

Abstract
Stathmin is a cytosolic protein that binds tubulin and destabilizes cellular microtubules, an activity regulated by phosphorylation. Despite its abundant expression in the developing mammalian nervous system and despite its high degree of evolutionary conservation, stathmin-deficient mice do not exhibit a developmental phenotype. 1 Here we report that aging stathmin−/− mice develop an axonopathy of the central and peripheral nervous systems. The pathological hallmark of the early axonal lesions was a highly irregular axoplasm predominantly affecting large, heavily myelinated axons in motor tracts. As the lesions progressed, degeneration of axons, dysmyelination, and an unusual glial reaction were observed. At the functional level, electrophysiology recordings demonstrated a significant reduction of motor nerve conduction velocity in stathmin−/− mice. At the molecular level, increased gene expression of SCG 10-like protein, a stathmin-related gene with microtubule destabilizing activity, was detected in the central nervous system of aging stathmin−/− mice. Together, these findings suggest that stathmin plays an essential role in the maintenance of axonal integrity.
Stathmin 2 is a 148-amino acid cytosolic phosphoprotein that has also been referred to as p19, 3 prosolin, 4 Lap18, 5 oncoprotein 18, 6 metablastin, 7 and Op18. 8 This protein undergoes phosphorylation, at four serine residues, in mammalian cells in response to a diverse group of extracellular factors. 9 Its expression is differentiation stage-specific, occurring in many immature cell types, most abundantly in the developing nervous system, 10-12 and is dramatically down-regulated during terminal differentiation. 7,12,13 In cultured cell lines, phosphorylation of stathmin fluctuates during the cell cycle and reaches its maximum during the M phase. 14-16 Overexpression of phosphorylation site mutants of stathmin results in cell cycle arrest at the G2/M interphase. 17,18 Therefore, the protein has been implicated in cell cycle regulation. However, although stathmin is expressed in a variety of transformed cell lines in culture, 10,11 and in some cancer cells in vivo, 19-23 expression in normal tissues is primarily restricted to postmitotic cells. 7,12,13,24,25
Recently, stathmin has been identified as a microtubule (MT)-destabilizing factor. In vitro, the protein binds to tubulin, 26-29 inhibits MT assembly, 9,27,29 and promotes MT catastrophes. 26,30 Consistent with this activity, overexpression of stathmin in cells leads to disassembly of MTs 9,27,31 and, hence, inability to enter mitosis. 17,18 Interestingly, the MT-destabilizing activity of stathmin is controlled by phosphorylation. 9,27,31,32 Therefore, the biological function of stathmin is possibly related to its MT-destabilizing activity.
The high degree of evolutionary conservation of the gene encoding stathmin 33-37 further suggests that it serves an essential function. It was surprising, therefore, that stathmin knockout mice did not exhibit any overt developmental abnormalities. 1 This is presumably because of expression of other members of the stathmin gene family, 33,38-40 which includes the genes encoding SCG10, Rb3 (and related proteins derived by alternative splicing), and SCG10-like protein (SCLIP). With respect to SCG10, we have found no evidence for up-regulation in young stathmin knockout mice (Schubart et al 1 and unpublished observations).
We have now elucidated a phenotype of stathmin−/− mice by pathological-anatomical, neurophysiological, and molecular studies. Aged stathmin−/− mice were found to develop a progressive axonopathy, suggesting that stathmin is essential for the integrity of the nervous system.
Materials and Methods
Animals
The generation of stathmin-deficient mice was described previously. 1 The line has been maintained by successively backcrossing heterozygotes (stathmin+/−) into the C57BL/6J strain. Homozygous wild-type (stathmin+/+) and mutant (stathmin−/−) mice were F1 mice obtained by crossing stathmin+/− littermates. Animals used for the initial studies were from the second and third backcrosses (N2 and N3). The neuropathological findings were subsequently recapitulated in mice from the sixth backcross (N6). Animals were subjected to clinical examinations as previously described 41 for neurological and behavioral assessment of mice with experimental autoimmune encephalomyelitis, an animal model for multiple sclerosis. Specifically, we examined cranial nerves, motor function and sensory perception, coordination, balance, and gait. The mice were bred and housed in the barrier facility maintained by an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility at the Albert Einstein College of Medicine. A total of 97 mice were analyzed, 58 for structural studies, 16 for Western and Northern blotting, and 23 for neurophysiology. Investigators were unaware of the genotype of the animals while carrying out the studies. All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Animal Institute Committees of the academic institutions involved.
Genotyping
Genotyping for the targeted stathmin gene was performed on tail DNA, using the polymerase chain reaction, which was performed in polymerase chain reaction buffer (Boehringer Mannheim Corp., Indianapolis, IN) containing 1.5 mmol/L MgCl2, deoxynucleotide triphosphates (1 mmol/L each), three primers, and Taq polymerase (0.05 U/μl) using 25 temperature cycles (94°C, 30 seconds; 57°C, 30 seconds; 72°C, 60 seconds). The primers used were a wild-type allele-specific forward primer (GAGAATCCATGATTGCCAGCAC, 1 μmol/L), corresponding to a region of intron III deleted in the mutant allele; a mutant allele-specific forward primer (CTAATGGCTATAGTTTCATGTTCC, 1 μmol/L), which anneals in the mPGK-1b 3′-flanking region 42 that is part of the PGKneo cassette used; 1 and a reverse primer (AGCAAAACCAAATTAAGGGCCAGC, 2 μmol/L), corresponding to a region of intron III of the stathmin gene that is retained in the mutant allele. The polymerase chain reaction products obtained from the wild-type and mutant alleles were 463 bp and 610 bp in size, respectively.
Neuropathology
Neuropathological studies were performed as previously described. 43,44 Briefly, mice were anesthetized with ether and perfused transcardially either with fixative [2.5% glutaraldehyde in Millonig’s buffer (phosphate-buffered saline [PBS] supplemented with 5.4g dextrose/L) or 10% formalin in PBS, pH 7.4], or PBS, pH 7.4. Fixative-perfused tissue was embedded in paraffin or epoxy (Epon 812; Ladd Research Industries, Williston, VT) and PBS-perfused tissue was used for frozen blocks in OCT/Tissuetek or for tissue homogenization and denaturation before immunoblot analysis.
Light Microscopy
Glutaraldehyde/osmium-tetroxide fixed epoxy sections were sectioned on a Reichert ultramicrotome at 1 μm and stained with toluidine blue. Both longitudinal and cross sections were obtained from the optic nerve, corpus callosum, and brain stem. At least four, in most cases six, different levels of spinal cord were sampled, in addition to spinal roots and sciatic nerve, the latter sampled at mid-thigh level.
Immunocytochemistry
Either 10-μm frozen or 1-μm epoxy sections were incubated with primary antibody and developed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA). The following primary antibodies were used: rabbit anti-stathmin C-terminal peptide, 12 1:1000; mouse anti-glial fibrillary acidic protein (GFAP) (Zymed, San Francisco, CA), 1:200; mouse anti-NFH-(strongly phosphorylated) SMI 35 (Sternberger Monoclonals, Baltimore, MD), 1:5000; mouse anti-NFH-(nonphosphorylated) SMI 32 (Sternberger Monoclonals), 1:2000; mouse anti-NFH-(intermediately phosphorylated) SMI 31 (Sternberger Monoclonals), 1:5000; mouse anti-NFL (Sigma Chemical Co., St. Louis, MO), 1:100.
