

Abstract
We examined presynaptic cholinergic markers and β-secretase activity during progressive central nervous system amyloidogenesis in Tg2576 Alzheimer mice (transgenic for human amyloid precursor protein Swedish mutation; hAPPswe). At 14, 18, and 23 months of age there were no significant differences between wild-type and transgenic mice in four distinct central nervous system cholinergic indices—choline acetyltransferase and acetylcholinesterase activities, and binding to vesicular acetylcholine transporter and Na+-dependent high-affinity choline uptake sites. A novel enzyme-linked immunosorbent assay measuring only the secreted human β-secretase cleavage product (APPsβswe) of APPswe also revealed no change with aging in Tg2576 mouse brain. In contrast, transgenic but not wild-type mice exhibited an age-dependent increase in soluble Aβ40 and Aβ42 levels and progressive amyloid deposition in brain. Thus, aging Tg2576 mice exhibited presynaptic cholinergic integrity despite progressively increased soluble Aβ40 and Aβ42 levels and amyloid plaque density in brain. Older Tg2576 mice may best resemble preclinical or early stages of human Alzheimer’s disease with preserved presynaptic cholinergic innervation. Homeostatic APPsβswe levels with aging suggest that progressive amyloid deposition in brain results not from increased β-secretase cleavage of APP but from impaired Aβ/amyloid clearance mechanisms.
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive dementia and the accumulation of neuritic plaques and neurofibrillary tangles in limbic and neocortical brain regions. Cholinergic deficits were the first neurochemical changes recognized in AD brain 1-3 as shown by decreases in choline acetyltransferase (ChAT; acetyl-CoA: choline O-acetyltransferase; EC 2.3.1.6) and acetylcholinesterase (AChE; acetylcholine acetylhydrolase; EC 3.1.1.7) activities with perhaps compensatory increases in Na+-dependent high-affinity choline uptake (SDHACU) sites. 4-6 The major cholinergic nuclei of brain affected are in the basal forebrain 7,8 that project to the neocortex, entorhinal area, and hippocampus. 9,10 To date, AChE inhibitors are the only class of drugs approved by the United States Food and Drug Administration for the treatment of cognitive deficits caused by AD.
Because basal forebrain cholinergic neurons project widely to hippocampus and neocortex, it was suggested that cholinergic denervation is linked to or even induces neurodegenerative changes, including amyloid deposition, in AD cortex. 11-13 This cholinergic hypothesis has been supplanted by recent genetic, biochemical, and pathological evidence supporting the amyloid hypothesis of AD pathogenesis. 14 Despite recent progress, however, the temporal course of cholinergic deficits in the clinical and pathological staging of human AD remains controversial. Some studies suggest that cortical cholinergic deficits occur early and in parallel with dementia and amyloid plaque deposition. 15,16 Other studies of human AD brain demonstrate cholinergic deficits only in very late stages of disease. 17,18
Studies of central nervous system (CNS) cholinergic markers in human amyloid precursor protein transgenic (hAPP tg) mouse models of AD are limited, and report only on immunohistochemical and microscopic analysis. For example, brain sections from hAPP Swedish mutation (hAPPswe) tg mice (not Tg2576) at 12 months of age exhibit AChE-immunoreactive dystrophic fibers in the vicinity of amyloid plaques. 19 Similarly, in 17- to 22-month hAPP V642I (London mutation) tg mice AChE-positive fibers are reorganized within the hippocampus and amyloid plaques are surrounded by dystrophic AChE-positive fibers. 20 In 8-month-old hAPPswe (Tg2576)/hPresenilin-1 M146L double-tg mice vesicular acetylcholine transporter (VAChT)-immunoreactive synapses are decreased in frontal cortex and the size of these synapses is reduced in hippocampus; at this age there is extensive amyloid plaque deposition in brain. In contrast, in Tg2576 mice at 8 months of age there are no amyloid plaques in brain and VAChT-immunostained synapses are increased in frontal and parietal cortex. 21
Based on this evidence, we pursued a macroscopic study of aging Tg2576 mouse brain by measuring four cholinergic markers—VAChT, SDHACU, ChAT, and AChE—during progressive CNS amyloidogenesis in 14-, 18-, and 23-month-old mice. With aging, these mice exhibit a partial AD-like phenotype that includes learning and memory deficits and pathological findings of amyloid plaque deposition, neuritic changes, gliosis, inflammatory responses, phosphorylated tau epitopes, α-synuclein-immunopositive (Lewy-like) neurites, and increased Aβ40 and Aβ42 levels, but not neuronal loss or neurofibrillary tangles. 22-40 VAChT sites are a specific marker of cholinergic terminals. 41-44 When labeled vesamicol is used with selective blockers, VAChT may be quantified and is useful for assessing loss of cholinergic projections in AD brain both in vitro and in vivo. 45-47 During AD progression in vivo VAChT binding is less affected than postmortem ChAT activity measurements would suggest. 45 ChAT is the hallmark of cholinergic fibers and is present in the striatum/cerebral cortex/cerebellum of adult rats in the ratios ∼26:9:1. 48 Hemicholinium-3 (HC-3) binds specifically to SDHACU sites present on cholinergic neurons; 49 choline uptake is the rate-limiting step in acetylcholine biosynthesis.
In addition to Aβ40 and Aβ42 enzyme-linked immunosorbent assay (ELISA) and histochemical analysis of brain sections, we used a novel ELISA that detects only the secreted β-secretase (BACE) cleavage fragment (APPsβswe) of human APPswe. 50 By measuring both secreted products of BACE cleavage of APPswe (Aβ peptides and APPsβswe) in Tg2576 mouse brain we may indirectly assess a hypothesized increase in BACE activity with aging.
