

Abstract
An important role for β-catenin pathways in colorectal carcinogenesis was first suggested by the protein’s association with adenomatous polyposis coli (APC) protein, and by evidence of dysregulation of β-catenin protein expression at all stages of the adenoma-carcinoma sequence. Recent studies have, however, shown that yet more components of colorectal carcinogenesis are linked to β-catenin pathways. Pro-oncogenic factors that also release β-catenin from the adherens complex and/or encourage translocation to the nucleus include ras, epidermal growth factor (EGF), c-erbB-2, PKC-βΙΙ, MUC1, and PPAR-γ, whereas anti-oncogenic factors that also inhibit nuclear β-catenin signaling include transforming growth factor (TGF)-β, retinoic acid, and vitamin D. Association of nuclear β-catenin with the T cell factor (TCF)/lymphoid enhancer factor (LEF) family of transcription factors promotes the expression of several compounds that have important roles in the development and progression of colorectal carcinoma, namely: c-myc, cyclin D1, gastrin, cyclooxygenase (COX)-2, matrix metalloproteinase (MMP)-7, urokinase-type plasminogen activator receptor (aPAR), CD44 proteins, and P-glycoprotein. Finally, genetic aberrations of several components of the β-catenin pathways, eg, Frizzled (Frz), AXIN, and TCF-4, may potentially contribute to colorectal carcinogenesis. In discussing the above interactions, this review demonstrates that β-catenin represents a key molecule in the development of colorectal carcinoma.
The protein β-catenin was first described in humans as a member of the cell membrane-bound adherens complex. 1 Comparative studies of signaling pathways in Xenopus and Drosophila subsequently led to the discovery of a second role for β-catenin in human cells; this cell-signaling role involves translocation of the protein from the cytoplasm into the nucleus. 2,3 There have been recent advances in characterizing regulators of β-catenin function in both its cell adhesion and cell signaling roles. However, the last 4 years have seen, in particular, a revelation of several important molecular pathways that may serve as downstream effectors of β-catenin signaling.
Among all human cancers, the molecular pathogenesis of colorectal adenocarcinoma (CRC) has been one of the most extensively studied. At least three pathways are thought to exist, namely the adenoma-carcinoma sequence (as initially proposed by Fearon and Vogelstein 4 ), the microsatellite instability pathway, and the ulcerative colitis dysplasia-carcinoma pathway. 4 As the first of these three pathways is accepted as being central to a majority of CRCs, 4 it will be the focus of this review and represents the process referred to by the term “colorectal carcinogenesis,” unless otherwise indicated.
Although several oncogenes and oncosuppressor genes are known to be involved in colorectal carcinogenesis, mutation of the adenomatous polyposis coli gene (APC) is regarded as being particularly crucial as an instigator of this process. 4,5 A potential role for β-catenin in colorectal carcinogenesis first seemed likely in view of the importance of disruption of cell to cell adhesion to cancer invasion and metastasis. 6 However, a more primary role in the pathogenesis of CRC soon became apparent with the discoveries that APC protein binds to cytosolic β-catenin, 7 and that APC mutation leads to nuclear accumulation of β-catenin, 8 a feature associated with progression along the adenoma-carcinoma sequence. 9 The significance of this primary role now seems to be ever increasing as several of the regulators and effectors of the β-catenin signaling pathway are being found to represent molecular pathways that have well-characterized links with colorectal carcinogenesis. The following review discusses these regulators and effectors and also abnormalities of components of the β-catenin signaling pathway that are or may potentially be involved in colorectal carcinogenesis. As a consequence, this review demonstrates that β-catenin may represent a crucial molecule linking several components of this process.
β-Catenin Pathways
The following summary (see also Figure 1 ▶ ) is intended to help place into context the following discussions of how regulators and components of the β-catenin pathways may relate to colorectal carcinogenesis. More detailed reviews of the β-catenin pathways are provided elsewhere. 2,3
Figure 1.

Summary of established β-catenin pathways. β-catenin may exist: 1) as part of the cell membrane-bound adherens complex with E-cadherin and α-catenin; 2) in the cytosol where binding with GSK-3β, APC, and AXIN promotes; 3) ubiquitination and, hence, degradation; or 4) in the nucleus where binding with a TCF leads to promotion of gene transcription. *, Recent data suggest an alternative pathway (involving Siah-1 protein) via which β-catenin may be targeted for ubiquitination without GSK-3β-mediated phosphorylation. Molecules that bind to β-catenin are shown in bold. The Wnt-Frz-Dvl regulatory pathway is shown in italics. Black broken arrows represent the potential subcellular movements of β-catenin. Dvl, disheveled protein.
β-catenin may be regarded as existing in three different subcellular forms: membrane-bound (as part of the adherens complex), cytosolic, and nuclear (Figure 2, a and e) ▶ . 2,3 Binding of the protein to other members of the adherens complex, ie, E-cadherin and α-catenin, is thought to be regulated by tyrosine phosphorylation. 10 Indeed, physical association of the complex with tyrosine kinases and phosphatases 10,11 is in keeping with tight regulation of this process. Tyrosine phosphorylation of β-catenin leads to its dissociation from the adherens complex 10 and probable transfer of the protein to the cytosol where it exists in a soluble, monomeric state. 2,3 Cytosolic β-catenin may subsequently be degraded or be translocated into the nucleus. The degradation of β-catenin involves binding of the protein to a complex involving APC protein, and two further proteins, AXIN and glycogen synthase kinase (GSK)-3β. 2,3 The latter serves to phosphorylate serine and threonine residues on β-catenin, a crucial step required to target the protein for ubiquitination and proteosomal degradation. Both APC and AXIN enhance this phosphorylation and are, therefore, promoters of β-catenin degradation. For APC protein to have this promoting effect, it must bind to β-catenin via two possible binding regions, one of which contains tandem repeats of 20 amino acids. 2,12 Phosphorylation of β-catenin is important in enabling binding to the F box protein β-TrCP and hence ubiquitin-mediated proteolysis. 2,13 However, it has recently been shown that β-catenin may also be targeted for such degradation independent of GSK-3β-mediated phosphorylation. 13,14 This putative alternative pathway requires interaction between β-catenin, APC, and a complex of proteins including the p53-inducible protein, Siah-1. 13,14
Figure 2.
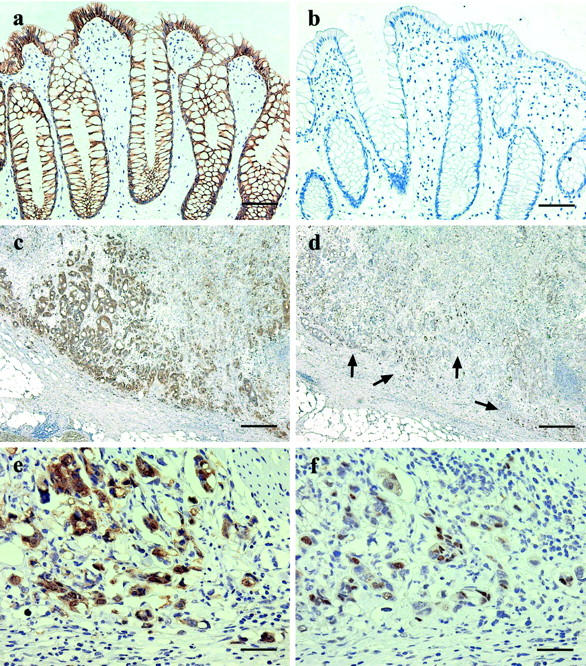
Sections of nonneoplastic colorectal mucosa (a and b) and colorectal carcinoma (c to f) immunostained for β-catenin (a, c, and e) and cyclin D1 (b, d, and f). The colorectal epithelial cells show strong membranous expression of β-catenin (a) and no detectable expression of cyclin D1 (b). The invasive edge of the carcinoma shows particularly prominent cytoplasmic and nuclear expression of β-catenin (c) and nuclear overexpression of cyclin D1 (d, arrows). Higher power views of the carcinoma show diffuse but heterogeneous nuclear expression of both proteins (e and f). The carcinoma cells also show prominent cytoplasmic β-catenin expression and loss of membranous expression (e). Scale bars: 150 μm (a and b); 600 μm (c and d); 75 μm (e and f).
An important regulator of GSK-3β activity is the Wnt family. Binding of these glycoproteins to their trans-membrane receptor, Frz, leads to increased activity of the protein Dishevelled (Dvl) that, in turn, inhibits GSK-3β phosphorylating activity. In the presence of increased cytosolic levels of β-catenin because of, for example, Wnt signal activation, the protein is translocated into the nucleus. Here, β-catenin binds with a member of the TCF/LEF family of transcription factors to form a complex that activates transcription of target genes by binding to their promoter sequences. Of this family of transcription factors, TCF-4, LEF-1, and TCF-1 have the most known relevance to colorectal carcinogenesis, as is discussed below. It was initially thought that all TCFs bind to β-catenin to promote gene transcription. However, at least two TCFs, LEF-1 and TCF-1, are now known to exist in truncated forms that repress β-catenin/TCF transcriptional activity. 15,16 Little is known about the fate of nuclear β-catenin. However, recent studies do suggest the protein may be translocated from the nucleus back to the cell membrane, 17 presumably through a soluble, cytosolic intermediate state. The regulation of this retrograde movement has not been extensively studied but is likely to at least involve E-cadherin, as is discussed later.
