

Abstract
We previously reported that a severe acquired immune deficiency syndrome-like disease develops in transgenic (Tg) mice expressing the human immunodeficiency virus-1 in its natural target cells: immature and mature CD4+ T cells and cells of the macrophage/dendritic lineage. Here, we show that these mice also develop cardiac disease, characterized most prominently by a focal myocytolysis, occasionally by myocarditis and by deposition of endogenous immunoglobulin on cardiomyocytes. Microfil perfusion demonstrated widespread coronary arteriospasm and echocardiographic analysis revealed depressed cardiac function in Tg mice. A higher (but still modest) level of cardiomyocyte apoptosis was detected in Tg as compared to non-Tg hearts. Tg expression was detected in some of the infiltrating mononuclear cells, but not in cardiomyocytes or in cells of the heart vessels, suggesting a human immunodeficiency virus-1-induced disease process mediated by cells of the immune system. The similarity of the heart disease observed in these Tg mice to that observed in acquired immune deficiency syndrome patients suggests a common pathogenesis.
Infection by human immunodeficiency virus (HIV)-1 leads to the development of a multiorgan disease, the acquired immune deficiency syndrome (AIDS). 1,2 Specific phenotypes have been observed in the lymphoid organs, 1-3 lung, 4,5 kidney, 6,7 bone marrow, 8 striated muscle, 9 peripheral 10 and central 11,12 nervous system, and heart. 13
Cardiac disease in AIDS was under-appreciated early in the course of the epidemic, but is now recognized as one of the most frequent complications of infection with HIV-1. 13-19 First described in 1986 by Cohen and colleagues, 20 the disease is characterized by a progressive, dilated cardiomyopathy that may be accompanied by a compensatory cardiac hypertrophy 21 as well as functional loss. 21 Histologically, a variety of lesions are observed, the most common being cardiomyocyte necrosis, often with fibrosis. 13,14,21 Cardiomyocyte pathology may or may not be accompanied by inflammatory infiltrates. 22 Epicardial lesions have also been documented. 17
The pathogenesis of the cardiac disease in AIDS remains obscure. Resident dendritic cells 23 or infiltrating mononuclear cells 24,25 as well as cardiomyocytes 24,26-29 have been reported to express HIV-1. However, cardiac disease also develops in the absence of obvious myocarditis or detectable infection of cardiomyocytes. 26,27,29,30 Indeed, in most of the studies implicating cardiomyocyte infection, no cell-type-specific markers were used and cells other than cardiomyocytes could have been scored as positive. Therefore, the conclusion that cardiomyocytes are infected remains tentative at present.
We recently developed a novel murine Tg model of AIDS (CD4C/HIV) in which wild-type or mutant HIV-1 genomes are expressed under the control of regulatory sequences (CD4C) comprising the murine CD4 gene enhancer and the promoter elements of the human CD4 gene. 31,32 Consequently, these CD4C/HIV Tg mice express HIV-1 gene products in the natural target cell populations of the virus, ie, in immature CD4+CD8+ T cells, in mature CD4+ T cells, and in cells of the macrophage/dendritic lineage, including peritoneal and alveolar macrophages, Kupffer cells, and dendritic cells. They also exhibit most of the phenotypes associated with this syndrome in human patients: weight loss/failure to thrive, wasting, early death, thymic atrophy, lymphadenopathy, preferential and progressive loss of CD4+ T cells, down-regulation of CD4 cell-surface expression, increase in CD8+ T cell and of B cell number, T cell activation, immunodeficiency, lymphocytic interstitial pneumonitis, interstitial nephritis. 31,32 Most recently, we have documented B cell activation, elevated levels of autoantibody production, and an impairment of germinal center formation in these mice. 33
In a mutational analysis of the HIV-1 genome, we determined that the expression of a single HIV-1 gene, nef, is both necessary and sufficient to cause this AIDS-like disease in Tg mice. 32 Indeed, Tg mice harboring three distinct mutant HIV transgenes, CD4C/HIVMutA (expressing Rev, Env, and Nef), CD4C/HIVMutB (expressing all HIV-1 gene products except Env), and CD4C/HIVMutG (expressing only Nef) all develop an indistinguishable AIDS-like disease, whereas Tg mice harboring a mutant genome lacking Nef expression (CD4C/HIVMutH) failed to develop the AIDS-like disease. 32 Nef is a structurally complex, multifunctional accessory protein encoded by primate lentiviruses [HIV-1, HIV-2, and simian immunodeficiency virus (SIV)]. 34-36 It plays a critical role in virus virulence. Individuals harboring HIV-1 with defects in the nef gene remain healthy after more than a decade of infection. 37,38 Similarly, Rhesus macaques infected with nef deleted SIV, fail to develop simian AIDS. 39 Therefore, it seems that Nef expression in CD4+ cells of the immune system of CD4C/HIV Tg mice mimics its action in human AIDS.