Materials and Methods
Animals
Three male Tg2576 mice (hAPPswe in a C57B6/SJL genetic background; 22 Mayo Clinical Ventures, Minneapolis, MN) were crossed with (C57B6 × SJL) F1 females (Jackson Laboratories, Bar Harbor, ME). At one month of age, progeny were screened for hAPPswe transgenesis by polymerase chain reaction of genomic DNA extracted from tails. The hAPP primers used were 5′-GTG GAT AAC CCC TCC CCC AGC CTA GAC CA-3′ and 5′-CTG ACC ACT CGA CCA GGT TCT GGG T-3′. In transgene-positive mice a 450-bp polymerase chain reaction product was detected on 4% agarose gels. All tg mice were hAPPswe hemizygous (homozygous is lethal). Nontransgenic littermates were used as wild-type (wt) controls. Mice were housed under specific pathogen-free conditions with a 12-hour light-dark cycle and food and water was provided ad libitum. Animal care was in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health. Housing facilities were accredited by the American Association for the Accreditation of Laboratory Animal Care. All experimental procedures were approved by the University of Michigan Committee on Use and Care of Animals.
Frozen and Paraformaldehyde-Fixed Brain
Six wt and six tg mice of three age groups (14, 18, and 23 months) were used in this study. All wt mice were male except for one female in the 23-month age group; all tg mice were female except for one male in the 18-month age group. Male tg mice are more aggressive than wt mice and must be singly-housed; thus, housing costs for aging tg males are prohibitive. Mice were sacrificed painlessly and removed brains were divided sagittally. The left half was frozen onto a gauze-covered glass microscope slide on dry ice with M-1 embedding matrix (Shandon Lipshaw, Pittsburgh, PA). The right half was fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4, at 4°C and processed for paraffin embedding. Sagittal sections of 20 μm were obtained from frozen hemi-brains for as long as striatum, hippocampus, and cerebellum were visible. Every five consecutive sections were grouped and numbered from 1 to 20. The sections from odd numbered groups were collected onto poly-l-lysine-treated glass slides and air-dried at room temperature for 8 hours to overnight. Even numbered sections were microdissected to obtain four brain regions: cortex, striatum, hippocampus, and cerebellum. Both microdissected tissues and cryostat sections were stored at −70°C until further analysis. Paraformaldehyde-fixed hemi-brains were sectioned coronally at 5- to 8-μm thickness and sections were processed for hematoxylin and eosin, Bielschowky silver, and Congo Red staining.
[3H]Vesamicol Binding to VAChT Sites
Tissue sections mounted onto glass slides were thawed at room temperature and prewashed for 5 minutes in PBS-ethylenediaminetetraacetic acid (EDTA), pH 7.4, at room temperature. Sections were then incubated with [3H]vesamicol (specific activity, 35.0 Ci/mmol; final concentration, 10 nmol/L; New England Nuclear, Boston, MA) in PBS-EDTA, pH 7.4, at room temperature for 60 minutes with intermittent agitation. To obtain nonspecific binding, sections were treated as above with the addition of 200 nmol/L (−)methylaminobenzovesamicol. Tissue sections were rinsed in PBS-EDTA, pH 7.4, at 4°C for 1 minute twice, washed in distilled water at 4°C for 5 seconds, air-dried overnight, and exposed to Hyperfilm (Amersham, Arlington Heights, IL) for 6 weeks.
[3H]Hemicholinium-3 (HC-3) Binding to SDHACU Sites
The [3H]HC-3 binding assay was performed as above except: 1) the final concentration of [3H]HC-3 (specific activity, 137.2 Ci/mmol; New England Nuclear) was 2 nmol/L, 2) nonspecific binding was performed with the addition of unlabeled 10 μmol/L HC-3, and 3) the incubation time was 2 hours instead of 1 hour.
Quantitation of autoradiograms from binding assays was performed with the MCID-M5+ image analysis system (Imaging Research Inc., St. Catharines, Ontario, Canada). Briefly, the image was captured by a charge-coupled device video camera and displayed on a computer screen. The total and nonspecific binding activities were measured by outlining the cortex, striatum, hippocampus, and cerebellum. Specific binding was calculated by subtracting nonspecific from total binding. Measures to minimize sampling errors and variability of autoradiography included quantitative image analysis of 10 (for HC-3 binding) or 20 (for vesamicol binding) consecutive tissue sections for each mouse hemi-brain, and by using a standardized [3H] calibration panel with each autoradiogram.
ChAT Activity
ChAT activity was measured by the radioisotopic method of Fonnum. 51 Briefly, 60 μl of 500 nmol/L NaCl, 83 nmol/L PBS (pH 7.4), 13 mmol/L choline bromide, 33 mmol/L EDTA, 0.17 mmol/L physostigmine, and 0.083% Triton X-100, was added to glass tubes at 4°C. Microdissected cortex, hippocampus, and cerebellum were homogenized in 0.5 ml, and striatum in 1 ml, of 100 mmol/L of PBS, pH 7.4, at 4°C with intermittent sonication. Twenty μl of samples and 20 μl of [14C]acetyl CoA (2.25 μCi/ml; final concentration, 0.2 mmol/L; New England Nuclear) were added to the tubes at 4°C. The reaction time was 15 minutes in a shaking water bath at 37°C. The reaction cocktail was washed twice with 2.5 ml of 10 mmol/L PBS, pH 7.4, at 4°C and collected in a scintillation vial. Two ml of acetonitrile/tetraphenylboron and 10 ml of toluene scintillant were added to the vial with repeated vigorous agitation. The samples were left overnight and counted in a scintillation counter. All ChAT assays were measured in triplicate. Protein was determined by a modified bicinchoninic acid method; 52 all samples were measured in triplicate.