β-Catenin Regulators and Colorectal Carcinogenesis
Most of the regulators of β-catenin function that are linked to colorectal carcinogenesis may be allocated to one of three groups according to their modes of action. There are, however, two molecules (PPAR-γ and MUC-1) that have roles in colorectal carcinogenesis and seem to regulate β-catenin function but have, as yet, unknown mechanisms underlying their relation to the protein; these two molecules are, therefore, considered separately.
Promoters of Tyrosine Phosphorylation
Colorectal carcinoma cells in vivo have been shown to express several growth factors, including EGF and platelet-derived growth factor, and to express receptors for these and other growth factors, for example, EGF-R, c-erbB-2, and c-met (receptor for hepatocyte growth factor). 18-20 Further, it is well known that EGF and hepatocyte growth factor treatment leads to transformation of colonic epithelial cells into a more malignant phenotype (eg, increased invasive qualities and higher proliferation rates). 21,22 This change has been, at least in part, attributed to the ability of both growth factors to promote tyrosine phosphorylation of β-catenin and, hence, disruption of the adherens complex. 23,24 It is uncertain whether this phosphorylation is performed by the growth factor receptors themselves or via soluble tyrosine kinases. However, the growth factor receptors, c-met 11 and c-erbB-2, 25 have both been shown to bind to β-catenin in vitro. Mutations of the ras oncogenes, which lead to overexpression of their gene products, are found in at least 50% of sporadic CRCs. 26 Cytoplasmic expression of the trefoil peptide TFF-3 (previously known as ITF), which has mitogenic and prokinetic properties, is significantly higher in CRC tissue compared with normal colonic mucosa. 27 Inducing overexpression of ras oncogenes in cell lines 28 or treatment with TFF-3 29 both have a similar effect in promoting tyrosine phosphorylation of β-catenin. From what is known of β-catenin pathways, release of β-catenin from its membrane-bound pool by tyrosine phosphorylation might be expected to increase nuclear translocation. However, in the case of TFF-3 treatment, increased tyrosine phosphorylation of β-catenin was seen without demonstrable nuclear accumulation of the protein. 29 It is possible that additional factors, eg, GSK-3β inhibition or loss of functioning APC protein (see below), are needed to transfer membrane-released β-catenin into the nucleus, although the studies that failed to show increased nuclear β-catenin with TFF-3 treatment had been performed in an APC mutated colorectal epithelial cell line. 29 Unfortunately, the aforementioned studies on growth factors, their receptors, or ras and β-catenin had not mentioned whether or not nuclear accumulation of the latter protein was seen.
Inhibitors of GSK-3β
The growth factor EGF and ras gene product are similar in having a second effect on β-catenin regulation. Both activate the proto-oncogene Akt, whose gene product has been shown to inactivate GSK-3β and, therefore, suppress degradation of cytosolic β-catenin. 30 Interestingly, this effect of ras gene product is mediated via the insulin-like growth factor receptor, 30 levels of which are overexpressed in CRC. 31 Two other molecules that have more recently been found to inhibit GSK-3β are the βΙΙ isoform of protein kinase C (PKC-βΙΙ) 32 and polycystin-1. 33 Intracellular PKC-βΙΙ levels are raised after exposure to secondary bile acids, which are thought to be carcinogenic in the large bowel, 34 and higher levels of the isoform are found in CRC tissue compared with normal colorectal mucosa. 35 Murray and colleagues 32 have recently described transgenic mice that overexpress PKC-βΙΙ and, when treated with azoxymethane, develop a greater number of aberrant crypt foci (the earliest morphologically recognizable stage in the adenoma-carcinoma sequence) than their similarly treated nontransgenic litter mates. Further, the colonic epithelium of the transgenic mice showed an expansion of the crypt proliferative compartment and increased levels of β-catenin, measured by Western blotting. 32 Polycystin-1 is the protein product of the gene whose mutation underlies type I adult polycystic kidney disease. 36 In vitro studies have shown that polycystin-1 inhibits GSK-3β, 33 forms a complex with β-catenin, 37 and also increases expression of target genes of the β-catenin/TCF complex. 33 In contrast to adult polycystic kidney disease, there are, as yet, no known human hereditary diseases that are characterized by overexpression of polycystin-1 and that could be studied for an increased risk of CRC. The existence of antibodies to the protein 37 will, however, permit an informative study of whether the protein is overexpressed in CRCs.
Inhibitors of β-Catenin/TCF Activity
Molecular pathways that interfere with β-catenin/TCF activity in the nucleus would be predicted to have an anti-oncogenic effect. Although TGF-β may have opposing effects in different cell types, 38 its main effects on colonic epithelial cells are to reduce proliferation and to induce differentiation. 39 One of the effector pathways of TGF-β involves TAK-1, which is a member of the MAP-kinase 3 family 38,40 and which promotes the activity of a second kinase, NLK. 41 NLK has been shown, in embryonic kidney cells, to co-localize with and to phosphorylate TCF-4, 41 which is the only TCF isoform consistently found in normal colorectal epithelium and every CRC cell line examined so far. 2 Phosphorylation of TCF-4 was subsequently associated with impaired binding of the β-catenin/TCF-4 complex to DNA. 41 In sharp contrast, smad4, another effector of TGF-β, has been shown to form a complex with β-catenin and LEF-1 and to increase β-catenin/LEF-1 transcriptional activity in a hepatoma cell line. 42 These dichotomous effects of TGF-β on the β-catenin signaling pathway may explain, at least in part, its differing effects in different cell types. We are aware of only one previous study of the effects of TGF-β treatment on β-catenin expression in a colorectal cell line. Ilyas and colleagues 43 reported an increase in β-catenin protein levels in the HCA46 cell line that also showed growth inhibition. Unfortunately, as β-catenin had only been studied using an enzyme-linked immunosorbent assay, shifts in the subcellular localization of the protein could not be assessed for.
Like TGF-β, both retinoic acid and vitamin D have been shown to have growth-inhibitory effects on colonic epithelial cells. 44,45 For these reasons, both compounds have been suggested as potential candidates for chemoprevention of CRC. 44,45 In a mutant APC colorectal cancer cell line, both retinoic acid and vitamin D reduced TCF reporter activity. 46 In the case of retinoic acid, its receptor was thought to compete with β-catenin for a common binding site on TCF-4. 46 Although retinoic acid did not seem to interfere with β-catenin ubiquitination or proteosomal degradation, the vitamin has been shown in breast carcinoma cells to increase cadherin expression, thereby decreasing cytosolic β-catenin and down-regulating β-catenin signaling by a second route. 46
Uncertain Mechanism
The mucin, MUC-1, is expressed at higher levels in CRC tissue than normal colorectal mucosa, and the quantity of MUC-1 expressed correlates positively with tumor stage. 47 This transmembrane molecule has a cytoplasmic tail with tyrosine phosphorylation sites. 48 MUC-1, through an as yet unknown mechanism, reduces the binding of β-catenin to E-cadherin, but this effect is abrogated by phosphorylation of its tail by GSK-3β. 49 Increasing MUC-1 expression alone failed to alter total levels or the subcellular location of β-catenin in an embryonic kidney epithelial cell line that expresses wild-type APC. 49 However, inhibition of GSK-3β, as may occur in vivo (for reasons discussed above), could allow for a stronger effect of MUC-1 in liberating membrane-bound β-catenin to enter the cytoplasmic-nuclear signaling pathway.
Much interest has recently focused on the role of peroxisome proliferative activated receptor (PPAR)-γ in colorectal carcinogenesis, especially because the molecule is activated by prostaglandins and, hence, increased COX-2 activity. 50 PPAR-γ is also activated by fatty acids 50 and a high-fat diet is recognized as a potential risk factor for CRC. 4,51,52 When transgenic mice heterogeneous for APC mutation (APCmin/+ mice) were treated with synthetic PPAR-γ activators, they showed greater numbers of colonic tumors, many of which were adenocarcinomas. 53 Further, colonic β-catenin levels, measured by immunoblot, were higher in the treated mice compared with untreated controls. 53 Unfortunately, no immunohistochemical studies were performed to determine whether this increase was associated with a shift in subcellular location. Although this and possible mechanisms linking PPAR-γ to β-catenin remain to be elucidated, it is of interest that treatment of APCmin/+ mice with the COX-2 inhibitor, sulindac, reduces both the number of intestinal tumors as well as nuclear β-catenin accumulation in the tumors. 54
Further, treatment of CRC cell lines with indomethacin, another inhibitor of COX-2, is associated with reduced nuclear β-catenin expression. 55
Components of β-Catenin Pathways and Colorectal Carcinogenesis
The following section discusses predominantly genetic abnormalities of components of the β-catenin pathways that have been shown to or may relate to colorectal carcinogenesis. Although the Wnt-Frz-Dvl pathway is, in essence, a regulator of β-catenin function, it is classically described as part of the β-catenin signaling pathway and is, therefore, included in this section.