On further investigation, we recently observed a novel feature of the AIDS-like disease in these Tg mice, namely the development of cardiac disease. We report here our study on this cardiac disease observed in Nef-expressing CD4C/HIVMutA and CD4C/HIVMutG Tg mice. We provide pathological and functional evidence of both focal as well as global cardiac disease including cardiac vasculature abnormalities. We conclude that the cardiac disease is similar to that documented in human AIDS, like other phenotypes of this severe murine AIDS-like disease and that it is mediated directly or indirectly by Nef-expressing cells of the immune system or factors produced by these cells.
Materials and Methods
Generation of Tg Mice
The generation and characterization of the CD4C/HIVWT, CD4C/HIVMutA, CD4C/HIVMutB, and CD4C/HIVMutG Tg mice have been described previously. 31,32 Tg and non-Tg littermates were housed together in the same cage, and were maintained on a standard 12-hour light/dark cycle. Mice were fed ad libitum on a diet of Purina Laboratory Chow (a 50%/50% mixture of Chow no. 5001/5015).
Tissue Sampling
For routine histological analysis, mice were killed by CO2 inhalation, and organs to be evaluated were dissected and fixed by overnight immersion in 3.7% formaldehyde buffered in phosphate-buffered saline (PBS). For all analyses of heart tissues in situ, the heart was divided by transverse sectioning into three equally sized pieces. The middle one third of the heart was used for analysis.
For terminal dUTP nick-end labeling (TUNEL) assay or immunohistochemistry (IHC), tissues were fixed by intracardiac perfusion. Mice anesthetized with Avertin were exsanguinated by PBS perfusion, followed by fixation with 4% paraformaldehyde or periodate-polylysine-paraformaldehyde fixative. 40 In detail, after anesthesia induction, a midline incision was made through the anterior abdominal wall ∼2 cm below the xyphoid process. The incision was carried superiorly to the xyphoid process and then laterally to the midaxillary lines, exposing the diaphragm. The diaphragm was punctured just below the xyphoid process and then rapidly cut away from the thoracic cage. Bilateral midaxillary incisions were made through the ribs beginning at the subcostal margin and terminating in the axilla. The rib flap was raised and a 26-gauge needle introduced into the left ventricle close to the apex of the heart. Simultaneously, the free wall of the right atrium was pierced with a sharp scissors. Perfusate was pumped at mean arterial pressure (∼100 mm Hg) using a peristaltic pump. Exsanguination (by PBS) was considered to be complete once the liver had blanched and the effluent PBS was clear. Fixative was then pumped for 5 minutes, at which point the limbs of the mice were rigidly fixed in place. Mice were postfixed by overnight immersion in fixative. After dissection, the organs were embedded in paraffin using a Shandon tissue infiltrator (Hypercenter XP, Pittsburgh, PA), sectioned at 5 μm, and stained with hematoxylin and eosin, as described previously 31 or processed for TUNEL or IHC.
Microscopic Analysis
Tg and control non-Tg tissues were assessed independently by two investigators (SJ and DGK). Myocarditis was scored following the Dallas criteria. 41
In Situ Hybridization
In situ hybridization was performed on paraffin-embedded tissues, using 35S- UTP-labeled anti-sense and control sense RNA probes as described previously. 31,32 Tissues from non-Tg control animals hybridized with anti-sense probes as well as Tg animal tissues hybridized with sense probes failed to exhibit any specific hybridization signal.