Acetylcholinesterase (AChE) Activity
The colorimetric assay for acetylcholinesterase activity by Ellman and colleagues 53 was used. A microtiter plate (Nunc 96-well VWR Scientific, West Chester, PA) was placed on ice and 300 μl of 0.1 mol/L phosphate buffer, pH 8.0, was added, followed by 2 μl of 0.075 mol/L acetylthiocholine iodide and 10 μl of 0.01 mol/L dithiobisnitrobenzoic acid. The brain region homogenates were added as 5-μl aliquots into triplicate wells. Also included were a nonenzymatic protein (bovine serum albumin) and no enzyme blank. The microtiter plate was removed from the ice and gently shaken at room temperature until the plate was read at 405 nm with a Beckman Biomek 1000. The resultant absorbance values were converted into enzyme activity (pmol/min/μg protein).
Aβ40, Aβ42, and APPsβswe ELISA
Brain homogenates for ELISA were the same samples used for measuring ChAT activity. Homogenates were centrifuged at 7840 × g for 5 minutes at 4°C to remove insoluble material. A sandwich ELISA was performed on the supernatants as described 54,55 using BAN50 as the capture antibody and either horseradish peroxidase-coupled BA-27 or BC-05 as the detection antibody for Aβ40 or Aβ42, respectively. BAN-50 is a monoclonal antibody specific for Aβ1-10. All samples were measured in duplicate. APPsβswe was measured with a novel ELISA specific to the β-secretase cleaved fragment of human APPswe. 50
Statistical Analysis
A repeated mixed model of three fixed factors (age, genotype, and region) was used to evaluate factor effects in ChAT and AChE activities, HC-3 binding, and vesamicol binding. Repeated mixed models of two fixed factors (age and region) were used to evaluate factor effects in Aβ40, Aβ42, and APPsβswe levels and the Aβ42/Aβ40 ratio in tg mice. We assumed compound symmetry for the covariance structure in the models. A significance level of 5% was set for each statistical test. Pairwise comparisons between age groups or brain regions were used if applicable. The notation A ∼ B ∼ C > D means that 1) the averages A, B, C, and D are arranged in decreasing order; 2) D is significantly different from the others (A, B, and C); and 3) no significant difference exists between A, B, and C. No adjustment of the significance level was made for multiple tests. GLM and MIXED procedures in the statistical software SAS (PC version 8.1; SAS Institute Inc., Cary, NC) were used for the analyses.
Results
Amyloidogenesis Increases with Age in Tg2576 Mouse Brain
The spatial and temporal progression of amyloid plaque deposition in Tg2576 mouse brain was cortex ≫ hippocampus > striatum = thalamus, but no plaques were found in cerebellum even at 23 months of age (Table 1) ▶ . Although amyloid plaques were rare in the cortex of the 14-month-old age group and not seen in other brain regions, there were significant levels of soluble Aβ40 and Aβ42 (see Figure 5 ▶ ), suggesting that elevations in Aβ40 and Aβ42 concentrations precede their deposition as amyloid. The levels of Aβ40 and Aβ42 increased progressively with age and in parallel in all brain regions from tg mice. At 14 months there were no significant differences in Aβ40 or Aβ42 levels between the four brain regions. At 18 and 23 months, the relative levels of soluble CNS Aβ40 were cortex > striatum ∼ hippocampus > cerebellum; for Aβ42, relative levels were cortex ∼ hippocampus ∼ striatum > cerebellum at 18 and 23 months, with cortex > hippocampus only at 23 months. Interestingly, although the striatum and hippocampus had similar levels of soluble Aβ40 and Aβ42, amyloid plaques were more abundant in the hippocampus. Aβ42 levels were consistently greater than Aβ40 at all ages, and the Aβ42/Aβ40 ratio significantly peaked at 18 months and declined at 23 months (P < 0.03; see Figure 5 ▶ ) suggesting that Aβ42 precedes Aβ40 deposition in brain. No amyloid plaques were detected in 14-, 18-, and 23-month-old wt mouse brain (data not shown). As expected for the human-specific ELISA, no soluble Aβ40 and Aβ42 was detected in wt mouse brain (data not shown).
Table 1.
Pathologic Findings in hAPPswe tg Mouse Brains Based on Bielschowsky Silver and Congo Red Staining of Brain Sections
| Age (months) | |||
|---|---|---|---|
| 14 | 18 | 23 | |
| Amyloid plaques | |||
| Cerebral cortex | + | ++ | +++ |
| Hippocampus | − | +/− | ++ |
| Striatum | − | − | + |
| Thalamus | − | − | + |
| Cerebellum | − | − | − |
| Vascular amyloid | +/− | +/− | + |
−, None; +/−, rare; +, occasional; ++, frequent; and +++, numerous plaques. Vascular amyloid was both meningeal and parenchymal.
Figure 5.