β-Catenin
The most common mutations that have been found in the human gene encoding β-catenin (CTNNB1) effect the protein’s serine and threonine residues that are targeted by GSK-3β. 56 As a result, the mutant protein is resistant to phosphorylation and escapes the β-TrCP-mediated ubiquitination/proteosomal degradation pathway; 56 such changes seem to be effected by the CTNNB1 mutations in a dominant manner. 57 Because APC mutations are thought to instigate colorectal carcinogenesis via their suppression of β-catenin degradation, 2,3,12 it may be speculated that CTNNB1 mutations have particular importance in the relatively small proportion of CRCs that lack APC mutations. This is, indeed, borne out by recent studies demonstrating that 50% of such CRCs show CTNNB1 mutations. 56 Sparks and colleagues 56 also found that APC and CTNNB1 mutations were mutually exclusive among 30 colorectal neoplasms, although the two have periodically been described in the same CRC cell line. 58 Further support for the importance of CTNNB1 mutations to intestinal carcinogenesis and how they substitute for APC mutation comes from two independent studies of transgenic mice that are similar in producing β-catenin that lacks binding sites for GSK-3β. 59,60 Both types of mice develop dysplastic lesions and polyps in the small intestine as are seen in APCmin/+ mice. 61 Although neither transgenic β-catenin mouse developed colorectal polyps, 59,60 this too is a recognized feature of APCmin/+ mice. 61
APC, GSK-3β, and AXIN
The occurrence and nature of APC mutations in CRCs are well described elsewhere 62,63 and will not, therefore, be discussed in detail here. In brief, the APC mutations found in these cancers almost always cause truncation of the central region of the protein containing the 20-amino acid β-catenin-binding site. 12 Subsequent loss of APC protein binding to β-catenin is thought to significantly reduce phosphorylation of β-catenin by GSK-3β, thereby leading to accumulation of cytosolic β-catenin. 2,3,12 The central role of GSK-3β in the regulation of β-catenin function would suggest mutations of the gene encoding the kinase might have a significant role in colorectal carcinogenesis. No mutations of the GSK-3β gene were found in three colorectal cancer cell lines 56 but, obviously, study of more cell lines and of CRCs in vivo is needed to comprehensively test this hypothesis. As described above, the protein AXIN functions by promoting degradation of β-catenin. Satoh and colleagues 64 have recently described nonsense mutations of the AXIN gene in hepatocellular carcinomas. These mutations produce truncated AXIN protein that shows reduced binding to β-catenin, and were associated with nuclear accumulation of β-catenin and increased β-catenin/TCF transcriptional activity. 64 Since then, two sequence variants of the AXIN gene have been reported in 4 out of 34 CRC cell lines tested. 65 Further, one of these variants (a leucine to methionine substitution at residue 396) interferes with binding of AXIN to GSK-3β protein 65 and could, therefore, promote β-catenin nuclear signaling. Finally, having first cloned the human homologue of AXIN (called AXIN2), 66 Liu and colleagues 67 recently demonstrated frameshift mutations of AXIN2 in 11 out of 45 CRCs showing microsatellite instability, compared with none of 60 microsatellite stable tumors. None of the 11 CRCs with AXIN2 mutations showed mutations of APC or CTNNB1. 67 Instead, AXIN2 mutations, a type of which was shown to activate TCF-dependent transcription in vitro, were suggested to explain the nuclear β-catenin accumulation seen in 10 of the 11 tumors. 67
Wnt, Frz, and TCF-4
Little is known about the role of the human Wnt-Frz-Dvl signaling pathway in adulthood. Aberrant activation of this pathway would, however, be predicted to have oncogenic effects through reduced degradation of cytosolic β-catenin and increased β-catenin nuclear translocation. Higher levels of Wnt2 mRNA are found in CRC tissue compared with normal colorectal mucosa, 68,69 but increased Wnt2 expression is not seen in colorectal cancer cell lines. 69 This discrepancy may, however, be explained by the recent, intriguing findings of Smith and colleagues. 69 Using in situ hybridization probes for Wnt2 and immunohistochemical markers for macrophages, this research group found that stromal macrophages were the source of the elevated Wnt2 mRNA levels in CRC tissue. A concentration of Wnt2 at the tumor/stromal interface, for the above reasons, would then provide at least one explanation for the prominence of nuclear β-catenin expression found at the invading front of CRC (Figure 2c) ▶ . 70
There are, thus far, seven known members of the Frz family. 71,72 Tanaka and colleagues 72 recently identified a novel Frz gene (FzE3) in 60% of esophageal squamous cell carcinomas and also in 21% (3 out of 14) of CRCs. Further, transfecting cells with the FzE3 gene led to increased nuclear accumulation of β-catenin, an effect that was potentiated by co-transfection with LEF-1. 72 Clarifying the role of FzE3 gene expression in colorectal carcinogenesis will be aided by much needed study of the roles of the different Frz proteins in normal and malignant colorectal epithelium.
Although different regulators of TCF-4 function have been well-studied (see above), only recently has information emerged regarding aberrations of the TCF-4 gene itself. From mutation screening of the entire TCF-4 gene in 24 colorectal cancer cell lines, Duval and colleagues 73 found DNA variants in 12 cases. 73 Of particular interest are mutations of the 3′ end of the gene found in half these cases. The mutant gene products were predicted to show reduced binding for c-terminal-binding protein, which is thought to repress TCF transcriptional activity. 73 Although these 3′ end TCF-4 mutations, therefore, provide a plausible explanation for increased β-catenin/TCF transcriptional activity, their relative contribution to colorectal carcinogenesis in vivo remains to be determined. The same group had earlier demonstrated a single base pair deletion in exon 4 in 39% and 50% of microsatellite unstable CRCs and cell lines, respectively, but in none of the microsatellite stable CRCs and cell lines studied. 74 However, this exon 4 mutation was thought unlikely to interfere with the binding regions of TCF-4. 74
β-Catenin Effectors and Colorectal Carcinogenesis
The effector pathways described below share, in common, induction by β-catenin/TCF activity and recognized involvement in colorectal carcinogenesis. Further, in all but two cases (uPAR and CD44), TCF-binding sites have been demonstrated in the promoter sequences of the genes encoding these effector molecules.
C-myc
C-myc was the first target gene of the β-catenin signaling pathway to be demonstrated in humans. 75 This discovery has helped to clarify two previously unexplained observations concerning the transcription factor. First, c-myc expression is suppressed by an extra copy of chromosome 5, 76 which, in turn, is known to contain the APC gene locus. 75 Second, overexpression of c-myc mRNA and protein is well-described in CRCs but rearrangement and/or amplification of the c-myc gene have rarely been found to explain these observations. 77 The relationship between β-catenin and c-myc was first clarified in vitro by demonstrating that expression of a mutant β-catenin protein resistant to the down-regulatory effects of APC protein increased c-myc expression via TCF-4 binding to the c-myc promoter. 75 Since then, the existence of this relationship has been demonstrated in vivo by Brabletz and colleagues, 78 who have reported a strong correlation between nuclear β-catenin and c-myc immunostaining among CRCs. There are also, however, recent data suggesting that γ-catenin may be equally if not more important in mediating the association between APC mutation and c-myc up-regulation in CRC. 79
Cyclin D1
As with c-myc, cyclin D1 protein had been known, for some time, to be overexpressed in CRC 80 yet, unlike in breast and esophageal carcinomas, genetic aberration of the cyclin D1 gene is rarely found in CRCs. 80,81 In 1999, two independent research groups published in vitro findings demonstrating that β-catenin regulates cyclin D1 expression. 82,83 Tetsu and McCormick 82 showed that both wild-type and mutant β-catenin increases transcription of the cell cycle protein, and that treatment with dominant-negative TCF-4 suppresses this effect. Further, Shtutman and colleagues 83 demonstrated that the β-catenin/LEF-1 complex binds to the cyclin D1 promoter, and the promotion of cyclin D1 expression by β-catenin is inhibited by transfection with wild-type APC and/or AXIN. Since then, β-catenin gene mutations have been shown to induce cyclin D1 overexpression in the azoxymethane rat model of CRC. 84 Finally, we have recently shown that, in human CRC in vivo, cyclin D1 overexpression always and only takes place in association with nuclear β-catenin expression (submitted for publication; Figure 2; a to f ▶ ).
Gastrin
Like cyclin D1, the gastrointestinal hormone gastrin is thought to have a role in driving cell proliferation in CRC. 85 Up-regulation of gastrin gene expression is seen in colorectal adenomas 86 and CRCs, 87 and the likelihood that this up-regulation is mediated by β-catenin nuclear signaling is supported by recent data from Koh and colleagues. 85 In keeping with TCF-4-binding sites on the gastrin gene promoter, co-transfection of HeLa cells with an active β-catenin expression construct leads to increased gastrin promoter activity, whereas induction of wild-type APC in CRC cell lines reduces gastrin mRNA levels. 85 Further, compared with control APCmin/+ mice, transgenic mice that overexpress glycine-extended gastrin show increased numbers of polyps whereas their gastrin-deficient counterparts show reduced numbers. 85 Confirmation that β-catenin nuclear signaling represents the primary mediator of gastrin up-regulation in CRC in vivo is awaited.
COX-2
The recent attention paid to the role of prostaglandin and COX-2 overexpression in colorectal carcinogenesis has particularly been fuelled by interest in using aspirin or more specific COX-2 inhibitors to reduce the risk of developing CRC. 88 Although COX-2 had been demonstrated at higher levels in human CRCs than in normal colonic mucosa, 89 the first indication of an interaction between COX-2 and β-catenin pathways came from studies on mutant APC mice: intestinal polyps from such mice show elevated levels of COX-2 whereas introducing a null mutation of the COX-2 gene to the animals reduces the number and size of intestinal tumors. 90 As discussed earlier, COX-2 may regulate β-catenin function via PPAR-γ activation. However, there are also recent data showing that β-catenin signaling pathways may, in turn, regulate the expression of COX-2. Addition of wild-type APC to mutant APC colorectal cell lines reduces COX-2 protein expression, 91 overexpression of β-catenin in murine mammary cell lines increases basal COX-2 protein and mRNA, 92 and both basal and inducible levels of COX-2 protein and mRNA are higher in mutant APC murine cell lines. 93 The changes in COX-2 described in the latter study model are also associated with increased β-catenin/LEF-1 complex formation. 93 Although these data suggest β-catenin/TCF transcriptional activity may directly induce COX-2 expression, evidence to confirm this is still awaited. The human COX-2 gene promoter has two potential TCF-binding sites but no increase in COX-2 promoter reporter activity has yet been demonstrated after overexpression of β-catenin in vitro. 92 It is possible, therefore, that β-catenin signaling and COX-2 regulation may be linked via an, as yet unidentified, intermediary.