Morphometry—Light Microscopy
Abnormal cellular aggregates in white matter, representing the end-stage of central nervous system (CNS) axonopathy (see Results), were counted in stathmin+/+ and stathmin−/− mice of different ages: 21 months of age, four of five mice each; 14 months of age, five of six mice each, and 6 weeks of age, six of six mice each. Matched cross-sections of cervical, thoracic, and upper lumbar spinal cord were scored at ×100 magnification.
Electron Microscopy and Ultrastructural Morphometry
Blocks of tissue were selected for electron microscopy after light microscopy of semithin sections. Thin sections (60 to 80 nm) of epon-embedded tissue were mounted on 200-mesh copper grids, stained with lead acetate (1% in H2O) and uranyl acetate (8.5% in ethanol), carbon-coated, and scanned in a Siemens 101 electron microscope. The procedure has been described in more detail elsewhere. 45
The basic outline of ultrastructural morphometry has been described elsewhere. 43 Briefly, matched cross-sections of spinal cord anterior column and sciatic nerve were scanned at ×7.500 magnification and printed at constant enlargement. Three axons each per spinal cord and per sciatic nerve were selected, n = 2 mice (21 months of age) per genotype. Myelinated axons were randomly selected for wild-type littermates. Only axons with a uniformly hyperdense axoplasm were analyzed for stathmin−/− mice. A square of 2.5-cm side-length was drawn into the micrograph and neurofilaments (NFs) and MTs were counted on the micrograph. Thus a total of six axons per mouse was analyzed.
Immunoblot Analysis
Spinal cord tissue of 17-month-old mice (four stathmin +/+ and four stathmin−/−), which had been perfused with ice-cold PBS in situ, was homogenized immediately in 8 mol/L urea. Protein was measured by the Bradford assay 46 using Bio-Rad reagents (Bio-Rad Laboratories, Hercules, CA). Aliquots (50 μg) were subjected to immunoblot analysis as previously described. 10 In addition to the antibodies described in the Immunocytochemistry section, the following antibodies were used: mouse anti-tubulin (Sigma Chemical Co.); and mouse anti-GFAP (Boehringer Mannheim Corp.). The blots were developed using ECL reagents (Amersham Life Science Inc., Arlington Heights, IL).
Northern Blot Analysis
Brain and spinal cord tissue was dissected from 19-month-old mice perfused with ice-cold PBS, pH 7.4, pretreated with diethyl pyrocarbonate 0.1%, and total RNA was isolated as described. 47 To average the abundance of expressed mRNAs, RNA from four knockout mice was pooled. Northern blot analysis was performed as previously described. 1,48 32P-labeled DNA probes for stathmin-related transcripts were generated by reverse transcriptase-polymerase chain reaction using normal mouse brain RNA as template. The primers used were: forward (stathmin) 5′-TCCCCGACCCCTTCCTAAATATCC; reverse (stathmin)5′-TACAGCACTAGCCATTAACCCAGC; forward (SCG10)5′-CTAGCTGCTATCATTGAACGTCTGC; reverse (SCG10) 5′-CTTCCCATACTGATATCGCATGATCC; for-ward (Rb3) 5′-AAAAGATTGACCAGTGAAGCCATCC; reverse (Rb3) 5′-GAACAGACGCACACAGACCTTAT-CC; forward (SCLIP) 5′-GTCTAGATGCGAATTTGTGCCTGC; reverse (SCLIP) 5′-GGTTGCCAACACATCCACTAAGAGG.
The amplified products were of the sizes predicted from the published sequences (GenBank accession numbers: AF105222 for Rb3, J04979 for stathmin, AU079912 for SCG10, AF069708 for SCLIP). Autoradiographs were developed with the phosphoimager system and software (Molecular Dynamics, Eugene, OR), and images were analyzed with the software program NIH image. Image analysis was performed on a Macintosh computer using the public domain NIH Image program (developed at the National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/).
Neurophysiology
Motor nerve conduction velocity in the sciatic nerve was measured by recording anti-dromic compound action potentials in lumbosacral dorsal roots of 23 mice (15 stathmin−/− and 8 stathmin+/+) anesthetized with ketamine (100 mg/kg, intramuscularly) and pentobarbital (30 mg/kg, intraperitoneally). Stimulus strengths from 0.02 to 1.0 mA (100 μsec duration, 1 to 2 Hz) were applied to the sciatic nerve to evoke anti-dromic action potentials in motor axons. The ventral root recordings consisted of population responses (compound action potentials). The conduction velocity for each root was calculated from the measured conduction distance (range, 27 to 35 mm) divided by the latency of the response at stimulus strengths that generated the maximal response amplitude. The latency was measured as the time from the onset of the stimulus to the beginning of the rapid upward deflection of the voltage trace. Individual conduction velocities measured from an animal were averaged to give a single value for each animal. For analysis, animals were grouped by genotype (stathmin−/− and stathmin+/+). Data (means ± SD) were compared using a two-sample t-test (Sigma Stat 2.0).
Results
Stathmin Knockout Mice Develop a Late-Onset Axonopathy—Lack of a Clinical Phenotype
With age, stathmin−/− mice develop a pan-nervous system axonopathy (see below) that did not result in an apparent clinical phenotype. On inspection, handling, and clinical examination of the mice 41,43,44 we could not detect any overt abnormalities between stathmin−/− mice and wild-type littermates. Body weight was lower in stathmin−/− mice, but this difference was not statistically significant.
Stathmin Knockout Mice Develop a Late-Onset Axonopathy—Light Microscopy Demonstrates an Involvement of Large-Caliber Myelinated Axons
With age, stathmin−/− mice develop a pan-nervous system axonopathy with structural and functional correlates. Affected axons were predominantly heavily myelinated and of large caliber. At the light microscope level, the earliest pathology was noted in the spinal cord and sciatic nerve of stathmin−/− animals 14 months of age, and the findings progressively worsened with age. This observation was in line with our previous report on stathmin knockout mice displaying no gross neuroanatomical abnormalities at birth and during the first months of life. 1 Semithin sections of spinal cord and sciatic nerve from 20-month-old knockout mice revealed a striking axonal degeneration of large-caliber axons (Figure 1; A to F ▶ ). The mildest finding consisted of loss of regularity of the axoplasm with preservation of myelin sheaths (Figure 1; A, B, D, and E ▶ ). In later stages, myelin pathology was observed in association with the axonal changes. The myelin sheaths were thinned and existed as isolated lamellae around a tightly packed, hyperdense axoplasm. In other nerve fibers, myelin debris surrounded tightly packed axonal remnants. In association with some of these axons, infiltrating cells were observed within the myelin sheaths and next to degenerating axons (Figure 1E) ▶ . None of these findings were observed in wild-type (stathmin+/+) littermates (Figure 1, C and F) ▶ . For quantitation of the lesions, see below.
Figure 1.
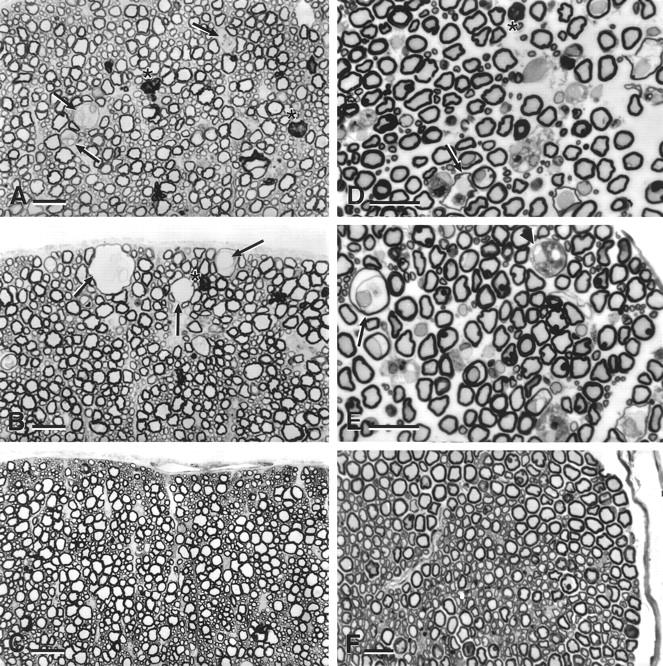
Light microscopy of stathmin−/− mice: pan-nervous system axonopathy. Semithin sections (1 μm) of anterior column of cervical spinal cord (A–C) and of sciatic nerve (D–F) of 20-month-old mice stained with toluidine blue. A, B, D, and E: Stathmin−/− mouse. C and F: Wild-type littermate (stathmin+/+ mouse). Note different stages of axonal degeneration in A, B, D, and E. Three subpial axons at different stages of advanced degeneration can be seen in B (arrows).