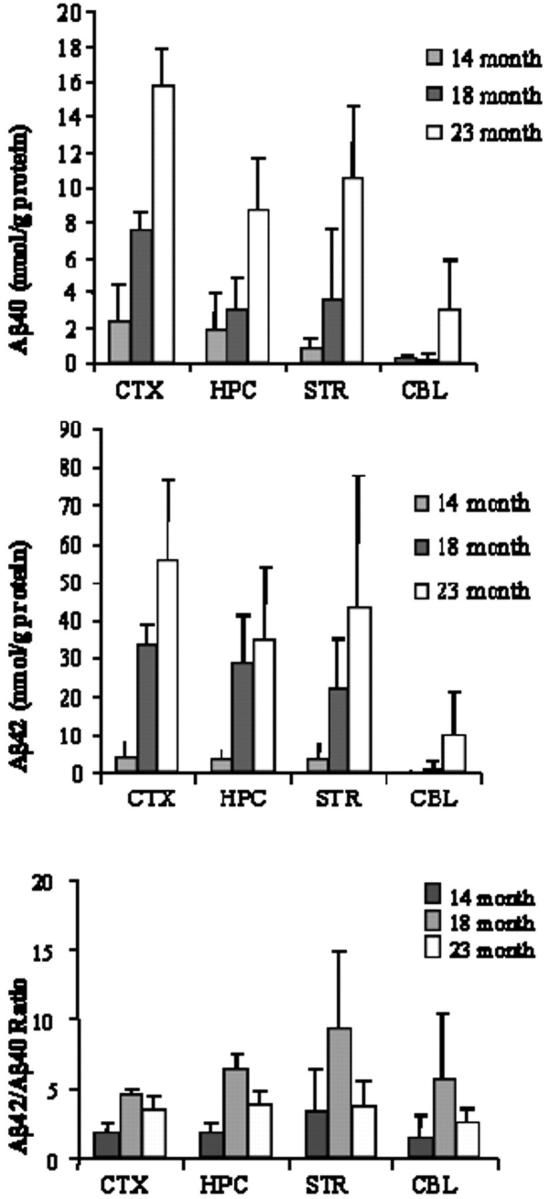
Soluble Aβ40 and Aβ42 levels and the Aβ42/Aβ40 ratio in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD, n = 3.
Cholinergic Markers Are Preserved in Aging Wt and Tg2576 Mouse Brain
In both wt and tg mouse brain, the order of vesamicol binding was striatum > hippocampus > cortex > cerebellum (P < 0.0001, Figure 1 ▶ ), whereas the order of HC-3 binding, ChAT activity, and AChE activity was striatum > hippocampus ∼ cortex > cerebellum (P < 0.001, Figures 2, 3, and 4 ▶ ▶ ▶ ). In hAPPswe mice, the 14-month age group had a higher level of ChAT activity in striatum than the 18- and 23-month age groups, but this difference was not statistically significant (Figure 3) ▶ . In fact, there were no significant differences between wt and tg mice for four cholinergic markers in three age groups and four brain regions. Thus, in the Tg2576 line neither hAPPswe transgenesis nor progressive amyloidogenesis with aging alter these cholinergic markers. Our results for four distinct cholinergic markers were consistent with each other and with the literature. 48,56,57
Figure 1.

Specific vesamicol (VAChT) binding in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD of dpm/μg protein, n = 4.
Figure 2.

Hemicholinium-3 binding (SDHACU) in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD of dpm/μg protein, n = 3. Similar results were obtained with a duplicate analysis, n = 3.
Figure 3.

ChAT activity in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD of nmol/h/mg protein, n = 6.
Figure 4.

Acetylcholinesterase (AChE) activity in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD of pmol/min/μg protein, n = 6.
The Secreted β-Secretase Product of hAPPswe Is Static in Aging Tg2576 Mouse Brain
By the use of a novel, sensitive, and specific ELISA for APPsβswe, 50 we may indirectly assess a hypothesized increase in BACE activity with aging. However, APPsβswe levels were static in all brain regions as follows: cortex ∼ hippocampus ∼ striatum > cerebellum (Figure 6) ▶ . In contrast, in the same samples Aβ40 and Aβ42 levels progressively increased with aging (Figure 5) ▶ . As expected for the human-specific ELISA, no APPsβswe was detected in wt mouse brain (data not shown). This ELISA detects neither mouse APP and its secreted α-/β-secretase fragments nor full-length human APPswe and its α-secretase fragment. 50
Figure 6.

Soluble APPsβswe levels in cortex (CTX), hippocampus (HPC), striatum (STR), and cerebellum (CBL) of aging Tg2576 mouse brain. Data shown are mean ± SD of absorbance, n = 3.
Discussion
The presynaptic cholinergic markers VAChT binding, SDHACU binding, and ChAT activity, as well as AChE activity, were preserved in Tg2576 mouse brain at 14, 18, and 23 months of age. In contrast, progressive CNS amyloidogenesis was demonstrated by increasing soluble Aβ40 and Aβ42 levels and by histochemical staining of sections. Because behavioral deficits precede amyloid deposition in brain, soluble Aβ peptides and oligomers may have synaptotoxic and neurotoxic effects independent of plaque formation. 58,59 In 14- to 23-month-old mice, however, the neurochemical basis for learning and memory deficits does not seem to be because of cholinergic denervation. Rather, glutamate receptor dysregulation has been implicated. 35
Our results extend published data on Tg2576 mice 22,36 by examination of Aβ40 and Aβ42 levels in brain regions instead of whole brain. Hsiao and colleagues 22 report higher levels of Aβ40 compared to Aβ42 in brains of tg mice only up to 12 months of age, but with aging the Aβ42 level increased 14-fold compared to fivefold for Aβ40. In contrast to Kawarabashi and colleagues, 36 we found that Aβ42 levels exceeded Aβ40 levels in old Tg2576 mouse brain. This discrepancy may reflect a difference in extraction methods. Similar to our mouse data, in human brain levels of the more fibrillogenic Aβ42 are consistently greater than Aβ40 in cortex from nondemented patients and through all stages of AD. 60,61 The Aβ42/Aβ40 ratio increased at 18 months (compared to 14 months) and then declined at 23 months of age. This pattern was observed in all brain regions, and suggests that Aβ42 preferentially accumulates earlier than Aβ40 in aging Tg2576 mouse brain. Finally, homeostatic soluble APPsβswe levels suggest that Aβ accumulates with aging not because of increased BACE activity but because of impaired Aβ/amyloid clearance mechanisms in aging brain. This finding is in agreement with static BACE expression in aging Tg2576 mouse brain. 37 A similar conclusion was suggested in a different hAPP tg mouse (hAPP V717I) by finding homeostatic APP catabolite levels except Aβ peptides in aging brain (3 to 15 months). 62 We cannot exclude, however, the possibility that equivalent changes in both APPsβswe generation and clearance result in static, and possibly regulated, levels with aging.