MMP-7
MMP-7 (matrilysin) is expressed in up to 90% of CRCs and is thought to be important in mediating stromal invasion 94 but may also have a role in earlier stages of carcinogenesis. In support of the latter, ablation of MMP-7 expression reduces the number of intestinal adenomas in APCmin/+ mice. 95 Co-localization of nuclear β-catenin and MMP-7 protein 96 and mRNA 97 has been demonstrated in human CRCs 96 and tumors from APCmin/+ mice, 97 respectively. The human MMP-7 gene promoter has at least two TCF-binding sites 96 but, unlike with COX-2, the β-catenin/TCF complex has been shown to increase MMP-7 promoter activity in vitro. 97 Further, this effect is abrogated by mutating a TCF-binding site on the MMP-7 promoter or by introducing a dominant-negative TCF mutant. 96
uPAR
Another factor that may mediate stromal invasion by CRC is the proteolytic protein plasmin, which is produced by cleavage of plasminogen. 98 Plasminogen is cleaved by the soluble factor urokinase-type plasminogen activator (uPA) but the transmembrane protein, uPAR, is also functionally involved in cleavage of plasminogen at the cell surface. 99 uPAR is overexpressed in CRCs 100,101 and, as might be expected, is particularly localized to the invasion fronts of these cancers; 100 incidentally, as mentioned earlier, Wnt2 production and β-catenin nuclear expression are also most prominent at these invasion fronts. The identification of uPAR as a target gene of the β-catenin signaling pathway resulted from a differential hybridization-based search for altered gene expression in colorectal cells after induction of β-catenin expression. 102 A subsequent in vivo study showed good correlation between β-catenin mRNA and uPAR protein and mRNA expression among CRCs. 102 The mechanism by which β-catenin induces uPAR expression is unclear but it has been suggested that the oncoproteins c-jun and fra-1, whose promoters bear TCF binding sites, may mediate this interaction. 102
CD44
CD44 represents a group of trans-membrane glycoproteins that are thought to function primarily as cell adhesion molecules. 103 The usual gene product of the CD44 gene is termed CD44s (standard), but differential splicing of the gene produces a variety of splice variants (CD44v), more than 1000 variants being theoretically possible. 103 CD44s and CD44v6 are expressed in 56% and 99% of CRCs, respectively, and the association of such expression with poorer prognosis suggests a possible pro-oncogenic role for these molecules. 104,105 The fact that aberrant expression of CD44 molecules occurs at as early a stage in colorectal carcinogenesis as APC mutations 106 led Wielenga and colleagues 107 to study the relationship between the two phenomena and the β-catenin signaling pathway. The tumors that develop in APCmin/+ mice were found to show increased expression of CD44s and CD44v6 protein and mRNA, whereas, in patients with familial adenomatous polyposis, increased expression of CD44s protein was seen in aberrant crypt foci. The above findings together with the fact that TCF-4 −/− mice show no evidence of gastrointestinal CD44 expression 107 were interpreted as suggesting β-catenin may regulate expression of CD44. Clearly, the in vitro and in vivo methods used to investigate other effector molecules of β-catenin will need to be applied to further test this hypothesis.
MDR-1/P-Glycoprotein (Pgp)
Increased transcription of the multidrug resistance-1 gene, MDR-1 and its product, Pgp, have been demonstrated in CRCs. 108 Pgp is thought to have a generic anti-apoptotic function and, in keeping with this, Pgp expression in CRC has been shown to correlate with markers of a more aggressive phenotype, namely vascular invasion, lymph node metastases and shorter disease-free survival. 108 Further, high expression of Pgp may mediate resistance of CRC to chemotherapeutic drugs based on naturally occurring compounds. 108 Analysis of cDNA expression profiles of a CRC cell line with suppressed TCF-4 activity has recently shown MDR-1 to be yet another target of nuclear β-catenin signaling. 109 MDR-1 promoter activity could be directly regulated by TCF-4 and close correlation between Pgp expression and cellular accumulation of β-catenin was demonstrated immunohistochemically in colorectal adenomas and CRCs from patients with familial adenomatous polyposis. 109 A particular significance of this interaction between β-catenin and MDR-1 is that it represents at least one possible explanation for the anti-apoptotic effects of nuclear β-catenin signaling (see below).
β-Catenin and Colorectal Carcinogenesis—Other Potential Links
This last, more heterogeneous group discusses components of β-catenin pathways and colorectal carcinogenesis that may be linked on more theoretical grounds. Further studies in these areas are, however, awaited to support or refute these suggested relationships.
β-Catenin and Regulators of Apoptosis
Dysregulation of apoptosis plays an important role in colorectal carcinogenesis. 110 Nuclear β-catenin signaling seems to have an anti-apoptotic effect in that mechanisms that reduce such signaling, eg, introduction of functioning APC or AXIN protein, 64 also induce apoptosis. Aside from regulation of MDR-1 expression, no plausible mechanisms have yet been suggested to underlie this effect of β-catenin. Although p53 is recognized as having an important oncosuppressor function with regards to colorectal carcinogenesis, there have been surprisingly few studies of the relationship between regulation of this protein and β-catenin. One such study showed that overexpression of nuclear β-catenin, in lung adenocarcinoma cells, led to accumulation of transcriptionally active p53 probably through inhibition of its degradation. 111 These findings seem, however, to contradict the aforementioned anti-apoptotic effect of nuclear β-catenin signaling. It will, therefore, be important to see whether the findings of Damalas and colleagues 111 are tissue-specific and, in particular, whether they can be reproduced in colorectal cell lines. There have been more recent data suggesting that p53 may in turn reduce expression of β-catenin by inducing Siah-1-mediated degradation of β-catenin. 13,14 These exciting findings provide at least a second explanation (ie, aside from reduced apoptosis) for why mutational loss of p53 function may promote colorectal carcinogenesis. Low levels of Siah-1 have been observed in SW480, 14 a CRC cell line known to express mutant p53. 112 Future studies, however, will be needed to quantify the exact contribution of loss of p53 and Siah-1 functions to β-catenin signaling in CRCs in vivo.
Another regulator of apoptosis whose dysregulation may be important to colorectal carcinogenesis is bcl-2. 110,113 We are unaware of any studies that have directly investigated the relationship between β-catenin and bcl-2, which is known to inhibit apoptosis. 113 However, COX-2 is known to induce bcl-2 expression 114 so it is possible that nuclear β-catenin signaling indirectly protects against apoptosis by promoting COX-2 expression, as described earlier. Although not directly supporting this hypothesis, it is also interesting that transgenic mutant β-catenin mice 59 share some similar, unexpected phenotypic changes with bcl-2 null mice, 115 eg, formation of polycystic kidneys.
β-Catenin and CDX1/CDX2
There has been recent interest in possible roles for the two homeobox proteins CDX1 and CDX2 in colorectal carcinogenesis. There are some data suggesting that, in relation to this process, CDX1 has oncogenic activity whereas CDX2 has a tumor suppressor function. Lorentz and colleagues 116 have recently shown increased ras expression leads to up-regulation of CDX1 but down-regulation of CDX2 in a CRC cell line. Further, CDX1 overexpression increases cell proliferation and inhibits apoptosis in vitro 117 whereas transgenic mice heterozygous for a null CDX2 mutation develop multiple gastrointestinal adenomatous polyps, particularly in the proximal colon. 118 These opposite effects of CDX1 and CDX2 are supported, in part, by recent data showing how the two proteins may interact with APC/β-catenin pathways. Wnt signaling has been shown to promote CDX1 expression in vitro, the CDX1 promoter has a binding site for the β-catenin/TCF complex, and TCF-4 knockout mice show reduced intestinal expression of CDX1. 119 By contrast, wild-type APC expression, and hence reduced nuclear β-catenin signaling, has been shown to increase CDX2 RNA levels in a colorectal cancer cell line. 120 There are also, however, data that contradict and complicate the suggested relationships between the two homeobox proteins and APC/β-catenin pathways in colorectal carcinogenesis. Immunohistochemical studies have shown that expression of CDX2 121 but also CDX1 122 is progressively reduced along the adenoma-carcinoma sequence. Further, an inactivating mutation of CDX2 is associated with reduced β-catenin/TCF transcriptional activity in the presence of intact APC and β-catenin genes, 120 suggesting that CDX2 may function to maintain nuclear β-catenin signaling. It is clear that further work will be required to characterize the exact interactions between CDX1, CDX2, and β-catenin pathways and to determine exactly how they relate to colorectal carcinogenesis.
Other Components of β-Catenin Pathways
In parallel with the discovery that nuclear β-catenin signaling has pro-oncogenic effects, greater insight has only recently been gained into the regulation of β-catenin activity and movement within and around the nucleus.
