominantly one brain region may reflect a stroke-like lesion. In a previous study it was shown that cases of CM from Vietnamese also show differential rates of sequestration within different areas of the brain. 22 In contrast to the AI findings, the cerebral cortices and cerebellum were preferentially affected when compared to mid-brain structures or the brainstem. This implies that AI is not solely related to the extent of sequestration.
Detection of β-APP staining is highly sensitive, but by no means specific for a particular type of injury. Unlike some reports of Alzheimer’s disease and human immunodeficiency virus, 10,23,24 the distribution of AI was not restricted to a perivascular distribution within the same plane of section. This does not rule out the effects of neighboring vessels that are not cut in the same plane. Leukocyte recruitment in some conditions is temporally and regionally correlated with acute neuronal degeneration. 25 However, there was no significant correlation between the presence or absence of intravascular leukocytes and CM or AI. AI was also found independently of edema, hemorrhage, and glial responses. However, the reverse was not always observed. Analyses of microglial responses were useful because they provided an estimation of the timing of the axonal lesions. In some patients, lesions to axonal bundles occurred simultaneously within the same brain region whereas in other patients multiple lesions occurred at different times within the same brain region, as shown by the different surrounding microglial responses. Further, some lesions appeared within the same time frame in remote sites of the brain whereas in other cases one brain region appeared to have been affected considerably before others. These findings further emphasize the inter- and intrapatient variability that has been previously detailed in other clinicopathological studies of fatal malaria. 2-5
Recent literature has raised the possibility that hypoxia, 26 raised intracranial pressure, 26-29 and hypoglycemia 30 can cause AI. We found no association between AI and intracranial pressure or hypoglycemia. However the relationship between systemic glucose levels and local cerebrovascular hypoglycemia is not well defined in CM. Further, there was no association with impairment of vital organ function such as renal failure, jaundice, or shock. There was no exacerbation of AI with increasing numbers of criteria of severity, which may imply that although these patients are increasingly ill and more likely to die, the extent of AI within the central nervous system is determined independently or early in the disease. However, there were correlations with plasma lactate, CSF protein, and Glasgow coma score. This latter finding is consistent with an earlier report of axonal damage in people who had undergone transient, reversible concussion as well as in those with more severe injuries associated with head trauma. 31 This suggests that axonal damage may be associated with clinically reversible coma both in trauma and malaria. AI also was most prominent in the brains of patients who died within 3 days of admission to hospital. Of the five patients with CM alone, without other organ complications, the average AI load was variable. This highlights the fact that AI can occur without other systemic effects of severe malaria, but that lesion volume does not necessarily correlate with clinical outcome (death in our series), in part because of the importance of lesion location. 8
In conclusion we have found a marker of potentially reversible axonal damage that is significantly associated with the incidence of CM in adults and that distinguishes CM from non-CM patients. This marker highlights the internal capsule and pons as areas of primary involvement in AI. Unlike other pathological correlates such as neuronal stress markers, AI does not seem to purely reflect the systemic contribution of severe malaria to the specific neurological syndrome of CM. Thus discerning the mechanisms of disturbed axonal flow is an important step in understanding the specific neurological complications associated with CM.
Acknowledgments
We thank the Central Oxford Research Ethics Committee and the relatives of patients included for permission to conduct these studies; the staff of the Malaria Research Ward, Center for Tropical Diseases, Ho Chi Minh City, Vietnam, for their contribution to this work; the director and staff of the Center for Tropical Diseases for their help and support for this study; and the members of the Histopathology Laboratory, The John Radcliffe Hospital, Oxford, for technical assistance with preparation of sections.
Footnotes
Address reprint requests to Dr. Isabelle Medana, Lloyd’s Tercentenary Fellow, Nuffield Department of Clinical Laboratory Sciences, Level 5 Lab, R 5501, The John Radcliffe Hospital, Oxford, OX3 9DU, UK. E-mail: isabelle.medana@ndcls.ox.ac.uk.
Supported by grant 044055/Z/95/Z/140 from the Wellcome Trust as part of the Wellcome-Viet Nam-Oxford Tropical Medicine Research Program and by the Lloyd’s of London Tercentenary Foundation (to I. M.).