TUNEL Assay
The detection of DNA fragmentation in cells undergoing apoptosis was made using the TUNEL assay in situ with a digoxigenin-derivitized UTP (Boehringer Mannheim, Montreal, Canada). The technique was essentially that recommended by the manufacturer. Only animals in which the tissue processing resulted in rapid and complete fixation of hearts and other positive control tissues (lymph nodes, intestine) were considered. Indeed, we have noted TUNEL-positive artifacts in control mice that were overanesthetized (poor respiratory effort, cyanotic appearance) before perfusion fixation, or in whom the perfusion resulted in an incomplete fixation. These artifacts were most notable in the central nervous system where a significant proportion of hippocampal and cortical neurons were TUNEL-positive, and in the heart where large numbers of myocyte nuclei were TUNEL-positive. However, TUNEL-positive cells were rarely observed in these control tissues (eg, zero to two positive cells per transsection of heart) when the perfusion fixation was sufficiently rapid and complete. Consequently, hypoxic or poorly fixed mice were excluded from TUNEL analysis.
Heart tissue analyzed was included in paraffin blocks together with several other tissues including small intestine and mesenteric lymph nodes. Because overfixation can result in a false-negative TUNEL signal, only hearts from blocks in which the internal positive controls (apical intestinal epithelial cells and clusters of lymph node follicular cells) were TUNEL-positive, were considered for analysis. From one to three sections were analyzed per heart.
Detection of Endogenous Ig Deposition
Endogenous Ig were visualized in Tg hearts by immunohistochemistry using anti-mouse Ig or IgM (Sigma) antibodies, essentially as previously described. 31,32
Experimental Autoimmune Myocarditis
Induction of experimental autoimmune myocarditis was performed essentially as described. 42 Briefly 2-month-old BALB/c mice were challenged twice, at 1week intervals, with 100 μg of cardiac myosin (M-0531, Sigma ) emulsified in Freund’s complete adjuvant. Control mice received PBS emulsified in adjuvant. Two weeks after the second injection, mice were anesthetized and perfusion-fixed with 4% paraformaldehyde, and hearts were dissected and processed into paraffin.
Microfil Perfusion
Microfil (Flow Tech, Carver, MA) perfusion (1 ml) was performed after thoracotomy under avertin anesthesia, via the apex of the left ventricle while the heart was still beating, as described. 43 For hypoxic stress, oxygen tension was automatically controlled using a digital O2 sensor (model VA201T, Vulcain Inc., Quebec, Canada) in airtight chambers. Oxygen tension was progressively lowered to 16%, 12% for 2 minutes each, and then to 8% for 4 minutes before Microfil perfusion. Hearts were fixed by immersion in 3.7% formaldehyde buffered with PBS, sliced into 1-mm-thick transverse sections, dehydrated through ethanol series, and cleared in methyl salicylate, as described. 43
Quantitation of Microfil-Infiltrated Vasculature
Digital images of cleared Microfil-infiltrated heart tissues were captured using a Leica stereo dissecting microscope (model MZ12) mated to a Sony color charge-coupled device camera (model DCX-950; Sony). Nonoverlapping images were obtained of the entire cross-sectional area of transverse heart sections of left and right ventricle, at two distinct planes of focus. Two heart sections were analyzed per heart. An average of 30 images was analyzed per heart. Images were captured and processed (see below) blindly from seven Tg and seven non-Tg animals. Image processing was accomplished with Photoshop (Adobe Systems Inc., San Jose, CA, version 5.1) using a combination of automatic thresholding of the Microfil-infiltrated vessels from the tissue background and manual deletion of tissue artifacts from incomplete clearing of the myocardium. Processed images were then quantitated using the threshold, followed by measurement of the total thresholded area functions of the Northern Eclipse image analysis software (Empire Imaging Inc., version 6).
IHC
IHC to detect the active form of caspase-3 (ab397; D. Nicholson, Merck Frost, Montreal, Canada), dystrobrevin (Novocastra Vector Laboratories (Canada) Inc.), α, ε (J. Sanes, Washington University, St. Louis, MO) and γ sarcoglycan (Novocastra) was performed on fresh frozen 10-μm heart sections. Sections were prepared from heart tissue removed from freshly killed mice. The middle third of the heart, cut transversally, was enrobed in OCT (Sakura Chemicals, Montreal, Canada) and plunged into isopentane prechilled in liquid nitrogen to the freezing point (viscous liquid). Incubations with primary antisera were overnight at 4°C with 1/1000 dilution (α-caspase-3) and 2 hours at room temperature using 1/500 dilution for all anti-sarcoglycan antibodies. After incubation with the appropriate secondary antibodies conjugated to horseradish peroxidase, immunoreaction was detected using diaminobenzidine as chromagen. Tissues were counterstained with hematoxylin.