There is increasing evidence that β-catenin/TCF transcriptional activity may be regulated by differential expressions of different isoforms of TCF-1 and LEF-1. The genes encoding these proteins each have two promoters. 15,123 Whereas full length TCF-1 and LEF-1 can both activate gene transcription, their shorter/truncated isoforms both lack β-catenin-binding sites and seem to repress β-catenin/TCF transcriptional activity, as mentioned earlier in this review. 15,16 Although transcription of both TCF-1 and LEF-1 is up-regulated by β-catenin nuclear signaling, 15,16 it is not yet clear how the differential expression of their isoforms is controlled. There are, however, recent data suggesting dysregulation of these putative positive and negative feedback loops may contribute to colorectal carcinogenesis. LEF-1, for example, is not found in any cell in normal colorectal tissue but is aberrantly expressed in CRC and always in its full-length, transcriptionally active, isoform. 15 By contrast, truncated TCF-1 seems to be the predominant isoform found in human cells. 16 A potential role for TCF-1 as a tumor suppressor gene is supported by the fact that TCF-1-deficient mice develop intestinal neoplasms akin to those seen in mutant APC mice, and crossbreeding these two strains leads to a greater number of neoplasms as well as the formation of colonic tumors. 16 Study of a handful of CRC cell lines has shown, if anything, overexpression of TCF-1 protein and mRNA, this being attributed to increased β-catenin nuclear signaling secondary to APC mutations or oncogenic mutations of β-catenin. 16 However, CRCs in vivo and more CRC cell lines will have to be studied before the possibility that TCF-1 down-regulation contributes to increased β-catenin transcriptional activity in colorectal carcinogenesis can be refuted.
Another means of reducing nuclear β-catenin activity involves the relocation of free protein from the cytosol to the cell membrane. Ben-Ze’ev 124 showed that transfection with N-cadherin can induce such retrograde movement and reduce β-catenin-directed transactivation while producing a better differentiated phenotype in colorectal carcinoma cells. Like TCF-1, however, it is unclear to what extent disorders of N-cadherin expression occur in CRCs in vivo. Regardless of the answer to the question, cadherin derivatives may still have a potential therapeutic and/or prophylactic role in suppressing nuclear β-catenin activity and, therefore, oncogenesis. Interestingly, another compound that promotes movement of β-catenin back to the cell membrane 125 is also thought to be the primary mediator of the CRC preventative effects of a high-fiber diet, ie, butyrate. 126 Colonocyte butyrate levels are elevated by such a diet and the compound increases apoptosis, induces differentiation, and promotes cell to cell adhesion in colorectal cancer cell lines. 126,127 Although butyrate has also been shown to reduce transcription of β-catenin, 125 its reduction of nuclear β-catenin levels is thought to be primarily because of encouraging movement of β-catenin to the cell membrane through promoting expression of E-cadherin. 125 The abilities of N- and E-cadherin to recruit β-catenin back to the cell membrane are most certainly related to both proteins sharing overlapping binding sites on β-catenin with APC protein and TCFs. 128 It has been known for some time now that the β-catenin/TCF complex binds to the 5′ end of the murine gene encoding E-cadherin. 129 Although we are unaware that such binding has been demonstrated in human cells, if β-catenin is found to induce E-cadherin transcription in these cells, this may represent yet another negative feedback loop. Therefore, the oncogenic effects of loss of E-cadherin in CRCs, for example, because of gene mutation 130 may not only relate to loss of cell to cell adhesion but also to loss of suppression of nuclear β-catenin activity.
As with E-cadherin, loss of α-catenin is frequently described in CRC and may be important in releasing β-catenin from a membrane-bound state. 131 However, recent studies in CRC cell lines have shown that α-catenin may have a second role in regulating β-catenin pathways. Having demonstrated ectopic expression of α-catenin in nuclei, Giannini and colleagues 132 proceeded to show that at this site, the protein may repress TCF transcriptional activity and inhibit the binding of the β-catenin/TCF complex to DNA.
A final component of the β-catenin pathways that deserves future study for potential roles in colorectal carcinogenesis is the cytosolic degradation of the protein. Inhibition of such degradation could be predicted to encourage nuclear translocation. Searches for epigenetic and genetic anomalies of the β-TrCP and the newly described Siah-1 13,14 degradation pathways in CRC would, therefore, be particularly informative.
Conclusions and Further Areas for Study
This review has shown that the protein β-catenin interacts with components of almost all stages and aspects of colorectal carcinogenesis via the adenoma-carcinoma sequence (Figure 3) ▶ . Nuclear β-catenin accumulation is seen in aberrant crypt foci and adenomas and, in the latter case, may explain, for example, the up-regulation of cyclin D1 and gastrin expression seen at this early stage of colorectal carcinogenesis. 80,86 This nuclear accumulation is retained through the sequence to primary adenocarcinoma and secondary deposits of CRC, where it may then play a role in stromal invasion by promoting MMP-7 and uPAR expression. In this way and by linking several components of colorectal carcinogenesis, β-catenin seems to act as a key and central molecule to this process (Figure 3) ▶ . Whether increased nuclear β-catenin signaling is de riguer for the development of CRC is uncertain as at least 20% of these cancers show no immunohistochemical evidence of nuclear accumulation of the protein. 9,133 Although different components of the β-catenin pathways may be deranged in microsatellite unstable CRCs (eg, less APC mutations, 134 more TCF-4 mutations, 74 more AXIN2 mutations 67 ), the rate of this nuclear accumulation is no higher in these cancers compared to their microsatellite-stable counterparts. 135 Finally, we (unpublished observations) and others 136 have recently found that dysregulation of β-catenin protein expression is less commonly observed in ulcerative colitis-related CRCs than in their sporadic counterparts. These observations have, however, all been based on semiquantitative analyses using immunohistochemical techniques and the sensitivity of these techniques in detecting activation of the β-catenin signaling pathway needs to be considered. Regardless of the extent to which CRCs may show abnormalities of the β-catenin pathways, it seems clear that their common pathway and the key to the protein’s oncogenic effects is the translocation of β-catenin into the nucleus and its interaction with DNA. If our current knowledge of β-catenin’s role in colorectal carcinogenesis is to be translated into therapeutic and/or preventative interventions, an obvious aim would, therefore, be to block this common pathway. Deciding how to target this pathway is being aided by emerging knowledge about β-catenin’s nuclear interactions. For example, the protein is now known to enter the nucleus by binding directly to the nuclear pore machinery, a process that is inhibited by an as yet unidentified but potentially therapeutically useful cytosolic factor. 137
Figure 3.

Summary of regulators, effectors, and genetic aberrations of components of β-catenin pathways, which relate to colorectal carcinogenesis. Those preceded by “?” represent pathways or changes that have yet to be unequivocally demonstrated in colorectal carcinoma. HGF, hepatocyte growth factor; PKC-βΙΙ, protein kinase C-βΙΙ; TFF-3, trefoil factor 3.
If β-catenin-directed interventions are to be developed, much more, however, needs to be learned about the physiological role of nuclear β-catenin signaling in human tissues, particularly if side effects are to be avoided. Learning more about β-catenin’s functions in other tissues may, in turn, further expand our understanding of colorectal carcinogenesis. For example, β-catenin pathways are thought to play an important role in maintaining endothelial cell survival as is required for angiogenesis. 138 Further, nuclear β-catenin signaling has recently been shown, in fibroblasts, to increase production of fibronectin, 139 an extracellular compound to which colorectal cancer cells adhere during invasion. 140 A final area for future study is the regulation of β-catenin gene transcription. Aside from the rare example of butyrate, 125 little is known about molecules and mechanisms that may influence the actual synthesis of β-catenin. Exploring whether and, if so, how β-catenin gene transcription interacts with its subcellular movements and its degradation is likely to further complicate what we know of the protein in colorectal carcinogenesis, but may eventually reveal other possible targets for therapeutic and/or preventative intervention.
Footnotes
Address reprint requests to Dr. Massimo Pignatelli, Division of Histopathology, Department of Pathology and Microbiology, Marlborough St., Bristol BS2 8HW, UK. E-mail: massimo.pignatelli@bristol.ac.uk.