I. M. is a Lloyd’s of London Tercentenary Fellow.
References
- 1.Turner GD, Morrison H, Jones M, Davis TM, Looareesuwan S, Buley ID, Gatter KC, Newbold CI, Pukritayakamee S, Nagachinta B, White N, Berendt A: An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol 1994, 145:1057-1069 [PMC free article] [PubMed] [Google Scholar]
- 2.Turner GDH: Cerebral malaria. Brain Pathol 1997, 7:569-582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchiafava E, Bignami A: On summer-autumnal malaria fevers. Malaria and the Parasites of Malaria Fevers. 1894, :pp 1-234 The New Sydenham Society, London [Google Scholar]
- 4.Clark H, Tomlinson W: The pathological anatomy of malaria. Boyd M eds. Malariology. 1945, :pp 874-903 W. B. Saunders, Philadelphia [Google Scholar]
- 5.Spitz S: The pathology of acute falciparum malaria. Military Surgeon 1946, 99:555-572 [PubMed] [Google Scholar]
- 6.Ferguson B, Matyszak MK, Esiri MM, Perry VH: Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120:393-399 [DOI] [PubMed] [Google Scholar]
- 7.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998, 338:278-285 [DOI] [PubMed] [Google Scholar]
- 8.Pendlebury ST, Lee MA, Blamire AM, Styles P, Matthews PM: Correlating magnetic resonance imaging markers of axonal injury and demyelination in motor impairment secondary to stroke and multiple sclerosis. Magn Reson Imaging 2000, 18:369-378 [DOI] [PubMed] [Google Scholar]
- 9.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW: Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 1993, 160:139-140 [DOI] [PubMed] [Google Scholar]
- 10.Umehara F, Abe M, Koreeda Y, Izumo S, Osame M: Axonal damage revealed by accumulation of β-amyloid precursor protein in HTLV-I-associated myelopathy. J Neurol Sci 2000, 176:95-101 [DOI] [PubMed] [Google Scholar]
- 11.Povlishock JT: Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 1992, 2:1-12 [PubMed] [Google Scholar]
- 12.Povlishock JT, Becker DP, Cheng CL, Vaughan GW: Axonal change in minor head injury. J Neuropathol Exp Neurol 1983, 42:225-242 [DOI] [PubMed] [Google Scholar]
- 13.Hien TT, Day NP, Nguyen HP, Nguyen TH, Tran TH, Pham PL, Dinh XS, Ly VC, Ha V, Waller D, Peto TE, White NJ: A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. N Engl J Med 1996, 335:76-83 [DOI] [PubMed] [Google Scholar]
- 14.: World Health Organization: Severe falciparum malaria. Trans R Soc Trop Med Hyg 2000, 94:S1-S90 [PubMed] [Google Scholar]
- 15.Medana IM, Mai NTH, Day NPJ, Hien TT, Bethell D, Phu NH, Farrar J, White NJ, Turner GDH: Cellular stress and injury responses in the brains of adult Vietnamese patients with fatal Plasmodium falciparum malaria. Neuropathol Appl Neurobiol 2001, 27:421-433 [DOI] [PubMed] [Google Scholar]
- 16.Gentleman SM, McKenzie JE, Royston MC, McIntosh TK, Graham DI: A comparison of manual and semi-automated methods in the assessment of axonal injury. Neuropathol Appl Neurobiol 1999, 25:41-47 [DOI] [PubMed] [Google Scholar]
- 17.Geddes JF, Vowles GH, Beer TW, Ellison DW: The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol 1997, 23:339-347 [PubMed] [Google Scholar]
- 18.Oehmichen M, Theuerkauf I, Meissner C: Is traumatic axonal injury (AI) associated with an early microglial activation? Application of a double-labeling technique for simultaneous detection of microglia and AI. Acta Neuropathol 1999, 97:491-494 [DOI] [PubMed] [Google Scholar]
- 19.Ransom BR, Fern R: Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia 1997, 21:134-141 [PubMed] [Google Scholar]
- 20.McKenzie KJ, McLellan DR, Gentleman SM, Maxwell WL, Gennarelli TA, Graham DI: Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol 1996, 92:608-613 [DOI] [PubMed] [Google Scholar]
- 21.Banati RB, Gehrmann J, Wiessner C, Hossmann KA, Kreutzberg GW: Glial expression of the beta-amyloid precursor protein (APP) in global ischemia. J Cereb Blood Flow Metab 1995, 15:647-654 [DOI] [PubMed] [Google Scholar]
- 22.Sein KK, Maeno Y, Thuc HV, Anh TK, Aikawa M: Differential sequestration of parasitized erythrocytes in the cerebrum and cerebellum in human cerebral malaria. Am J Trop Med Hyg 1993, 48:504-511 [DOI] [PubMed] [Google Scholar]
- 23.Raja F, Sherriff FE, Morris CS, Bridges LR, Esiri MM: Cerebral white matter damage in HIV infection demonstrated using beta-amyloid precursor protein immunoreactivity. Acta Neuropathol 1997, 93:184-189 [DOI] [PubMed] [Google Scholar]
- 24.Gray F, Belec L, Chretien F, Dubreuil-Lemaire ML, Ricolfi F, Wingertsmann L, Poron F, Gherardi R: Acute, relapsing brain oedema with diffuse blood-brain barrier alteration and axonal damage in the acquired immunodeficiency syndrome. Neuropathol Appl Neurobiol 1998, 24:209-216 [DOI] [PubMed] [Google Scholar]
- 25.Bell MD, Perry VH: Adhesion molecule expression on murine cerebral endothelium following the injection of a proinflammagen or during acute neuronal degeneration. J Neurocytol 1995, 24:695-710 [DOI] [PubMed] [Google Scholar]
- 26.Kaur B, Rutty GN, Timperley WR: The possible role of hypoxia in the formation of axonal bulbs. J Clin Pathol 1999, 52:203-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams JH, Graham DI, Jennett B: The neuropathology of the vegetative state after an acute brain insult. Brain 2000, 123:1327-1338 [DOI] [PubMed] [Google Scholar]
- 28.Graham DI, Lawrence AE, Adams JH, Doyle D, McLellan DR: Brain damage in non-missile head injury secondary to high intracranial pressure. Neuropathol Appl Neurobiol 1987, 13:209-217 [DOI] [PubMed] [Google Scholar]
- 29.Dolinak D, Smith C, Graham DI: Global hypoxia per se is an unusual cause of axonal injury. Acta Neuropathol 2000, 100:553-560 [DOI] [PubMed] [Google Scholar]
- 30.Dolinak D, Smith C, Graham DI: Hypoglycaemia is a cause of axonal injury. Neuropathol Appl Neurobiol 2000, 26:448-453 [DOI] [PubMed] [Google Scholar]
- 31.Oppenheimer DR: Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry 1968, 31:299-306 [DOI] [PMC free article] [PubMed] [Google Scholar]