Cholinergic receptor agonists are among many pharmacological agents that regulate APP metabolism and down-regulate Aβ generation in vitro and in vivo. 63-65 Thus, the putative toxicity of amyloid on human cholinergic neurons in the basal forebrain may: 1) enhance Aβ generation and amyloidogenesis in projection fields; 2) promote a vicious cycle of cholinergic loss and amyloidogenesis in AD brain; and thus 3) resurrect, in part, the cholinergic hypothesis of AD. Interestingly, despite similar levels of soluble Aβ peptides in tg mouse hippocampus and striatum and the highest ratio of Aβ42/Aβ40 found in the striatum, amyloid plaques were more abundant in the hippocampus than striatum—a brain region with dense cholinergic innervation. Again invoking a vicious cycle, Aβ peptides inhibit K+-evoked acetylcholine release from slice preparations of rat hippocampus and cortex but not striatum. 66
In the frontal cortex of 8-month-old Tg2576/hPresenilin-1 M146L double-tg mice immunohistological analysis reveals altered cholinergic boutons/terminals in the vicinity of amyloid plaques. 21 Thus, amyloid plaques may cause microscopic but not macroscopic morbidity of neuronal processes and terminals leading to cholinergic deficits and synaptic loss. The relationships between clinical dementia, AD pathology, and synaptic protein levels were recently re-examined in clinically staged human AD brains obtained at autopsy. The loss of synaptic proteins is found only after the emergence of moderate to severe dementia clinically and the full spectrum of amyloid and tau pathology in the neocortex (Braak stages 5 and 6) but is not found in the presence of amyloid pathology alone. Minimal and mild clinical stages of AD are associated with either unchanged or increased levels of synaptic proteins in the neocortex, suggesting regenerative sprouting. 67 An increased density of AChE-positive fibers in the CA1 region and dentate gyrus of 17- to 22-month-old hAPP V642I (London mutation) tg mice also suggests compensatory sprouting. 20 In fact, amyloid-induced axonal sprouting is found in a different hAPPswe tg mouse line (not Tg2576). 68 This may explain the increased VAChT immunoreactivity seen in Tg2576 mice at 8 months of age 21 and the slight (but not significant) increase in ChAT activity found in striatum of tg but not wt mice at 14 months of age (Figure 3) ▶ .
Normal aging does not affect cholinergic systems in mouse and rat brain. 69-71 ChAT activity is maximal at 12 months of age in the telencephalon, brainstem, and cerebellum but there are no significant differences in telencephalic ChAT activity in mice from 6 to 30 months of age. 69 Similarly, we found that aging (from 14 to 23 months) does not affect CNS cholinergic markers in wt C57B6/SJL mice. This agrees with recent data that C57B6 mice do not exhibit age-related deficits in spatial learning or hippocampal structure. 72 In contrast, in autopsied human brains ChAT activity in the hippocampus is maximal in the fourth decade and declines during normal aging. 73
We recognize that because the wt group was mostly male and the tg group mostly female, a difference between groups may have resulted from gender alone. However, we found no difference in cholinergic markers between the two groups of mice. The limited data on gender-specific cholinergic markers in aging rats suggests that alterations either require ovariectomy 74 or are slower to develop in females. 75 Finally, because female Tg2576 mice develop significantly (three times) more CNS amyloid plaque compared to males, 39 we may less likely detect a hypothesized amyloid-induced difference in cholinergic markers in older tg males.
If cholinergic deficits occur early in human AD, then aging Tg2576 mice fail to reproduce this aspect in addition to the lack of neuronal loss and neurofibrillary tangle pathology. However, progressive amyloidogenesis in brain in a temporal and spatial pattern similar to human AD as well as many other pathological features are recapitulated in older Tg2576 mice. 22-40 This data favors the consensus that amyloidogenesis is necessary but not sufficient to cause AD. The mediators of cholinergic denervation in human AD brain may be neuronal loss and neurofibrillary tangle formation but not Aβ or amyloid. Thus, it would be of interest to examine cholinergic markers in hAPP/hτ double-tg mice that exhibit both amyloid plaques and neurofibrillary tangles in aging brain. 76 If cholinergic deficits do not occur until late stages of human AD, then older Tg2576 mice may best resemble preclinical and early stages. Thus, Tg2576 mice may be useful for testing hypotheses and therapeutic strategies based on progressive CNS amyloidogenesis but not on cholinergic deficits.