Sirius Red Staining
Collagen staining of paraffin-embedded heart tissue was performed as described 44 using a 0.1% solution of Sirius Red (Direct Red 80, Aldrich, Toronto, Canada) in Bouin’s solution. Quantitation of Sirius Red staining was performed on transections of hearts of nine Tg and seven non-Tg mice. High-density digital images acquired using a Zeiss Cool Pix digital camera were processed using the thresholding function of Photoshop (version 6, Adobe). Collagen deposition was expressed as a percentage of the total tissue area occupied by Sirius Red-stained tissue.
Echocardiography
Left ventricular dimensions, wall thickness, and cardiac function were evaluated by echocardiography and compared between CD4C/HIVMutA Tg mice and age- and sex-matched non-Tg controls that had been bred for at least 10 generations on C3H background. Mice were lightly sedated with Avertin (1.25% ∼9 ml/kg). Echocardiography was performed using a Sonos 5500 (Hewlett Packard, Agilent Technologies, Andover, MA) equipped with a 15-MHZ linear-array transducer. Two-dimensional directed M-mode images were obtained in both parasternal long-axis and short-axis views at the level of the papillary muscle, and used for the measurement of wall thickness and ventricular dimensions. Aortic outflow tract velocity was measured with pulsed-wave Doppler in the parasternal long-axis view. All measurements were performed according to the American Society of Echocardiography recommended guidelines. 45 Left ventricular fractional shortening (FS%) was calculated from M-mode left ventricular dimensions; Ejection fraction (EF%) was calculated from the two-dimensional left ventricular cross-sectional areas (short-axis view); Stroke volume (SV) and cardiac output (CO) were calculated from aortic Doppler echocardiograms, respectively, according to established standard equations.
Results
Cardiac Pathology Observed in CD4/HIV Tg Mice
CD4C/HIVWt Mice
CD4C/HIVWt Tg mice express all HIV-1 open reading frames (ORFs) and died of a severe AIDS-like disease very early, within 36 days of birth. 31 However, no gross or microscopic cardiac pathology was observed in any of the 14 Tg and 8 non-Tg mice analyzed.
CD4C/HIVMutA Tg Mice
CD4C/HIVMutA Tg mice, expressing three HIV-1 proteins, Env, Rev, and Nef developed a severe AIDS-like disease affecting many organs, characterized in part by a premature death. 32 Significant pathological changes were observed in the hearts of a number of CD4C/HIVMutA Tg mice, at different frequency dependent on the extent of inbreeding on C3H background (Table 1) ▶ .
Table 1.
Cardiac Histopathology in CD4C/HIVMutA Tg Mice
| Tg line | Tg expression | No. of generations | Penetrance of pathology | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at death | Heart disease in absence of: | ||||||||
| Range | Mean* | Heart | Lung | Kidney | Kidney disease | Lung disease | |||
| 21407 | High | 2–4 | 21–82 days | 45 ± 21 days | 4/8† | 4/8 | 4/8 | 2/4 | 2/4 |
| 21388 | Medium | 2–4 | 4–8.5 months | 6.5 ± 1.3 months | 14/16 | 8/11 | 10/10 | 0/10 | 3/10 |
| 21380 | Medium | 2–4 | 5–16 months | 9.4 ± 4.1 months | 6/9 | 3/4 | 5/5 | 0/5 | 1/4 |
*Mean ± SD.
†No. of animals with pathology/no. of tissues analyzed.
Gross pathological changes assessed during early backcrossing were noted in 30% (17 of 57) of Tg animals from two medium expressor lines (F21380 and F21388), but in none of the 40 age-matched non-Tg control littermates. The most frequently observed pathological lesions were the presence of small white spots on the surface of the heart (14 of 17). The white spots are likely to represent calcifications observed histologically (see below, Figure 1C ▶ ). Heart enlargement was the second most common phenotype. It was seen in 6 of 17 Tg mice predominantly or exclusively on the left side, and was often no longer apparent after exsanguination. We have not evaluated heart weights in these Tg animals because they are subject to both stunted growth and wasting, 31,32 rendering heart weight measurements (normalized to either body weight or femur length) suspect. More recently, we noticed that gross pathological changes were rarely present in Tg mice more extensively inbred (>10 generations) on C3H. This observation is consistent with recent echocardiographic data (see below). In contrast, no gross pathological changes were noted in any of the 23 Tg animals from the F21407 Tg line that expressed high levels of the Tg and that had to be sacrificed very early in life (45 ± 22 days) because of severe illness. 32
Figure 1.