With regard to topology, the described lesions were most prominent in the anterior and lateral columns of the spinal cord (Figure 1, A and B) ▶ , predominantly, but not exclusively, in motor tracts. Similar abnormalities were also observed in optic nerve and in the peripheral nervous system (sciatic nerve; Figure 1, D and E ▶ ). In the spinal cord, lesions could not be detected in the dorsal columns. The axonal changes in spinal roots were not milder than those observed in the sciatic nerve. Because the sciatic nerve was sampled at mid-thigh level, a distance of ∼2 cm lay between spinal roots and sciatic nerve. Lack of an appreciable difference in axonopathy between roots and sciatic nerve does not argue for a markedly distal axonopathy. This does not, however, exclude a subtler distal axonopathy. In all areas, the abnormal findings were restricted to larger, heavily myelinated axons. The corpus callosum and other hemispheric white matter tracts were not affected by the axonopathy. Unmyelinated and small, thinly myelinated fibers were spared. Furthermore, gray matter of brain, cerebellum, and spinal cord appeared normal and there was no evidence of ventricular enlargement. There were also no signs of nervous system inflammation or irregularities in vascularization.
The presence of a pan-nervous system axonopathy in stathmin−/− mice prompted us to examine skeletal muscle for changes consistent with a neurogenic myopathy. We found subtle, yet consistent irregularities in fiber diameter and overly large fibers displaying nuclear hypertrophy and, occasionally, centric nuclei (results not shown). Such changes were present in aged stathmin−/− mice at 14 to 16 months of age, but not in wild-type littermate controls or in young mice of either genotype. The findings are suggestive of a subtle neurogenic late-onset myopathy in stathmin−/− mice.
Stathmin Knockout Mice Develop a Late-Onset Axonopathy—Ultrastructural Changes
The unmistakable hallmarks of the ultrastructural findings are depicted in Figure 2 ▶ . Large, heavily myelinated axons showed first signs of degeneration at the age of 12 months, preceding light-microscopic changes by 2 months. The axoplasm appeared disorganized with an irregular texture and abnormal distribution of cytoskeletal elements (Figure 2A) ▶ . In contrast to the normal, regular axoplasm of wild-type littermates (Figure 2B) ▶ , stathmin−/− mice exhibited a highly irregular pattern of tightly packed as well as diffuse axoplasm in the same axon (Figure 2A ▶ and Figure 3B ▶ ). In addition, we noted marked heterogeneity between axons, where axons with tightly packed axoplasm could be observed next to others that exhibited a low density of MTs and neurofilaments (NFs) (Figure 2A ▶ and Figure 3B ▶ ). The inhomogeneity of the changes in the axoplasm precluded a standardized morphometric analysis of NFs and MTs in individual axons (see below). Longitudinal sections of thoracic spinal cord anterior column did not reveal discontinuities of MTs or NFs (data not shown). There was also no evidence of axonal swelling associated with accumulation of NFs, a feature observed in other axonopathies, such as in motor neuron degeneration of amyotrophic lateral sclerosis, 49 or in transgenic mouse models, such as those overexpressing NF subunits, 50,51 and those expressing human Cu/Zn superoxide dismutase (SOD1) with a G93A mutation. 52 The distribution and localization of axonal mitochondria appeared normal in stathmin−/− mice, suggesting that axonal transport was not grossly altered (Figure 2A ▶ and Figure 3B ▶ ). In the oldest stathmin−/− mice examined, advanced stages of axonal decay were seen with dys- and demyelination of degenerating axons (Figure 2, C and D ▶ ; Figure 3, B and C ▶ ). In some cases, infiltrating cells (resembling microglia in the CNS) could be observed between myelin sheaths and axons (Figure 2D) ▶ . Interestingly, the somata and proximal dendrites of neurons throughout the CNS were not affected. Our ultrastructural focus in this respect was spinal cord motoneurons. MTs in these nerve cells were equally abundant and regularly distributed in somata and proximal dendrites of stathmin−/− mice and wild-type littermate controls of all ages (Figure 2, E and F ▶ ; data not shown). With respect to specific MT pathology in axons, we could not detect anomalies of shape of MT. Apart from an altered density of all axoplasmic cytoskeletal components (as described above), there was no anomaly of abundance, aggregation, and number of MT in stathmin−/− mice. With respect to motoneuron somata, these cells were examined for chromatolysis, which could not be detected.
Figure 2.
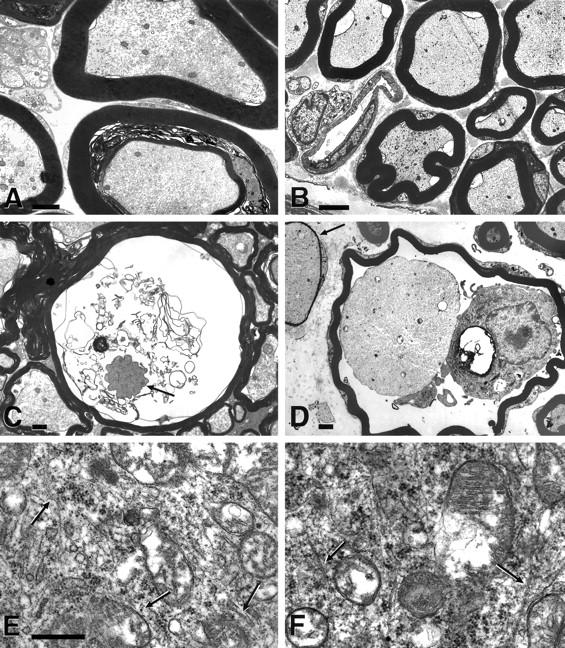
Ultrastructural hallmarks of pan-nervous system axonopathy in stathmin−/− mice. Transmission electron microscopic images of sciatic nerve (A, B, and D), spinal cord anterior column (C), and anterior horn (E and F). A: Stathmin−/− mouse, 14 months of age. B: Stathmin+/+ littermate, 14 months of age. C, D, and E: Stathmin−/− mouse, 20 months of age. F: Stathmin+/+ littermate, 20 months of age. Note inhomogeneous appearance of the axoplasm and heterogeneity between individual axons in A and C. Also note a decaying axon with tightly packed axoplasm surrounded by dissolving myelin lamellae and a thin myelin sheath indicating the original diameter of this axon in C. In D, a myelin macrophage is seen inside a myelin sheath of a degenerating axon displaying a tightly packed axoplasm. An adjacent axon also has a tightly packed axoplasm and a very thin myelin sheath, suggestive of remyelination. Also note the normal abundance and distribution of microtubules in cross- and longitudinal section (arrows) in somata of motoneurons of the lumbar spinal cord both in stathmin−/− mouse and wild-type littermate (E and F). Scale bars: 1 μm (A and C); 2 μm (B); 0.5 μm (D); 0.2 μm (E and F).