Acknowledgments
We thank Drs. L. C. Walker, W. J. Lipinski, and colleagues (Pfizer, Ann Arbor, MI), and L. L. Austin, K. Cherian, T. J. Desmond, and Y. Lu for expert assistance; and Dr. N. Suzuki (Takeda Chemical Co., Japan) for antibodies BAN-50, BA-27, and BC-05, Dr. K. Hsiao-Ashe (University of Minnesota), and Mayo Clinical Ventures (Minneapolis, MN) for breeding stock of Tg2576 mice.
Footnotes
Address reprint requests to Dr. R. Scott Turner, VAMC GRECC, 2215 Fuller Rd., Ann Arbor, MI 48105. E-mail: raymondt@umich.edu.
Supported by a Paul Beeson Physician Faculty Scholar in Aging Research Award from the American Federation for Aging Research, United States Public Health Service grant P50 AG08671, and the Veterans Affairs Medical Center Geriatrics Research Education and Clinical Center.
References
- 1.Davies P, Maloney AJF: Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2:1403. [DOI] [PubMed] [Google Scholar]
- 2.Bowen DM, Smith CB, White P, Davison AN: Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99:459-496 [DOI] [PubMed] [Google Scholar]
- 3.Perry EK, Perry RH, Blessed G, Tomlinson BE: Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1977, 1:189. [DOI] [PubMed] [Google Scholar]
- 4.Bowen DM, Benton JS, Spillane JA, Smith CC, Allen SJ: Choline acetyltransferase activity and histopathology of frontal neocortex from biopsies of demented patients. J Neurol Sci 1982, 57:191-202 [DOI] [PubMed] [Google Scholar]
- 5.Slotkin TA, Nemeroff CB, Bissette G, Seidler FJ: Overexpression of the high affinity choline transporter in cortical regions affected by Alzheimer’s disease, evidence from rapid autopsy studies. J Clin Invest 1994, 94:696-702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bissette G, Seidler FJ, Nemeroff CB, Slotkin TA: High affinity choline transporter status in Alzheimer’s disease tissue from rapid autopsy. Ann NY Acad Sci 1996, 777:197-204 [DOI] [PubMed] [Google Scholar]
- 7.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR: Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 1981, 10:122-126 [DOI] [PubMed] [Google Scholar]
- 8.Perry EK, Johnson M, Kerwin JM, Piggott MA, Court JA, Shaw PJ, Ince PG, Brown A, Perry RH: Convergent cholinergic activities in aging and Alzheimer’s disease. Neurobiol Aging 1992, 13:393-400 [DOI] [PubMed] [Google Scholar]
- 9.Rudelli RD, Ambler MW, Wisniewski AT: Morphology and distribution of Alzheimer neuritic (senile) and amyloid plaques in striatum and diencephalon. Acta Neuropathol (Berlin) 1984, 64:273-281 [DOI] [PubMed] [Google Scholar]
- 10.Selden N, Geula C, Hersh L, Mesulam M-M: Human striatum: chemoarchitecture of the caudate nucleus, putamen and ventral striatum in health and Alzheimer’s disease. Neuroscience 1994, 60:621-636 [DOI] [PubMed] [Google Scholar]
- 11.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, DeLong MR: Alzheimer disease and senile dementia: loss of neurons in the basal forebrain. Science 1982, 215:1237-1239 [DOI] [PubMed] [Google Scholar]
- 12.Pearson RC, Sofroniew MV, Cuello AC, Powell TP, Eckenstein F, Esiri MM, Wilcock GK: Persistence of cholinergic neurons in the basal nucleus in a brain with senile dementia of the Alzheimer’s type demonstrated by immunohistochemical staining for choline acetyltransferase. Brain Res 1983, 289:375-379 [DOI] [PubMed] [Google Scholar]
- 13.Arendash GW, Millard WJ, Dunn AJ, Meyer EM: Long-term neuropathologic and neurochemical effects of nucleus basalis lesions in the rat. Science 1987, 238:952-956 [DOI] [PubMed] [Google Scholar]
- 14.Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399(Suppl):A23-A31 [DOI] [PubMed] [Google Scholar]
- 15.Shinotoh H, Namba H, Fukishi K, Nagatsuka S, Tanaka N, Aotsuka A, Ota T, Tanada S, Irie T: Progressive loss of cortical acetylcholinesterase activity in association with cognitive decline in Alzheimer’s disease: a positron emission tomography study. Ann Neurol 2000, 48:194-200 [PubMed] [Google Scholar]
- 16.Beach TG, Kuo Y-M, Spiegel K, Emmerling MR, Sue LI, Kokjohn K, Roher AE: The cholinergic deficit coincides with Aβ deposition at the earliest histopathologic stages of Alzheimer disease. J Neuropath Exp Neurol 2000, 59:308-313 [DOI] [PubMed] [Google Scholar]
- 17.Davis KL, Mohs R, Marin D, Purohit D, Perl D, Lantz M, Austin G, Haroutunian V: Cholinergic markers in elderly patients with early signs of Alzheimer disease. J Am Med Assoc 1999, 281:1401-1406 [DOI] [PubMed] [Google Scholar]
- 18.Gilmor ML, Erickson JD, Varoqui H, Hersch LB, Bennett DA, Cochran EJ, Mufson EJ, Levey AI: Preservation of nucleus basalis neurons containing choline acetyltransferase and the vesicular acetylcholine transporter in the elderly with mild cognitive impairment and early Alzheimer’s disease. J Comp Neurol 1999, 411:693-704 [PubMed] [Google Scholar]
- 19.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B: Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 1997, 94:13287-13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bronfman FC, Moechars D, Van Leuven F: Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein London mutant transgenic mice. Neurobiol Dis 2000, 7:152-168 [DOI] [PubMed] [Google Scholar]
- 21.Wong TP, Debeir T, Duff K, Cuello AC: Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci 1999, 19:2706-2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 23.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT: AppSw transgenic mice develop age-related Abeta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 1997, 56:965-973 [DOI] [PubMed] [Google Scholar]