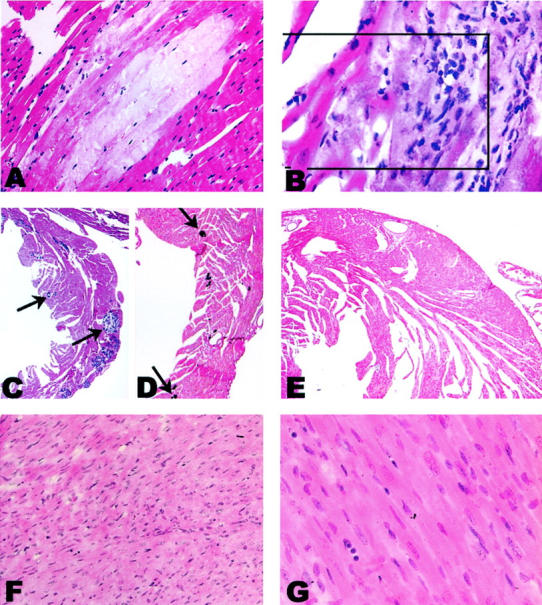
Cardiac pathology in CD4C/HIVMutA Tg mice. A: Focus of myocytolysis in the left ventricle. Note the absence of a myocarditis surrounding the lesion. B: Myocarditis consisting of mononuclear cells with associated myocytolysis. The boxed region is shown at higher power in Figure 3A ▶ . C: Multiple bluish calcifications of varying sizes (for reference, some are denoted by arrows) in the left ventricle. D: The tissue has been stained by the Von Kossa technique highlighting the calcium as deposits (arrows). E: Non-Tg control stained by the Von Kossa technique. F and G: Absence of cardiac histopathological lesions in CD4C/HIVWT. All panels show transverse sections. Original magnifications: ×350 (A); ×420 (B and G); ×50 (C, D, and E); ×170 (F). Counterstain, H&E: A–C, F, G.
The penetrance and severity of cardiac histopathological lesions appear greater during early breeding (Table 1) ▶ and in mice surviving for a longer time because Tg mice dying early had no (CD4C/HIVWT) or fewer (CD4C/HIVMutA F21407) lesions than those dying at an older age (CD4C/HIVMutA F21380, F21388). A variety and a gradient of severity of lesions were observed in these Tg hearts. Regardless of their extent, the most obvious lesions were focal in nature. In less severely affected mice, the ventricular walls (both left and right) contained small focal areas of myocytolysis (Figure 1A) ▶ , sometimes but not necessarily associated with a myocarditis (Figure 1B) ▶ . Often, small calcifications were observed in regions of myocytolysis. More severely affected hearts had larger foci of myocytolysis associated with micro to massive calcifications (Figure 1, B and D) ▶ occasionally with associated fibrous tissue deposition (see below; Figure 2, E and F ▶ ), which may represent a later stage of evolution of the cardiac disease. Occasionally, foci of myocarditis were observed in the absence of any other discernable cardiac changes. Rarely epicardial changes consisting of small aggregates of mononuclear inflammatory cells were present (not shown). Interestingly, we found no evidence of calcification in any other tissue than the myocardium in these Tg mice, ruling out metastatic calcification as a possible sequelae to the kidney disease that was observed in many of these mice.
Figure 2.