Figure 3.
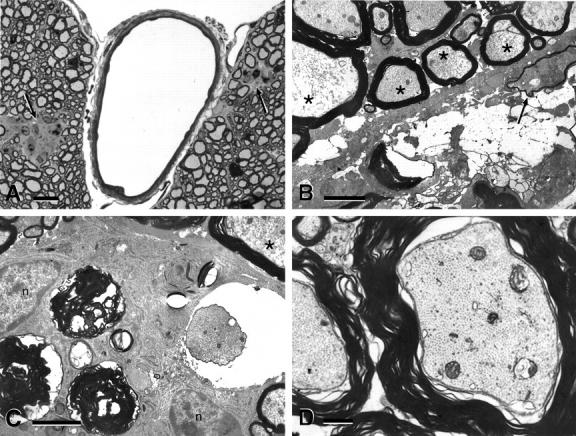
Advanced stages and reactive changes in CNS of stathmin−/− mice. A: Semithin section of lumbar spinal cord anterior column of 18-month-old stathmin−/− mouse stained with toluidine blue. B, C, and D: Transmission electron micrographs of spinal cord anterior column of 20-month-old stathmin−/− mouse (B and C) and wild-type littermate (D). Note white matter aggregates (nodules) in both anterior columns in A (arrows). In B, note myelin debris, a decayed axon, a degenerating axon with thinned myelin suggestive of remyelination (arrow), and severe axoplasmatic inhomogeneity (axons labeled with asterisk). Note, in C, axoplasmic inhomogeneity (asterisk) and myelin debris, in addition to two cells resembling reactive astrocytes next to myelin debris (nucleus labeled with n). D: Wild-type littermate control. Scale bars: 10 μm (A); 1 μm (B, C, and D).
Reactive Changes in the CNS in Advanced Lesions and Morphometry
Yet another feature of stathmin−/− mice was the presence of cellular aggregates in the CNS white matter (Figure 3A) ▶ . The size and abundance of these aggregates (nodules) increased with age and, remarkably, they were absent in wild-type littermates. The aggregates of large cells resembled hypertrophic astrocytes and microglia in association with demyelinated and shrunken axons. The ultrastructure of these lesions suggested that they represent clean-up reactions of the white matter with debris of degenerated axons and phagocytosed myelin surrounded by microglial cells and astrocytes (Figure 3, B and C) ▶ . To obtain quantitative parameters validly reflecting the neuropathological differences between wild-type littermates and stathmin−/− mice, we performed a morphometric analysis of the white matter reactive changes in the spinal cord. These white matter changes probably represented the end-stages of axonopathy. As can be appreciated in Table 1 ▶ , no such changes were observed in the wild-type, whereas their number increased in stathmin−/− mice as they aged from 14 to 21 months (2.6 lesions to 12.5 lesions counted per spinal cord).
Table 1.
Morphometric Analysis of White Matter Changes in Stathmin−/− Mice and Wild-Type Littermates
| Age of mice | Genotype | |||
|---|---|---|---|---|
| Stathmin+/+ | Stathmin−/− | |||
| Nodules* | n/animal† | Nodules* | n/animal† | |
| 6 weeks | 0 | 6 | 0 | 6 |
| 14 months | 0 | 6 | 2.6 ± 1.5 | 5 |
| 21 months | 0 | 5 | 12.5 ± 1.7 | 4 |
*Number of nodules found (mean +/− SD) per animal.
†Number of animals studied (four spinal cord sections per animal).
Morphometric data in addition to the white matter aggregate counts would be desirable. With respect to earlier lesions, an evaluation of the percentage of degenerated axons of all axons was greatly impeded by the different stages of the lesions present in any given cross-section of eg, spinal cord, sciatic nerve. With respect to a quantitative evaluation of ultrastructural axoplasmic changes, axons with varying degrees of density of the axoplasmic components could not be evaluated for number and density of cytoskeletal elements. This was only possible in axons with a uniformly tightly packed axoplasm. We have analyzed a limited number of such axons in matched sections of spinal cords from stathmin−/− mice and wild-type littermates (see Materials and Methods). The ratio of MT to NF per given area of an axon did not differ between stathmin−/− mice and wild-type littermates. A total number of 229 MTs and 2910 NFs (=7.8 MTs per 100 NFs) was counted in stathmin−/− mice versus 75 MTs and 930 NFs (=8.1 MTs per 100 NFs) in wild-type littermates.
Gene Expression of Stathmin and Related Genes in the CNS of Aged Mice
To gain further insight into the axonal abnormalities of aging stathmin knockout mice, we studied the expression of stathmin in spinal cord and retina of aged mice using immunocytochemistry, using a stathmin-specific antiserum. 12 Stathmin immunoreactivity was present in neurons with large somata in spinal cord gray matter and in retinal ganglion cells of aged wild-type mice (Figure 4; A to F ▶ ). The intracellular distribution of stathmin immunoreactivity in these neurons was restricted to the somata and proximal dendrites (Figure 4C) ▶ . In contrast, large myelinated axons of the white matter or the peripheral nervous system did not show detectable stathmin immunoreactivity in aged wild-type mice. These findings demonstrated that the neurons extending the large myelinated axons that exhibit axonal abnormalities in stathmin knockout mice normally expressed stathmin. In aging mice, we did not detect stathmin immunoreactivity in oligodendrocytes and Schwann cells. From a previous study, it was known that oligodendrocytes in early postnatal mice expressed stathmin. 12
Figure 4.
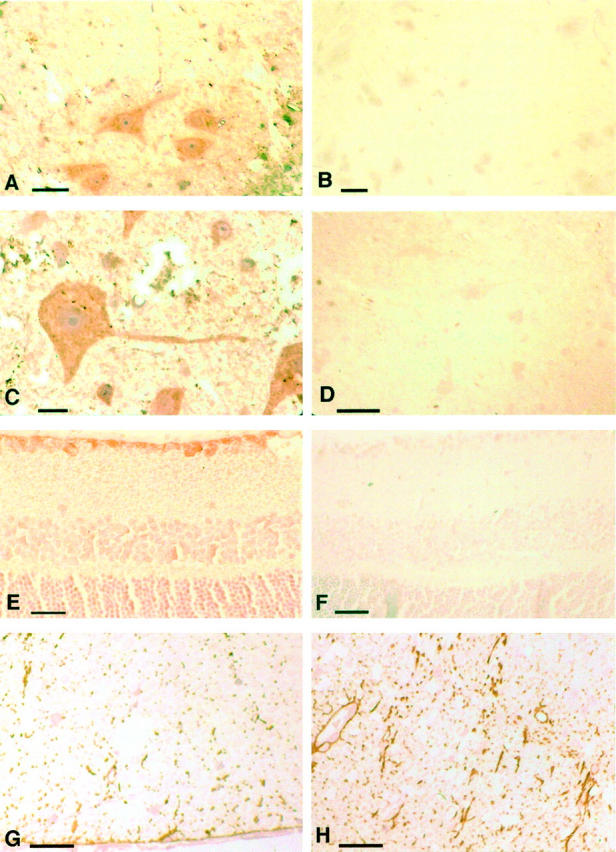
Immunocytochemistry. A: Wild-type littermate control, 18 months of age, spinal cord anterior horn and adjacent white matter. Immunocytochemistry on a 1-μm epon section, stained specifically for stathmin. Stathmin expression is depicted in large-soma motoneurons and their dendrites. Light counterstain with hematoxylin. B: Same as in A, absorption of the stathmin antiserum with antigen confirms specificity of the anti-stathmin peptide immunoserum used. C: As in A, higher magnification demonstrating stathmin immunoreactivity in the soma and dendrite of this motoneuron. D: Stathmin−/− mouse, stathmin antiserum as in A to C, lack of specific staining, counterstain with hematoxylin. E: Wild-type littermate control mouse 14 months of age, retina, 5-μm paraffin section, specific stain with anti-stathmin peptide immunoserum. Retinal ganglion cells (top layer) stain positive for stathmin. F: Stathmin−/− mouse, 14 months of age, retina, absence of stathmin immunoreactivity in retinal ganglion cells (top layer), see also comment for D. G: Wild-type littermate control, 18 months of age, spinal cord anterior column. Immunocytochemistry on epon 1-μm section, stain specific for GFAP. Note regular abundance of GFAP immunoreactivity in this aged mouse. H: Stathmin−/− mouse, 18 months of age, spinal cord anterion column. Note increased GFAP immunoreactivity as compared to wild-type littermate, reactive astrogliosis. Scale bars: 10 μm (A, G, and H); 15 μm (B); 5 μm (C); 20 μm (D); 25 μm (E).