- 24.Frautschy SA, Yang F, Irizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 1998, 152:307-317 [PMC free article] [PubMed] [Google Scholar]
- 25.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K: Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998, 4:97-100 [DOI] [PubMed] [Google Scholar]
- 26.Hsiao K: Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol 1998, 33:883-889 [DOI] [PubMed] [Google Scholar]
- 27.Pappolla MA, Chyan YJ, Omar RA, Hsiao K, Perry G, Smith GA, Bozner P: Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer’s disease: a chronic oxidative paradigm for testing antioxidant therapies in vivo. Am J Pathol 1998, 152:871-877 [PMC free article] [PubMed] [Google Scholar]
- 28.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlack SL, Tabaton M, Perry G: Amyloid-β deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 1998, 70:2212-2215 [DOI] [PubMed] [Google Scholar]
- 29.Benzing WC, Wujek JR, Ware EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR: Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 1999, 20:581-589 [DOI] [PubMed] [Google Scholar]
- 30.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK: Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 1999, 2:271-276 [DOI] [PubMed] [Google Scholar]
- 31.King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ: Progressive and gender-dependent cognitive impairment in the APPsw transgenic mouse model for Alzheimer’s disease. Behav Brain Res 1999, 103:145-162 [DOI] [PubMed] [Google Scholar]
- 32.Mehlhorn G, Hollborn M, Schliebs R: Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci 2000, 18:423-431 [DOI] [PubMed] [Google Scholar]
- 33.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T: Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 2000, 57:331-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang F, Ueda K, Chen P, Ashe KH, Cole CM: Plaque-associated alpha-synuclein (NACP) pathology in aged transgenic mice expressing amyloid precursor protein. Brain Res 2000, 853:381-383 [DOI] [PubMed] [Google Scholar]
- 35.Cha JH, Farrell LA, Ahmed SF, Fray A, Hsiao-Ashe KK, Young AB, Penney JB, Locascio JJ, Hyman BT, Irizarry MC: Glutamate receptor dysregulation in the hippocampus of transgenic mice carrying the mutated human amyloid precursor protein. Neurobiol Dis 2001, 8:90-102 [DOI] [PubMed] [Google Scholar]
- 36.Kawarabashi T, Younkin LH, Saido TC, Shoji M, Hsiao Ashe K, Younkin SG: Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 2001, 21:372-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irizarry MC, Locascio JJ, Hyman BT: β site APP cleaving enzyme mRNA expression in APP transgenic mice. Am J Pathol 2001, 158:173-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christie RH, Bacskai BJ, Zipfel WR, Williams RM, Kajdasz ST, Webb WW, Hyman BT: Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J Neurosci 2001, 21:858-864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC: Augmented senile plaque load in aged female β-amyloid precursor protein-transgenic mice. Am J Pathol 2001, 158:1173-1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner RS: Alzheimer’s disease in man and transgenic mice: females at higher risk. Am J Pathol 2001, 158:797-801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marien MR, Parsons SM, Altar CA: Quantitative autoradiography of brain binding sites for the vesicular acetylcholine transport blocker 2-(4-phenylpiperidino)cyclohexanol (AH5183). Proc Natl Acad Sci USA 1987, 84:876-880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weihe E, Tao-Cheng J-H, Schafer MKH, Erickson JD, Eiden LE: Visualization of the vesicular acetylcholine transporter in cholinergic nerve terminals and its targeting to a specific population of small synaptic vesicles. Proc Natl Acad Sci USA 1996, 93:3547-3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, Levey AI: Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci 1996, 16:2179-2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulholland GK, Wieland DM, Kilbourn MR, Frey KA, Sherman PS, Carey JE, Kuhl DE: [18F]Fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular acetylcholine transporter and cholinergic synapses. Synapse 1998, 30:263-274 [DOI] [PubMed] [Google Scholar]
- 45.Kuhl DE, Minoshima S, Fessler JA, Frey KA, Foster NL, Ficaro EP, Wieland DM, Koeppe RA: In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease and Parkinson’s disease. Ann Neurol 1996, 40:399-410 [DOI] [PubMed] [Google Scholar]
- 46.Jung YW, Frey KA, Mulholland GK, del Rosario R, Sherman PS, Raffel DM, Van Dort ME, Kuhl DE, Gildersleeve DL, Wieland DM: Vesamicol receptor mapping of brain cholinergic neurons with radioiodine-labeled positional isomers of benzovesamicol. J Med Chem 1996, 39:3332-3342 [DOI] [PubMed] [Google Scholar]
- 47.Efange SWN, Garland EM, Staley JK, Khare AB, Mash DC: Vesicular acetylcholine transporter density and Alzheimer’s disease. Neurobiol Aging 1997, 18:407-413 [DOI] [PubMed] [Google Scholar]
- 48.Stavinoha WB, Weintraub ST, Modak AT: Regional concentrations of choline and acetylcholine in the rat brain. J Neurochem 1974, 23:885-886 [DOI] [PubMed] [Google Scholar]
- 49.Manaker S, Wieczorek CM, Rainbow TC: Identification of sodium-dependent, high-affinity choline uptake sites in rat brain with [3H]hemicholinium-3. J Neurochem 1986, 46:483-488 [DOI] [PubMed] [Google Scholar]