References
- 1.Kemler R, Ozawa M: Uvomorulin-catenin complex: cytoplasmic anchorage of a Ca2+-dependent cell adhesion molecule. Bioessays 1989, 11:88-91 [DOI] [PubMed] [Google Scholar]
- 2.Morin PJ: β-Catenin signaling and cancer. Bioessays 1999, 21:1021-1030 [DOI] [PubMed] [Google Scholar]
- 3.Kikuchi A: Regulation of β-catenin signaling in the Wnt pathway. Biochem Biophys Res Commun 2000, 268:243-248 [DOI] [PubMed] [Google Scholar]
- 4.Potter JD: Colorectal cancer: molecules and populations. J Natl Cancer Inst 1999, 91:916-932 [DOI] [PubMed] [Google Scholar]
- 5.Ilyas M, Tomlinson IPM: The interactions of APC, E-cadherin and β-catenin in tumour development and progression. J Pathol 1997, 182:128-137 [DOI] [PubMed] [Google Scholar]
- 6.Behrens J, Birchmeier W: Cell-cell adhesion in invasion and metastasis of carcinomas. Cancer Treat Res 1994, 71:251-266 [DOI] [PubMed] [Google Scholar]
- 7.Su LK, Vogelstein B, Kinzler KW: Association of the APC tumor suppressor protein with catenins. Science 1993, 262:1731-1734 [DOI] [PubMed] [Google Scholar]
- 8.Hülsken J, Birchmeier W, Behrens J: E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol 1994, 127:2061-2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hao X, Tomlinson I, Ilyas M, Palazzo JP, Talbot IC: Reciprocity between membranous and nuclear expression of β-catenin in colorectal tumors. Virchows Arch 1997, 431:167-172 [DOI] [PubMed] [Google Scholar]
- 10.Müller T, Choidas A, Reichmann E, Ullrich A: Phosphorylation and free pool of β-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J Cell Biol 1999, 274:10173-10183 [DOI] [PubMed] [Google Scholar]
- 11.Hiscox S, Jiang WG: Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun 1999, 261:406-411 [DOI] [PubMed] [Google Scholar]
- 12.Polakis P, Hart M, Rubinfeld B: Defects in the regulation of β-catenin in colorectal cancer. Adv Exp Med Biol 1999, 470:23-32 [DOI] [PubMed] [Google Scholar]
- 13.Matsuzawa S, Reed JC: Siah-1, SIP and Ebi collaborate in a novel pathway for β-catenin degradation linked to p53 responses. Mol Cell 2001, 7:915-926 [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N: Siah-1 mediates a novel β-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell 2001, 7:927-936 [DOI] [PubMed] [Google Scholar]
- 15.Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF, Waterman ML: Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet 2001, 28:53-57 [DOI] [PubMed] [Google Scholar]
- 16.Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, Logtenberg T, Clevers H: Synergy between tumor suppressor APC and the β-catenin-Tcf4 target Tcf1. Science 1999, 285:1923-1926 [DOI] [PubMed] [Google Scholar]
- 17.Sadot E, Simcha I, Shtutman M, Ben-Ze’ev A, Geiger B: Inhibition of β-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci USA 1998, 95:15339-15344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscox SE, Hallett MB, Puntis MC, Nakamura T, Jiang WG: Expression of the HGF/SF receptor, c-met, and its ligand in human colorectal cancers. Cancer Invest 1997, 15:513-521 [DOI] [PubMed] [Google Scholar]
- 19.Nakae S, Shimada E, Urakawa T: Study of c-erbB-2 protein and epidermal growth factor receptor expression and DNA ploidy pattern in colorectal carcinoma. J Surg Oncol 1993, 54:246-251 [DOI] [PubMed] [Google Scholar]
- 20.Ito M, Yoshida K, Kyo E, Ayhan A, Nakayama H, Yasui W, Ito H, Tahara E: Expression of several growth factors and their receptor genes in human colon carcinomas. Virchows Arch 1990, 59:173-178 [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarty S, Rajagopal S, Huang S: Expression of antisense epidermal growth factor receptor mRNA downmodulates the malignant behaviour of human colon cancer cells. Clin Exp Metastasis 1995, 13:191-195 [DOI] [PubMed] [Google Scholar]
- 22.Jiang WG, Lloyds D, Puntis MC, Nakamura T, Hallett MB: Regulation of spreading and growth of colon cancer cells by hepatocyte growth factor. Clin Exp Metastasis 1993, 11:235-242 [DOI] [PubMed] [Google Scholar]
- 23.Hazan RB, Norton L: The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 1998, 273:9078-9084 [DOI] [PubMed] [Google Scholar]
- 24.Hiscox S, Jiang WG: Hepatocyte growth factor/scatter factor disrupts epithelial tumour cell-cell adhesion: involvement of β-catenin. Anticancer Res 1999, 19:509-518 [PubMed] [Google Scholar]
- 25.Kanai Y, Ochiai A, Shibata T, Oyama T, Ushijima S, Akimoto S, Hirohashi S: c-erbB-2 gene product directly associates with β-catenin and plakoglobin. Biochem Biophys Res Commun 1995, 208:1067-1072 [DOI] [PubMed] [Google Scholar]
- 26.Bos JL: p21ras: an oncoprotein functioning in growth factor-inducing signal transduction. Eur J Cancer 1995, 31A:1051-1054 [DOI] [PubMed] [Google Scholar]
- 27.Taupin D, Ooi K, Yeomans N, Giraud A: Conserved expression of intestinal trefoil factor in the human colonic adenoma-carcinoma sequence. Lab Invest 1996, 75:25-32 [PubMed] [Google Scholar]
- 28.Kinch MS, Clark GJ, Der CJ, Burridge K: Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia. J Cell Biol 1995, 130:461-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Efstathiou JA, Noda M, Rowan A, Dixon C, Chinery R, Jawhari A, Hattori T, Wright NA, Bodmer WF, Pignatelli M: Intestinal trefoil factor controls the expression of the adenomatous polyposis coli-catenin and the E-cadherin-catenin complexes in human colon carcinoma cells. Proc Natl Acad Sci USA 1998, 95:3122-3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter AC, Vaillancourt RR: Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene 1998, 16:1343-1352 [DOI] [PubMed] [Google Scholar]
- 31.Hakam A, Yeatman TJ, Lu L, Mora L, Marcet G, Nicosia SV, Karl RC, Coppola D: Expression of insulin-like growth factor-1 receptor in human colorectal cancer. Hum Pathol 1999, 30:1128-1133 [DOI] [PubMed] [Google Scholar]
- 32.Murray NR, Davidson LA, Chapkin RS, Gustafson WC, Schattenberg DG, Fields AP: Overexpression of protein kinase C βII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J Cell Biol 1999, 145:699-711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Grüning W, Sokol SY, Drummond I, Walz G: The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem 1999, 274:4947-4953 [DOI] [PubMed] [Google Scholar]
- 34.Pongracz J, Clark P, Neoptolemos JP, Lord JM: Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer 1995, 61:35-39 [DOI] [PubMed] [Google Scholar]
- 35.Davidson LA, Jiang YH, Derr JN, Aukema HM, Lupton JR, Chapkin RS: Protein kinase isoforms in human and rat colonic mucosa. Arch Biochem Biophys 1994, 312:547-553 [DOI] [PubMed] [Google Scholar]
- 36.: International Polycystic Kidney Disease Consortium: Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell 1995, 81:289-298 [DOI] [PubMed] [Google Scholar]
- 37.Huan Y, van Adelsberg J: Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest 1999, 104:1459-1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alevizopoulos A, Mermod N: Transforming growth factor-beta: the breaking open of a black box. Bioessays 1997, 19:581-591 [DOI] [PubMed] [Google Scholar]
- 39.Chakrabarty S, Fan D, Varani J: Modulation of differentiation and proliferation of human colon carcinoma cells by transforming growth factor beta 1 and beta 2. Int J Cancer 1990, 46:493-499 [DOI] [PubMed] [Google Scholar]
- 40.Yamaguchi K: Identification of a member of the MAPKKK family as a potential mediator of TGFβ signal transduction. Science 1995, 270:2008-2011 [DOI] [PubMed] [Google Scholar]
- 41.Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, Matsumoto K: The TAK1-NLK-MAPK-related pathway antagonizes signaling between β-catenin and transcription factor TCF. Nature 1999, 399:798-802 [DOI] [PubMed] [Google Scholar]
- 42.Nishita M, Hiashimoto M, Ogata S, Laurent MN, Ueno N, Shibuya H, Cho KWY: Interaction between Wnt and TGFβ signaling pathways during formation of Spemann’s organizer. Nature 2000, 403:781-784 [DOI] [PubMed] [Google Scholar]
- 43.Ilyas M, Efstathiou JA, Straub J, Kim HC, Bodmer WF: Transforming growth factor β stimulation of colorectal cancer cell lines: type II receptor bypass and changes in adhesion molecule expression. Proc Natl Acad Sci USA 1999, 96:3087-3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee MO, Han SY, Jiang S, Park JH, Kim SJ: Differential effects of retinoic acid on growth and apoptosis in human colon cancer cell lines associated with the induction of retinoic acid receptor β. Biochem Pharmacol 2000, 59:485-496 [DOI] [PubMed] [Google Scholar]
- 45.Thomas MG, Tebbutt S, Williamson RCN: Vitamin D and its metabolites inhibit cell proliferation in human rectal mucosa and a colon cancer cell line. Gut 1992, 33:1660-1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Easwaran V, Pishvaian M, Salimuddin, Byers S: Cross-regulation of β-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol 1999, 9:1415-1418 [DOI] [PubMed] [Google Scholar]