To investigate the different stages of NF phosphorylation, sections were also stained with antibodies specific for NF components. NF immunoreactivity was essentially indistinguishable in sections of spinal cord of aged wild-type and knockout mice (data not shown). Furthermore, staining with antibodies specific for phosphorylated epitopes of NF components did not show consistent differences between stathmin−/− mice and wild-type littermate controls. In contrast, staining for the astroglial marker, GFAP, demonstrated an increase in GFAP immunoreactivity, indicative of a reactive astrocytosis in aging stathmin−/− animals (Figure 4, G and H) ▶ . In general, the magnitude of this finding correlated with the severity of the CNS axonopathy. Immunoblot analysis of spinal cord extracts of aged mice confirmed the immunocytochemical findings. The abundance of tubulin and of NF subunits and their phosphorylated forms was unchanged between stathmin−/− mice and wild-type controls. In contrast, GFAP was slightly more abundant in the knockout mice (data not shown).
The absence of a developmental phenotype and the protracted time course of the abnormalities in stathmin−/− mice made it likely that related genes were up-regulated, substituting for the deleted gene. This substitution could not be verified for SCG10 in earlier stages, as previously reported. 1 To investigate this intriguing question, we performed gene expression studies by Northern blotting on RNA isolated from brain and spinal cord of aging mice for SCG10, Rb3, and SCLIP. Stathmin and the latter three genes constitute the stathmin gene family in which each member has a similar effect on MT stability and catastrophe rate. 53 The experiments revealed that the three known stathmin-related genes were expressed at relatively low levels in aged mice. Interestingly, although expression of SCG10 and Rb3 was not significantly altered, that of SCLIP was slightly increased in brain and greatly in spinal cord (Figure 5) ▶ of aged stathmin knockout mice. SCLIP was expressed at higher level in the brain relative to spinal cord.
Figure 5.
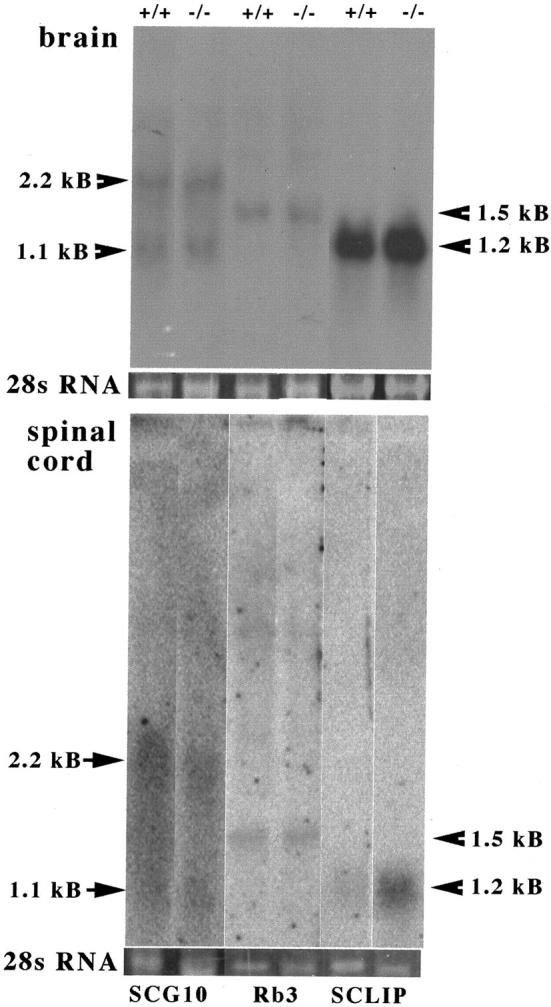
Expression of the stathmin-related genes SCG10, Rb3, and SCLIP in brain and spinal cord of aged stathmin knockout mice. Total brain (top) and spinal cord (bottom) RNA of wild-type (+/+) and stathmin-deficient mice (−/−) was subjected to Northern blot analysis (see Materials and Methods). RNA was pooled from four animals per group. Binding of 32P-labeled DNA probes was revealed by autoradiography. Note arrows and the respective size of the transcripts. The 28S ribosomal RNA band stained with ethidium bromide is shown for each lane (28S).
Functional Analysis by Neurophysiology—Impaired Motor Nerve Conduction
The structural pathology prompted us to investigate the neurophysiology in aging stathmin−/− mice. Aged knockout mice did not exhibit overt weakness or impaired locomotor or grooming behavior. Motor axon conduction velocity was significantly reduced in stathmin−/− mice as compared to wild-type littermates (mean age, 19 ± 2 months). Representative recordings from a stathmin−/− mouse and a stathmin+/+ littermate are depicted in Figure 6 ▶ . Of note is that the waveforms and amplitudes of the ventral root recordings were similar in each case. However, both the latency and peak of the potential in stathmin−/− mice were delayed. Interestingly, there seemed to be even greater slowing of the later components of the response than of the short latency components, suggesting that smaller axons may also be functionally affected (Figure 6) ▶ . The results of the conduction velocity recordings are summarized in Table 2 ▶ .
Figure 6.
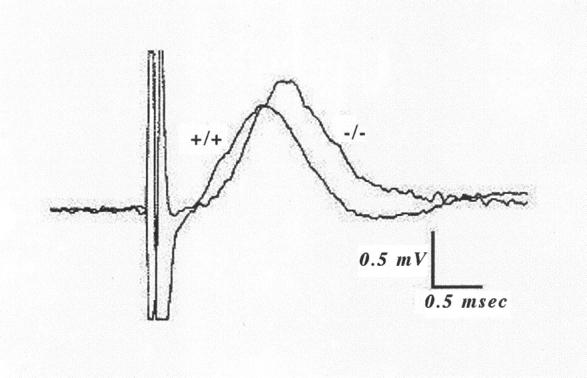
Motor nerve conduction velocity in stathmin−/− mice. Superimposed representative recordings from the sciatic nerve of a 20-month-old stathmin−/− mouse (−/−) and a wild-type littermate (+/+). The conduction distance was 29 mm in each case. Both records were evoked by stimuli that were ∼5× threshold, yielding a maximal response. The measured latencies for these examples (measured at 10% of peak amplitude) were 0.562 and 0.679 msec for wild-type and stathmin−/−, respectively, corresponding to a 20% decrease in the stathmin−/− mouse. Note that both the latency and peak of the stathmin−/− potential are delayed.
Table 2.