- 50.Steinhilb ML, Turner RS, Gaut JR: The protease inhibitor, MG132, blocks maturation of the amyloid precursor protein Swedish mutant preventing cleavage by β-secretase. J Biol Chem 2001, 276:4476-4484 [DOI] [PubMed] [Google Scholar]
- 51.Fonnum F: A rapid radiochemical method of the determination of choline acetyltransferase. J Neurochem 1975, 24:407-409 [DOI] [PubMed] [Google Scholar]
- 52.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC: Measurement of protein using bicinchoninic acid. Anal Biochem 1985, 150:76-85 [DOI] [PubMed] [Google Scholar]
- 53.Ellman GL, Courtney KD, Andres V, Jr, Featherstone RM: A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961, 7:88-95 [DOI] [PubMed] [Google Scholar]
- 54.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG: An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264:1336-1340 [DOI] [PubMed] [Google Scholar]
- 55.Turner RS, Suzuki N, Chyung ASC, Younkin SG, Lee VM-Y: Amyloids β40 and β42 are generated intracellularly in cultured human neurons and their secretion increases with maturation. J Biol Chem 1996, 271:8966-8970 [DOI] [PubMed] [Google Scholar]
- 56.Gordon MN, Finch CE: Topochemical localization of choline acetyltransferase and acetylcholinesterase in mouse brain. Brain Res 1984, 308:364-368 [DOI] [PubMed] [Google Scholar]
- 57.Kilbourn MR, Jung Y-W, Haka MS, Gildersleeve DL, Kuhl DE, Wieland DM: Mouse brain distribution of a carbon-11 labeled vesamicol derivative: presynaptic marker of cholinergic neurons. Life Sci 1990, 47:1955-1963 [DOI] [PubMed] [Google Scholar]
- 58.Hsia AY, Masliah E, McConlogue L, Yu G-Q, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 1999, 96:3228-3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko K, McConlogue L: High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 2000, 20:4050-4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD: Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. J Am Med Assoc 2000, 283:1571-1577 [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Dickson DW, Trojanowski JQ, Lee VM-Y: The levels of soluble versus insoluble brain Aβ distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 1999, 158:328-337 [DOI] [PubMed] [Google Scholar]
- 62.Dewachter I, Van Dorpe J, Smeijers L, Gilis M, Kuiperi C, Laenen I, Caluwaerts N, Moechars D, Checler F, Vanderstichele H, Van Leuven F: Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin-1. J Neurosci 2000, 20:6452-6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V: The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog Neurobiol 1998, 56:541-569 [DOI] [PubMed] [Google Scholar]
- 64.Lin L, Georgievska B, Mattsson A, Isacson O: Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc Natl Acad Sci USA 1999, 96:12108-12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mills J, Reiner PB: Regulation of amyloid precursor protein cleavage. J Neurochem 1999, 72:443-460 [DOI] [PubMed] [Google Scholar]
- 66.Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R: Amyloid beta-peptide inhibits high affinity choline uptake and acetylcholine release in rat hippocampal slices. J Neurochem 1998, 70:2179-2187 [DOI] [PubMed] [Google Scholar]
- 67.Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, Brayne C, Huppert FA, Paykel ES, McGee M, Jakes R, Honer WG, Harrington CR, Wischik CM: Staging of cytoskeletal and β-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol 2000, 157:623-636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M: Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci 1999, 19:8552-8559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Unsworth BR, Fleming LH, Caron PC: Neurotransmitter enzymes in telencephalon, brain stem and cerebellum during the entire life span of the mouse. Mech Aging Dev 1980, 13:205-217 [DOI] [PubMed] [Google Scholar]
- 70.Strong R, Hicks P, Hsu L, Bartus RT, Enna SJ: Age-related alterations in the rodent brain cholinergic system and behavior. Neurobiol Aging 1980, 1:59-63 [DOI] [PubMed] [Google Scholar]
- 71.Baxter MG, Frick KM, Price DL, Breckler SJ, Markowska AL, Gorman LK: Presynaptic markers of cholinergic function in the rat brain: relationship with age and cognitive status. Neuroscience 1999, 89:771-780 [DOI] [PubMed] [Google Scholar]
- 72.Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M: Hippocampal neurons and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging 1998, 19:599-606 [DOI] [PubMed] [Google Scholar]
- 73.Perry RH, Candy JM, Perry EK, Irving D, Blessed G, Fairbairn AF, Tomlinson BE: Extensive loss of choline acetyltransferase activity is not reflected by neuronal loss in the nucleus of Meynert in Alzheimer’s disease. Neurosci Lett 1982, 33:311-315 [DOI] [PubMed] [Google Scholar]
- 74.Gibbs RB: Impairment of basal forebrain cholinergic neurons associated with aging and long-term loss of ovarian function. Exp Neurol 1998, 151:289-302 [DOI] [PubMed] [Google Scholar]
- 75.Lukoyanov NV, Andrade JP, Dulce Madeira M, Paula-Barbosa MM: Effects of age and sex on the water maze performance and hippocampal cholinergic fibers in rats. Neurosci Lett 1999, 269:141-144 [DOI] [PubMed] [Google Scholar]
- 76.Lewis J, Dickson DW, Lin W-L, Chisholm L, Corral A, Jones G, Yen S-H, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E: Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293:1487-1491 [DOI] [PubMed] [Google Scholar]