- 47.Nakamori S, Ota DM, Cleary KR, Shirotani K, Irimura T: MUC1 mucin expression as a marker of progression and metastasis of human colorectal carcinoma. Gastroenterology 1994, 106:353-361 [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto M, Bharti A, Li Y, Kufe D: Interaction of the DF3/MUC1 breast carcinoma-associated antigen and β-catenin in cell adhesion. J Cell Biol 1997, 272:12492-12494 [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Bharti A, Chen D, Gong J, Kufe D: Interaction between glycogen synthase kinase 3β with the DF3/MUC1 carcinoma-associated antigen and β-catenin. Mol Cell Biol 1998, 18:7216-7224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fajas L, Auboeuf D, Raspe E, Schoojans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, Vidal-Puig A, Flier J, Briggs MR, Staels B, Vidal H, Auwerx J: The organization, promoter analysis, and expression of the human PPARγ gene. J Biol Chem 1997, 272:18779-18789 [DOI] [PubMed] [Google Scholar]
- 51.Giovanucci E, Willett WC: Dietary factors and risk for colon cancer. Ann Med 1994, 26:443-452 [DOI] [PubMed] [Google Scholar]
- 52.Wasan HS, Novelli M, Bee J, Bodmer WF: Dietary fat influences on polyp phenotype in multiple intestinal neoplasia mice. Proc Natl Acad Sci USA 1997, 94:3308-3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lefebvre A-M, Chen I, Desreumaux P, Najib J, Fruchart J-C, Geboes K, Briggs M, Heyman R, Auwerx J: Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCmin/+ mice. Nat Med 1998, 4:1053-1057 [DOI] [PubMed] [Google Scholar]
- 54.McEntee MF, Chiu C-H, Whelan J: Relationship of β-catenin and Bcl-2 expression to sulindac-induced regression of intestinal tumors in Min mice. Carcinogenesis 1999, 20:635-640 [DOI] [PubMed] [Google Scholar]
- 55.Smith ML, Hawcroft G, Hull MA: The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: evidence of different mechanisms of action. Eur J Cancer 2000, 36:664-674 [DOI] [PubMed] [Google Scholar]
- 56.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998, 58:1130-1134 [PubMed] [Google Scholar]
- 57.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancer by mutations of β-catenin or APC. Science 1997, 275:1787-1790 [DOI] [PubMed] [Google Scholar]
- 58.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF: β-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA 1997, 94:10330-10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Romagnolo B, Berrebi D, Saadi-Keddoucci S, Porteu A, Pichard A, Peuchmaur M, Vandewalle A, Kahn A, Perret C: Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated β-catenin. Cancer Res 1999, 59:3875-3879 [PubMed] [Google Scholar]
- 60.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM: Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J 1999, 18:5931-5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoemaker AR, Gould KA, Luongo C, Moser AR, Dove WF: Study of neoplasia in the Min mouse. Biochim Biophys Acta 1997, 1332:F25-F48 [DOI] [PubMed] [Google Scholar]
- 62.O’Sullivan MJ, McCarthy TV, Doyle CT: Familial adenomatous polyposis. From bedside to benchside. Am J Clin Pathol 1998, 109:521-526 [DOI] [PubMed] [Google Scholar]
- 63.Polakis P: The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta 1997, 1332:F127-F147 [DOI] [PubMed] [Google Scholar]
- 64.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishoguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y: AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 2000, 24:245-250 [DOI] [PubMed] [Google Scholar]
- 65.Webster MT, Rozycka M, Sara E, Davis E, Smalley M, Young N, Dale TC, Wooster R: Sequence variants of the axin gene in breast, colon, and other cancers: an analysis of mutations that interfere with GSK3 binding. Gene Chromosom Cancer 2000, 28:443-453 [PubMed] [Google Scholar]
- 66.Mai M, Qian C, Yokomizo A, Smith DI, Liu W: Cloning of the human homolog of conductin (AXIN2), a gene mapping to chromosome 17q23–q24. Genomics 1999, 55:341-344 [DOI] [PubMed] [Google Scholar]
- 67.Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C, Christensen E, Schmidt SS, Roche PC, Smith DI, Thibodeau SN: Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet 2000, 26:146-147 [DOI] [PubMed] [Google Scholar]
- 68.Vider B-Z, Zimber A, Chastre E, Prevot S, Gespach C, Estlein D, Wolloch Y, Tronick SR, Gazit A, Yaniv A: Evidence for the involvement of the Wnt 2 gene in human colorectal cancer. Oncogene 1996, 12:153-158 [PubMed] [Google Scholar]
- 69.Smith K, Bui TD, Poulsom R, Kaklamanis L, Williams G, Harris AL: Up-regulation of macrophage wnt gene expression in adenoma-carcinoma progression of human colorectal cancer. Br J Cancer 1999, 81:496-502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brabletz T, Jung A, Hermann K, Gunther K, Hohenberger W, Kirchner T: Nuclear overexpression of the oncoprotein β-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 1998, 194:701-704 [DOI] [PubMed] [Google Scholar]
- 71.Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R: A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382:225-230 [DOI] [PubMed] [Google Scholar]
- 72.Tanaka S, Akiyoshi T, Mori M, Wands JR, Sugimachi K: A novel frizzled gene identified in human esophageal carcinoma mediates APC/β-catenin signals. Proc Natl Acad Sci USA 1998, 95:10164-10169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duval A, Rolland S, Tubacher E, Bui H, Thomas G, Hamelin R: The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res 2000, 60:3872-3879 [PubMed] [Google Scholar]
- 74.Duval A, Gayet J, Zhou X-P, Iacopetta B, Thomas G, Hamelin R: Frequent frameshift mutations of the TCF-4 gene in colorectal cancers with microsatellite instability. Cancer Res 1999, 59:4213-4215 [PubMed] [Google Scholar]
- 75.He T-C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-MYC as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 76.Rodriquez-Alfageme C, Stanbridge EJ, Astrin SM: Suppression of deregulated c-MYC expression in human colon carcinoma cells by chromosome 5 transfer. Proc Natl Acad Sci USA 1992, 89:1482-1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith DR, Myint T, Goh HS: Over-expression of the c-myc proto-oncogene in colorectal carcinoma. Br J Cancer 1993, 68:407-413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T: Expression of nuclear β-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol 2000, 156:865-870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kolligs FT, Kolligs B, Hajra KM, Hu G, Tani M, Cho KR, Fearon ER: γ-Catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of β-catenin: Genes Dev 2000, 14:1319-1331 [PMC free article] [PubMed] [Google Scholar]
- 80.Arber N, Hibshoosh H, Moss MF, Sutter T, Zhang Y, Begg, Wang S, Weinstein B, Holt PR: Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996, 110:669-674 [DOI] [PubMed] [Google Scholar]
- 81.Sutter T, Doi S, Carnevale KA, Arber N, Weinstein IB: Expression of cyclins D1 and E in human colon adenocarcinomas. J Med 1997, 28:285-309 [PubMed] [Google Scholar]
- 82.Tetsu O, McCormick F: β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398:422-426 [DOI] [PubMed] [Google Scholar]
- 83.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A: The cyclin D1 is the target gene of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999, 96:5522-5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bissonnette M, Khare S, von Lintig FC, Wali RK, Nguyen L, Zhang Y, Hart J, Skarosi S, Varki N, Boss GR, Brasitus TA: Mutational and nonmutational activation of p21ras in rat colonic azoxymethane-induced tumors: effects on mitogen-activated protein kinase, cyclooxygenase-2, and cyclin D1. Cancer Res 2000, 60:4602-4609 [PubMed] [Google Scholar]
- 85.Koh TJ, Bulitta CJ, Fleming JV, Dockray GJ, Varro A, Wang TC: Gastrin is a target of the β-catenin/TCF-4 growth-signaling pathway in a model of intestinal polyposis. J Clin Invest 2000, 106:533-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watson SA, Michaeli D, Morris RM, McWilliams D, Smith AM: The role of gastrin in the adenoma: carcinoma sequence in the colon. Digestion 1999, 60:600A [Google Scholar]
- 87.Van Solinge WW, Nielsen FC, Friis-Hansen L, Falkmer UG, Rehfeld JF: Expression but incomplete maturation of progastrin in colorectal carcinomas. Gastroenterology 1993, 104:1099-1107 [DOI] [PubMed] [Google Scholar]
- 88.Giovannucci E: The prevention of colorectal cancer by aspirin use. Biomed Pharmacother 1999, 53:303-308 [DOI] [PubMed] [Google Scholar]
- 89.Kargman SL, O’Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S: Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res 1995, 55:2556-2559 [PubMed] [Google Scholar]
- 90.Oshima M, Dinchuk JE, Kargmann SL, Oshima H, Hancock B, Kwong E, Trzasko JM, Evans JF, Taketo MM: Suppression of intestinal polyposis in APCΔ716 knockout mice by inhibition of cyclooxygenase 2. Cell 1996, 87:803-809 [DOI] [PubMed] [Google Scholar]
- 91.Hsi LC, Angerman-Stewart J, Eling TE: Introduction of full-length APC modulates cyclooxygenase-2 expression in HT-29 human colorectal carcinoma cells at the translational level. Carcinogenesis 1999, 20:2045-2049 [DOI] [PubMed] [Google Scholar]
- 92.Howe LR, Subbaramaiah K, Chung WJ, Dannenberg AJ, Brown AMC: Transcriptional activation of Cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res 1999, 59:1572-1577 [PubMed] [Google Scholar]
- 93.Mei JM, Hord NG, Winterstein DF, Donald SP, Phang JM: Differential expression of prostaglandin endoperoxide H synthase-2 and formation of activated β-catenin-LEF-1 transcription complex in mouse colonic epithelial cells contrasting in Apc. Carcinogenesis 1999, 20:737-740 [DOI] [PubMed] [Google Scholar]
- 94.Newell KJ, Witty JP, Rodgers WH, Matrisian LM: Expression and localization of matrix-degrading metalloproteinases during colorectal carcinogenesis. Mol Carcinog 1994, 10:199-206 [DOI] [PubMed] [Google Scholar]