Nerve Conduction Velocities in the Sciatic Nerve Ventral Roots of 17- to 21-Month-Old Mice
| Genotype | Stathmin+/+ | Stathmin−/− |
|---|---|---|
| Body weight (g) | 34.6 ± 9.5 | 28.9 ± 5.6* |
| Conduction velocity (m/sec) [mean± SD] | 67.37 ± 12.3 | 52.46 ± 7.08† |
| Conduction velocity (m/sec) [range] | 49.4–87.5 | 40.0–67.7 |
| n | 8 | 15 |
*P > 0.05;
†P < 0.002; for differences from stathmin+/+ mice.
The observed 22% reduction represents primarily a decrease in conduction velocity of the most rapidly conducting (ie, largest) motor axons in the ventral root/sciatic nerve, a finding in line with the observed neuropathological changes. However, because we could not reliably record from individual axons spanning the range of size/conduction velocity, it was not possible to unambiguously conclude whether only the largest axons were affected or whether more slowly conducting axons exhibited parallel reductions in conduction velocity. It was also not exactly known if there was an additional decrease in conduction velocity in the axon proximal to the site of recording in the ventral roots. Spinal cord recordings were extremely difficult to obtain because of respiratory and cardiac motion in these small animals, but on the rare occasions when anti-dromic field potentials were recorded, their latencies seemed to be considerably slowed compared to the population response recorded a few mm from the point of exit of the ventral roots, suggesting that there was indeed further slowing of conduction in the most proximal portions of the motor axons.
Chromosomal Mapping Position of the Human Stathmin Gene—Lack of Adjacent Disease Markers
We investigated the mapping position of the human stathmin gene by querying publicly and privately held databanks (http://www.ncbi.nlm.nih.gov/entrez, OMIM section; http://www.celera.com). The stathmin gene is localized on chromosome 1, 1p36.1-1p35. In this interval, no unassigned disease loci have been mapped. Thus, no neurodegenerative disease, in which an axonopathy could possibly be involved, could be linked to the chromosomal mapping position of the stathmin gene.
Discussion
Here we report a phenotype of stathmin−/− mice that was not apparent on investigation of younger animals. With age, stathmin−/− mice showed a progressive pan-nervous system axonopathy with striking structural damage and associated functional impairment. Although unable to identify a definite molecular mechanism, we were nevertheless able to arrive at the following conclusions based on our experiments.
Age-Dependent Axonopathy in Stathmin-Deficient Mice
Our neuroanatomical and neurophysiological studies demonstrated that stathmin−/− mice develop, with age, a progressive axonopathy of large-caliber, heavily myelinated central and peripheral axons. Neuropathology consisted of primary degenerative changes in the axoplasm. The neuropathological phenotype could be unambiguously ascribed to the absence of stathmin expression because the pathology remained unaltered after several backcrosses into the C57BL/6 strain. Consistent with this conclusion was our finding that neurons giving rise to the affected axons in stathmin−/− mice did express the protein in aged wild-type mice. The decrease in compound motor nerve conduction velocity of stathmin−/− mice could very likely be regarded as the functional correlate of the structural abnormalities present in these axons.
The pathological lesions described were distinct from age-related changes that may develop in normal mice. 54,55 They also did not resemble findings from previously characterized age-related mouse models of neurodegeneration, such as age-related atrophic changes in motor axons of mice deficient in the mid-sized NF subunit, 56 or acquired changes seen in toxic or metabolic neuropathies.
Stathmin Is Essential for Maintaining Axonal Integrity, Suggesting a Novel Function, Possibly Distinct from its Microtubule Destabilizing Activity
The progressive axonopathy that we have described in aging stathmin−/− mice suggests that the protein plays an essential role in maintaining axonal integrity. We speculate that, whereas other genes may compensate for lack of stathmin expression during development, adaptive changes fail later in life. Although we have found that the expression of one of the stathmin-related genes, namely the gene encoding SCLIP, is up-regulated in stathmin-deficient mice, this apparent adaptation does not seem to compensate fully for the loss of stathmin. With respect to whether the axonopathy in stathmin−/− mice is a distal or proximal process, the following circumstantial evidence favors a distal axonopathy: 1) age-related decay of axons in stathmin−/− mice occurred in axons corresponding to those arising from neurons that demonstrate somatic, but not axonal stathmin expression in wild-type littermates; 2) lack of neuropathological changes in somata of these neurons from stathmin−/− mice; 3) predominant involvement of large-caliber axons from long tracts in stathmin−/− mice. However, comparison of spinal roots versus sciatic nerve showed no convincing evidence indicative of a distal worsening.
Defects in other genes have been shown to lead to axonal degeneration 52,56-59 or to attenuated repair after axonal injury. 60,61 However, the pathology observed in these mouse models is distinct from the axonopathy that develops in stathmin−/− mice. Furthermore, the axonopathy in aged stathmin−/− mice did not resemble the pathology associated with known human diseases of the aging nervous system, such as amyotrophic lateral sclerosis, Alzheimer’s disease, and Huntington’s disease. 49,62 Stathmin, therefore, seems to be a key gene for the maintenance of axonal integrity. Whether a similar axonopathy caused by a deficient stathmin gene exists in humans, remains a subject of future investigations.
The biochemical activity of stathmin that may be responsible for its axon-maintaining function is not known at present. In particular, the nervous system abnormalities uncovered in this study are not readily explained by the previously described pattern of expression and functional properties of stathmin. The MT-destabilizing activity of stathmin suggests that reversible phosphorylation of stathmin participates in the mechanism by which extracellular factors control MT dynamics in neurons, particularly at early stages of neuronal morphogenesis, when stathmin is most abundantly expressed. The lack of developmental defects of the nervous system in stathmin−/− mice was surprising, therefore, but may be explained by abundant expression of other members of the stathmin gene family. 39,40,63,64 It seems unlikely that lack of the known MT-destabilizing function of stathmin is responsible for the axonal changes that we have observed. This conclusion is based on the lack of abnormalities, in aged stathmin−/− mice, of the MT cytoskeleton in motoneurons, and on the lack of gross alterations in axonal transport in these animals. Thus, our studies suggest that stathmin serves a function essential for maintaining axonal integrity, that may be distinct from its MT-destabilizing activity. The elucidation of molecular pathways related to stathmin’s functions will be the subject of future gene expression studies that will use cDNA microarrays, subtractive mRNA hybridization, and differential display. 65-67
Acknowledgments
We thank Miriam Pakingam, Earl Swanson, and Bo Chao for providing technical assistance; Drs. J. M. Friedman, A. J. Hudspeth, G. Blobel, and R. Darnell Jr., all at The Rockefeller University, New York, NY, for their encouraging and insightful discussions; and Drs. R. Kucherlapati and W. Edelmann, Albert Einstein College of Medicine, Bronx, NY.
Footnotes
Address reprint requests to Dr. W. Liedtke, Laboratory of Molecular Genetics, The Rockefeller University, Box 305, 1230 York Ave., New York, NY, 10021. E-mail: wolfgang.liedtke@mail.rockefeller.edu.
Supported by United States Public Health grants RO1 NS 25547 (to R. E. W. F.), NS 08952 and NS 11920 (to C. S. R.), and RO1 NS 26333 (to U. K. S.); and in part by a Feodor Lynen Fellowship of the Alexander von Humboldt Foundation, Bonn, Germany (to W. L.).
References
- 1.Schubart UK, Yu J, Amat JA, Wang Z, Hoffmann MK, Edelmann W: Normal development of mice lacking metablastin (P19), a phosphoprotein implicated in cell cycle regulation. J Biol Chem 1996, 271:14062-14066 [DOI] [PubMed] [Google Scholar]
- 2.Sobel A, Boutterin MC, Beretta L, Chneiweiss H, Doye V, Peyro-Saint-Paul H: Intracellular substrates for extracellular signaling: characterization of a ubiquitous, neuron-enriched phosphoprotein (stathmin). J Biol Chem 1989, 264:3765-3772 [PubMed] [Google Scholar]
- 3.Pasmantier R, Danoff A, Fleischer N, Schubart UK: p19, a hormonally regulated phosphoprotein of peptide hormone-producing cells: secretagogue-induced phosphorylation in AtT-20 mouse pituitary tumor cells and in rat and hamster insulinoma cells. Endocrinology 1986, 119:1229-1238 [DOI] [PubMed] [Google Scholar]
- 4.Cooper HL, McDuffie E, Braverman R: Human peripheral lymphocyte growth regulation and response to phorbol esters is linked to synthesis and phosphorylation of the cytosolic protein, prosolin. J Immunol 1989, 143:956-963 [PubMed] [Google Scholar]
- 5.Ferrari AC, Seuanez HN, Hanash SM, Atweh GF: A gene that encodes for a leukemia-associated phosphoprotein (p18) maps to chromosome bands 1p35–36.1. Genes Chromosom Cancer 1990, 2:125-129 [DOI] [PubMed] [Google Scholar]
- 6.Melhem RF, Strahler JR, Hailat N, Zhu XX, Hanash SM: Involvement of Op18 in cell proliferation. Biochem Biophys Res Comm 1991, 179:1649-1655 [DOI] [PubMed] [Google Scholar]
- 7.Schubart UK, Xu J, Fan W, Cheng G, Goldstein H, Alpini G, Shafritz DA, Amat JA, Farooq M, Norton WT, Owen TA, Lian JB, Stein GS: Widespread, differentiation stage-specific expression of the gene encoding phosphoprotein p19 (Metablastin) in mammalian cells. Differentiation 1992, 51:21-32 [DOI] [PubMed] [Google Scholar]
- 8.Brattsand G, Roos G, Marklund U, Ueda H, Landberg G, Nanberg E, Sideras P, Gullberg M: Quantitative analysis of the expression and regulation of an activation-regulated phosphoprotein (oncoprotein 18) in normal and neoplastic cells. Leukemia 1993, 7:569-579 [PubMed] [Google Scholar]
- 9.Horwitz SB, Shen HJ, He L, Dittmar P, Neef R, Chen J, Schubart UK: The microtubule-destabilizing activity of metablastin (p19) is controlled by phosphorylation. J Biol Chem 1997, 272:8129-8132 [DOI] [PubMed] [Google Scholar]
- 10.Schubart UK: Expression of phosphoprotein p19 in brain, testis, and neuroendocrine tumor cells. Developmental regulation in rat brain. J Biol Chem 1988, 263:12156-12160 [PubMed] [Google Scholar]
- 11.Koppel J, Boutterin MC, Doye V, Peyro-Saint-Paul H, Sobel A: Developmental tissue expression and phylogenetic conservation of stathmin, a phosphoprotein associated with cell regulations. J Biol Chem 1990, 265:3703-3707 [PubMed] [Google Scholar]
- 12.Amat JA, Fields KL, Schubart UK: Distribution of phosphoprotein p19 in rat brain during ontogeny: stage-specific expression in neurons and glia. Brain Res Dev Brain Res 1991, 60:205-218 [DOI] [PubMed] [Google Scholar]
- 13.Amat JA, Fields KL, Schubart UK: Stage-specific expression of phosphoprotein p19 during spermatogenesis in the rat. Mol Reprod Dev 1990, 26:383-390 [DOI] [PubMed] [Google Scholar]
- 14.Strahler JR, Lamb BJ, Ungar DR, Fox DA, Hanash SM: Cell cycle progression is associated with distinct patterns of phosphorylation of Op18. Biochem Biophys Res Commun 1992, 185:197-203 [DOI] [PubMed] [Google Scholar]
- 15.Luo XN, Mookerjee B, Ferrari A, Mistry S, Atweh GF: Regulation of phosphoprotein p18 in leukemic cells. Cell cycle regulated phosphorylation by p34cdc2 kinase. J Biol Chem 1994, 269:10312-10318 [PubMed] [Google Scholar]
- 16.Brattsand G, Marklund U, Nylander K, Roos G, Gullberg M: Cell-cycle-regulated phosphorylation of oncoprotein 18 on Ser16, Ser25 and Ser38. Eur J Biochem 1994, 220:359-368 [DOI] [PubMed] [Google Scholar]
- 17.Marklund U, Osterman O, Melander H, Bergh A, Gullberg M: The phenotype of a Cdc2 kinase target site-deficient mutant of oncoprotein 18 reveals a role of this protein in cell cycle control. J Biol Chem 1994, 269:30626-30635 [PubMed] [Google Scholar]
- 18.Larsson N, Melander H, Marklund U, Osterman O, Gullberg M: G2/M transition requires multisite phosphorylation of oncoprotein 18 by two distinct protein kinase systems. J Biol Chem 1995, 270:14175-14183 [DOI] [PubMed] [Google Scholar]
- 19.Hanash SM, Strahler JR, Kuick R, Chu EHY, Nichols D: Identification of a polypeptide associated with the malignant phenotype in acute leukemia. J Biol Chem 1988, 263:12813-12815 [PubMed] [Google Scholar]
- 20.Roos G, Brattsand G, Landberg G, Marklund U, Gullberg M: Expression of oncoprotein 18 in human leukemias and lymphomas. Leukemia 1993, 7:1538-1546 [PubMed] [Google Scholar]
- 21.Nylander K, Marklund U, Brattsand G, Gullberg M, Roos G: Immunohistochemical detection of oncoprotein 18 (Op18) in malignant lymphomas. Histochem J 1995, 27:155-160 [DOI] [PubMed] [Google Scholar]
- 22.Friedrich B, Gronberg H, Landstrom M, Gullberg M, Bergh A: Differentiation-stage specific expression of oncoprotein 18 in human and rat prostatic adenocarcinoma. Prostate 1995, 27:102-109 [DOI] [PubMed] [Google Scholar]
- 23.Bieche I, Lachkar S, Becette V, Cifuentes-Diaz C, Sobel A, Lidereau R, Curmi PA: Overexpression of the stathmin gene in a subset of human breast cancer. Br J Cancer 1998, 78:701-709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Himi T, Okazaki T, Wang H, McNeill TH, Mori N: Differential localization of SCG10 and p19/stathmin messenger RNAs in adult rat brain indicates distinct roles for these growth-associated proteins. Neuroscience 1994, 60:907-926 [DOI] [PubMed] [Google Scholar]
- 25.Sugiura Y, Mori N: SCG10 expresses growth-associated manner in developing rat brain, but shows a different pattern to p19/stathmin or GAP-43. Brain Res Dev Brain Res 1995, 90:73-91 [DOI] [PubMed] [Google Scholar]
- 26.Belmont LD, Mitchison TJ: Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 1996, 84:623-631 [DOI] [PubMed] [Google Scholar]
- 27.Larsson N, Marklund U, Gradin HM, Brattsand G, Gullberg M: Control of microtubule dynamics by oncoprotein 18: dissection of the regulatory role of multisite phosphorylation during mitosis. Mol Cell Biol 1997, 17:5530-5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curmi PA, Andersen SS, Lachkar S, Gavet O, Karsenti E, Knossow M, Sobel A: The stathmin/tubulin interaction in vitro. J Biol Chem 1997, 272:25029-25036 [DOI] [PubMed] [Google Scholar]
- 29.Jourdain L, Curmi P, Sobel A, Pantaloni D, Carlier MF: Stathmin: a tubulin-sequestering protein which forms a ternary T2S complex with two tubulin molecules. Biochemistry 1997, 36:10817-10821 [DOI] [PubMed] [Google Scholar]