- 95.Wilson CL, Heppner KJ, Labosky PA, Hogan BLM, Matrisian LM: Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA 1997, 94:1402-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T: β-Catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 1999, 155:1033-1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crawford HC, Fingleton BM, Rudolph-Owen LA, Heppner Goss KJ, Rubinfeld B, Polakis P, Matrisian LM: The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene 1999, 18:2882-2891 [DOI] [PubMed] [Google Scholar]
- 98.Reuning U, Magdolen V, Wilhelm O, Fischer K, Lutz V, Graeff H, Schmitt M: Multifunctional potential of the plasminogen activation system in tumor invasion and metastasis. Int J Oncol 1998, 13:893-906 [DOI] [PubMed] [Google Scholar]
- 99.Ellis V, Behrendt N, Dano K: Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem 1991, 266:12752-12758 [PubMed] [Google Scholar]
- 100.Buo L, Meling GI, Karlsrud TS, Johansen HT: Aasen AO: Antigen levels of urokinase plasminogen activator and its receptor at the tumor-host interface of colorectal adenocarcinomas are related to tumor aggressiveness. Hum Pathol 1995, 26:1133-1138 [DOI] [PubMed] [Google Scholar]
- 101.Nakata S, Ito K, Fujimori M, Shingu K, Kajikawa S, Adachi W, Matsuyama I, Tsuchiya S, Kuwano M, Amano J: Involvement of vascular endothelial growth factor and urokinase-type plasminogen activator receptor in microvessel invasion in human colorectal cancers. Int J Cancer 1998, 79:179-186 [DOI] [PubMed] [Google Scholar]
- 102.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C: Target genes of β-catenin-T cell factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999, 96:1603-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goodison S, Urquidi V, Tarin D: CD44 cell adhesion molecules. Mol Pathol 1999, 52:189-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Imazeki F, Yokosuka O, Yamaguchi T, Ohto M, Isono K, Omata M: Expression of variant CD44-messenger RNA in colorectal adenocarcinomas and adenomatous polyps in humans. Gastroenterology 1996, 110:362-368 [DOI] [PubMed] [Google Scholar]
- 105.Mulder JW, Kruyt PM, Sewnath M, Oosting J, Seldenrijk CA, Weidema WF, Offerhaus GJ, Pals ST: Colorectal cancer prognosis and expression of exon-v6-containing CD44 proteins. Lancet 1994, 344:1470-1472 [DOI] [PubMed] [Google Scholar]
- 106.Kim H, Yang XL, Rosada C, Hamilton S, August T: CD44 expression in colorectal adenomas is an early event occurring prior to K-ras and p53 mutation. Arch Biochem Biophys 1994, 310:504-507 [DOI] [PubMed] [Google Scholar]
- 107.Wielenga VJM, Smits R, Korinek V, Smit L, Kielman M, Fodde R, Clevers H, Pals ST: Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol 1999, 154:515-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Linn SC, Giaccone G: MDR1/P-glycoprotein expression in colorectal cancer. Eur J Cancer 1995, 31:1291-1294 [DOI] [PubMed] [Google Scholar]
- 109.Yamada T, Takaoka AS, Naishiro Y, Hayashi R, Maruyama K, Maesawa C, Ochiai A, Hirohashi S: Transactivation of the multidrug resistance 1 gene by T-cell factor 4/β-catenin complex in early colorectal carcinogenesis. Cancer Res 2000, 60:4761-4766 [PubMed] [Google Scholar]
- 110.Butler LM, Hewett PJ, Fitridge RA, Cowled PA: Deregulation of apoptosis in colorectal carcinoma: theoretical and therapeutic implications. Aust NZJ Surg 1999, 69:88-94 [DOI] [PubMed] [Google Scholar]
- 111.Damalas A, Ben-Ze’ev A, Simcha I, Shtutman M, Fernando J, Leal M, Zhurinsky J, Geiger B, Oren M: Excess β-catenin promotes accumulation of transcriptionally active p53. EMBO J 1999, 18:3053-3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abarzua P, LoSardo JE, Gubler ML, Neri A: Microinjection of monoclonal antibody PAb421 into human SW480 colorectal carcinoma cells restores the transcription activation function to mutant p53. Cancer Res 1995, 55:3490-3494 [PubMed] [Google Scholar]
- 113.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17:3247-3259 [DOI] [PubMed] [Google Scholar]
- 114.Tsujii M, DuBois RN: Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 1995, 83:493-501 [DOI] [PubMed] [Google Scholar]
- 115.Sorenson CM: Nuclear localization of beta-catenin and loss of apical brush border actin in cystic tubules of bcl-2 −/− mice. Am J Physiol 1999, 276:F210-F217 [DOI] [PubMed] [Google Scholar]
- 116.Lorentz O, Cadoret A, Duluc I, Capeau J, Gespach C, Cherqui G, Freund JN: Downregulation of the colon tumour-suppressor homeobox gene Cdx-2 by oncogenic ras. Oncogene 1999, 18:87-92 [DOI] [PubMed] [Google Scholar]
- 117.Soubeyran P, Andre F, Lissitzky JC, Mallo GV, Moucadel V, Roccabianca M, Rechreche H, Marvaldi J, Dikic I, Dagorn JC, Iovanna JL: Cdx1 promotes differentiation in a rat intestinal epithelial cell line. Gastroenterology 1999, 117:1326-1338 [DOI] [PubMed] [Google Scholar]
- 118.Chawengsaksophak K, James R, Hammond V, Kontgen F, Beck F: Homeosis and intestinal tumours in Cdx2 mutant mice. Nature 1997, 386:84-87 [DOI] [PubMed] [Google Scholar]
- 119.Lickert H, Domon C, Huls G, Wehrle C, Duluc I, Clevers H, Meyer BI, Freund JN, Kemler R: Wnt/β-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development 2000, 127:3805-3813 [DOI] [PubMed] [Google Scholar]
- 120.da Costa LT, He TC, Yu J, Sparks AB, Morin PJ, Polyak K, Laken S, Vogelstein B, Kinzler KW: CDX2 is mutated in a colorectal cancer with normal APC/β-catenin signaling. Oncogene 1999, 18:5010-5014 [DOI] [PubMed] [Google Scholar]
- 121.Ee HC, Erler T, Bhathal PS, Young GP, James RJ: Cdx-2 homeodomain protein expression in rat and human colorectal adenoma and carcinoma. Am J Pathol 1995, 147:586-592 [PMC free article] [PubMed] [Google Scholar]
- 122.Silberg DG, Furth EE, Taylor JK, Schuck T, Chiou T, Traber PG: CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology 1997, 113:478-486 [DOI] [PubMed] [Google Scholar]
- 123.van de Wetering M, Oosterwegel M, Holstege F, Dooyes D, Suijkerbuijk R, Geurts van Kessel A, Clevers H: The human T cell transcription factor-1 gene. Structure, localization, and promoter characterization. J Biol Chem 1992, 267:8530-8536 [PubMed] [Google Scholar]
- 124.Ben-Ze’ev A: The dual role of cytoskeletal anchor proteins in cell adhesion and signal transduction. Ann NY Acad Sci 1997, 886:37-47 [DOI] [PubMed] [Google Scholar]
- 125.Barshishat M, Polak-Charcon S, Schwartz B: Butyrate regulates E-cadherin transcription, isoform expression and intracellular position in colon cancer cells. Br J Cancer 2000, 82:195-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Velazquez OC, Lederer HM, Rombeau JL: Butyrate and the colonocyte. Implications for neoplasia. Dig Dis Sci 1996, 41:727-739 [DOI] [PubMed] [Google Scholar]
- 127.Heerdt BG, Houston MA, Augenlicht LH: Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res 1995, 54:3288-3293 [PubMed] [Google Scholar]
- 128.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R: E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J Cell Sci 1999, 112:1237-1245 [DOI] [PubMed] [Google Scholar]
- 129.Huber O, Korn R, McLaughlin J, Oshugi M, Hermann BG, Kemler R: Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech Dev 1996, 59:3-11 [DOI] [PubMed] [Google Scholar]
- 130.Ilyas M, Tomlinson IP, Hanby A, Talbot IC, Bodmer WF: Allele loss, replication errors and loss of expression of E-cadherin in colorectal cancers. Gut 1997, 40:654-659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hirohashi S: Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol 1998, 153:333-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Giannini AL, Vivanco MD, Kypta RM: α-Catenin inhibits β-catenin signaling by preventing formation of a β-catenin*T-cell factor*DNA complex. J Biol Chem 2000, 275:21883-21888 [DOI] [PubMed] [Google Scholar]
- 133.Hugh TJ, Dillon SA, O’Dowd G, Getty B, Pignatelli M, Poston GJ, Kinsella AR: β-Catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer 1999, 82:504-511 [DOI] [PubMed] [Google Scholar]
- 134.Toft NJ, Arends MJ: DNA mismatch repair and colorectal cancer. J Pathol 1998, 185:123-129 [DOI] [PubMed] [Google Scholar]
- 135.Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y, Koike M, Shitara N, Iwama T, Kuroki T: Frequent mutation of β-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res 1999, 59:4506-4509 [PubMed] [Google Scholar]
- 136.Aust DE, Baretton GB, Waldman FM, Löhrs U: Molecular carcinogenesis in ulcerative colitis-related and sporadic colorectal cancer—differences and similarities. Verh Dtsch Ges Path 1999, 83:130-138 [PubMed] [Google Scholar]
- 137.Fagotto F, Glück U, Gumbiner BM: Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of β-catenin. Curr Biol 1998, 8:181-190 [DOI] [PubMed] [Google Scholar]
- 138.Carmeliet P, Lampugnani MG, Moons L, Breviaro F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E: Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 1999, 98:147-157 [DOI] [PubMed] [Google Scholar]
- 139.Gradl D, Kuhl M, Wedlich D: The Wnt/Wg signal transducer β-catenin controls fibronectin expression. Mol Cell Biol 1999, 19:5576-5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ebert EC: Mechanisms of colon cancer binding to substratum and cells. Dig Dis Sci 1996, 41:1551-1556 [DOI] [PubMed] [Google Scholar]