- 30.Howell B, Larsson N, Gullberg M, Cassimeris L: Dissociation of the tubulin-sequestering and microtubule catastrophe-promoting activities of oncoprotein 18/stathmin. Mol Biol Cell 1999, 10:105-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marklund U, Larsson N, Gradin HM, Brattsand G, Gullberg M: Oncoprotein 18 is a phosphorylation-responsive regulator of microtubule dynamics. EMBO J 1996, 15:5290-5298 [PMC free article] [PubMed] [Google Scholar]
- 32.Melander Gradin H, Marklund U, Larsson N, Chatila TA, Gullberg M: Regulation of microtubule dynamics by Ca2+/calmodulin-dependent kinase IV/Gr-dependent phosphorylation of oncoprotein 18. Mol Cell Biol 1997, 17:3459-3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schubart UK, Banerjee MD, Eng J: Homology between the cDNAs encoding phosphoprotein p19 and SCG10 reveals a novel mammalian gene family preferentially expressed in developing brain. DNA 1989, 8:389-398 [DOI] [PubMed] [Google Scholar]
- 34.Zhu X-X, Kozarsky K, Strahler JR, Eckerskorn C, Lottspeich F, Melhem R, Lowe J, Fox DA, Hanash SM, Atweh GF: Molecular cloning of a novel human leukemia-associated gene. Evidence of conservation in animal species. J Biol Chem 1989, 264:14556-14560 [PubMed] [Google Scholar]
- 35.Doye V, Soubrier F, Bauw G, Boutterin M-C, Beretta L, Koppel J, Vanderkerkhove J, Sobel A: A single cDNA encodes two isoforms of stathmin, a developmentally regulated neuron-enriched phosphoprotein. J Biol Chem 1989, 264:12134-12137 [PubMed] [Google Scholar]
- 36.Melhem RF, Zhu XX, Hailat N, Strahler JR, Hanash SM: Characterization of the gene for a proliferation-related phosphoprotein (oncoprotein 18) expressed in high amounts in acute leukemia. J Biol Chem 1991, 266:17747-17753 [PubMed] [Google Scholar]
- 37.Maucuer A, Moreau J, Méchali M, Sobel A: Stathmin gene family: phylogenetic conservation and developmental regulation in Xenopus. J Biol Chem 1993, 268:16420-16429 [PubMed] [Google Scholar]
- 38.Okazaki T, Yoshida BN, Avraham KB, Wang H, Wuenschell CW, Jenkins NA, Copeland NG, Anderson DJ, Mori N: Molecular diversity of the SCG10/stathmin gene family in the mouse. Genomics 1993, 18:360-373 [DOI] [PubMed] [Google Scholar]
- 39.Ozon S, Maucuer A, Sobel A: The stathmin family: molecular and biological characterization of novel mammalian proteins expressed in the nervous system. Eur J Biochem 1997, 248:794-806 [DOI] [PubMed] [Google Scholar]
- 40.Ozon S, Byk T, Sobel A: SCLIP: a novel SCG10-like protein of the stathmin family expressed in the nervous system. J Neurochem 1998, 70:2386-2396 [DOI] [PubMed] [Google Scholar]
- 41.Brown A, McFarlin DE, Raine CS: Chronic neuropathology of relapsing experimental allergic encephalomyelitis in the mouse. Lab Invest 1982, 46:171-185 [PubMed] [Google Scholar]
- 42.Boer PH, Potten H, Adra CN, Jardine K, Mullhofer G, McBurney MW: Polymorphisms in the coding and noncoding regions of murine Pgk-1 alleles. Biochem Genet 1990, 28:299-308 [DOI] [PubMed] [Google Scholar]
- 43.Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS: Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol 1998, 152:251-259 [PMC free article] [PubMed] [Google Scholar]
- 44.Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS: GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 1996, 17:607-615 [DOI] [PubMed] [Google Scholar]
- 45.Raine CS, Bornstein MB: Experimental allergic neuritis. Ultrastructure of serum-induced myelin aberrations in peripheral nervous system cultures. Lab Invest 1979, 40:423-432 [PubMed] [Google Scholar]
- 46.Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72:248-254 [DOI] [PubMed] [Google Scholar]
- 47.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 48.Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S: Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103:525-535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirano A: Cytopathology of amyotrophic lateral sclerosis. Rowland LP eds. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. 1991, :pp 91-101 Raven Press, New York [Google Scholar]
- 50.Xu Z, Cork LC, Griffin JW, Cleveland DW: Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 1993, 73:23-33 [DOI] [PubMed] [Google Scholar]
- 51.Cote F, Collard JF, Julien JP: Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 1993, 73:35-46 [DOI] [PubMed] [Google Scholar]
- 52.Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM: Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol 1997, 139:1307-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charbaut E, Curmi PA, Ozon S, Lachkar S, Redeker V, Sobel A: Stathmin family proteins display specific molecular and tubulin binding properties. J Biol Chem 2001, 276:16146-16154 [DOI] [PubMed] [Google Scholar]
- 54.Peters A, Vaughn D: Central nervous system. Johnson JJ eds. Aging and Cell Structure. 1981, :pp 1-34 Plenum Publishing Corp., New York [Google Scholar]
- 55.Spencer P, Ochoa J: The mammalian peripheral nervous system in old age. Johnson JJ eds. Aging and Cell Structure. 1981, :pp 35-103 Plenum Publishing Corp., New York [Google Scholar]
- 56.Elder GA, Friedrich VL, Jr, Margita A, Lazzarini RA: Age-related atrophy of motor axons in mice deficient in the mid-sized neurofilament subunit. J Cell Biol 1999, 146:181-192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giese KP, Martini R, Lemke G, Soriano P, Schachner M: Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell 1992, 71:565-576 [DOI] [PubMed] [Google Scholar]
- 58.Bronson RT, Sweet HO, Spencer CA, Davisson MT: Genetic and age related models of neurodegeneration in mice: dystrophic axons. J Neurogenet 1992, 8:71-83 [DOI] [PubMed] [Google Scholar]
- 59.Boison D, Stoffel W: Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc Natl Acad Sci USA 1994, 91:11709-11713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schrank B, Gotz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M: Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 1997, 94:9920-9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD: Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 1996, 13:43-47 [DOI] [PubMed] [Google Scholar]
- 62.Rowland LP: Amyotrophic lateral sclerosis and other motor neuron diseases. Advances in Neurology, 1991, Vol. 56.:p 569 Raven Press, New York [PubMed] [Google Scholar]
- 63.Stein R, Mori N, Matthews K, Lo LC, Anderson DJ: The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron 1988, 1:463-476 [DOI] [PubMed] [Google Scholar]
- 64.Matsuo N, Kawamoto S, Matsubara K, Okubo K: A novel SCG10-related gene uniquely expressed in the nervous system. Gene 1998, 215:477-481 [DOI] [PubMed] [Google Scholar]
- 65.Chen Y, Talmage DA: Subtractive cDNA cloning and characterization of genes induced by all-trans retinoic acid. Ann NY Acad Sci 1999, 886:225-228 [DOI] [PubMed] [Google Scholar]
- 66.Velculescu VE, Vogelstein B, Kinzler KW: Analysing uncharted transcriptomes with SAGE. Trends Genet 2000, 16:423-425 [DOI] [PubMed] [Google Scholar]
- 67.Serafini T: Of neurons and gene chips. Curr Opin Neurobiol 1999, 9:641-644 [DOI] [PubMed] [Google Scholar]