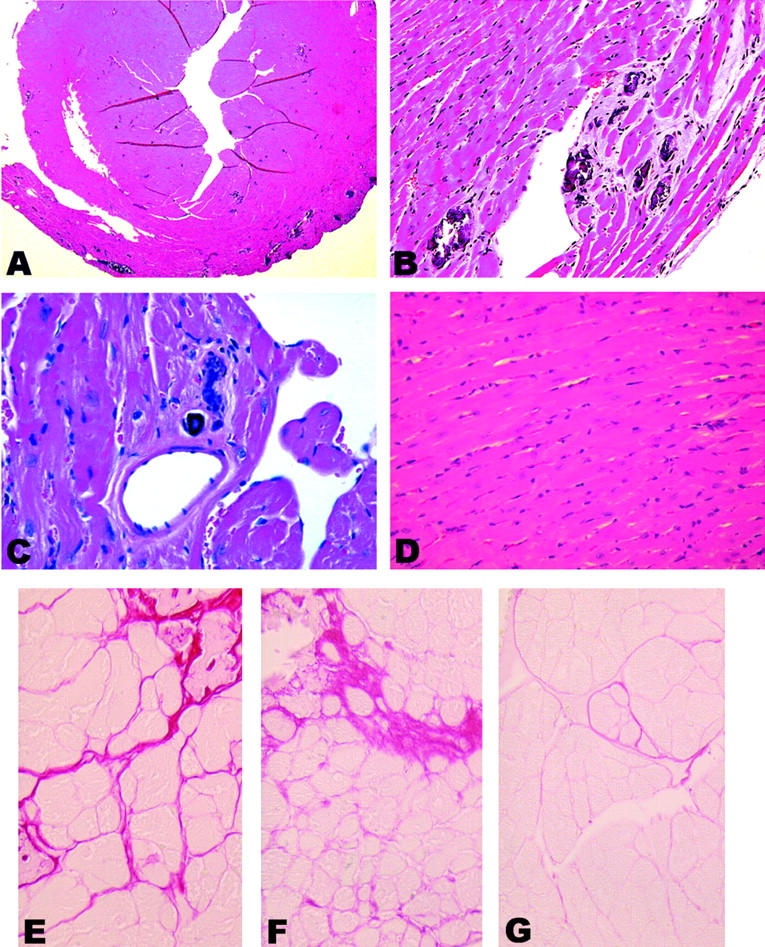
Cardiac pathology in CD4C/HIVMutG Tg mice. A: Multifocal disease is seen throughout the myocardium at low power. B and C: Higher magnification: myocytolysis is observed and is accompanied by calcifications as well as a mild diffuse myocarditis. D: Non-Tg control tissue. E, F, and G: Sirius Red staining of transverse sections of hearts. Tg heart (E and F) show evidence of replacement fibrosis, as well as increased interstitial fibrosis in the absence of any other obvious disease. G: No such fibrosis is observed in non-Tg control. Original magnifications: ×25 (A); ×250 (B and D); ×350 (C, E, F, and G). Counterstain, H&E: A–D.
Together, these results indicated that the development of a variety of cardiac lesions is a frequent phenotype in Tg mice expressing HIV-1 genes in CD4+ T cells and in cells of the macrophage/dendritic lineage. 32 Interestingly, these cardiac lesions are variable in nature and resemble closely many of the lesions observed in HIV-1 infection in humans. 13-19
Expression of Nef in Tg Mice Is Sufficient to Induce Cardiac Lesions
To determine which of the genes (env, rev, and nef) expressed in CD4C/HIVMutA Tg mice were responsible for the cardiac phenotype, we studied other mutant Tg mice expressing all of the genes of HIV-1 except env (CD4C/HIVMutB) or coding for only Nef (CD4C/HIVMutG). We previously reported that these Tg mice develop the same AIDS-like disease as CD4C/HIVWT or CD4C/HIVMutA Tg mice. 32 Cardiac changes indistinguishable from those observed in CD4C/HIVMutA Tg mice and at similar frequency were also observed in mice from both CD4C/HIVMutB (data not shown) and CD4C/HIVMutG (Figure 2) ▶ Tg lines. Multifocal lesions seen at low power (Figure 2A) ▶ included calcification and myocytolysis (Figure 2B) ▶ , myocarditis (Figure 2C) ▶ , as well as fibrosis. This fibrosis, detected by Sirius Red staining, consisted of both the replacement and interstitial types (Figure 2E) ▶ . The increased interstitial fibrosis was distributed diffusely in proximity to foci of replacement fibrosis, as well as apparently not associated with focal lesions (Figure 2, E and F) ▶ . Quantitation of Sirius Red staining of heart transections revealed an increased collagen deposition in the Tg as compared to the non-Tg mice. This differential staining was more striking in the inner one third of the myocardium, a region exhibiting pronounced perfusion deficit (see Figure 6 ▶ ). In this region, despite the fact that it did not reach statistical significance, there was a 50% increase in the tissue area occupied by collagen in Tg mice (3.1 ± 0.53%, n = 9), relative to age-matched non-Tg control mice (2.0 ± 0.31%, n = 7; mean ± SEM). Similar increases in Sirius Red staining have been reported in a SIV model of AIDS cardiac disease. 46
Figure 6.
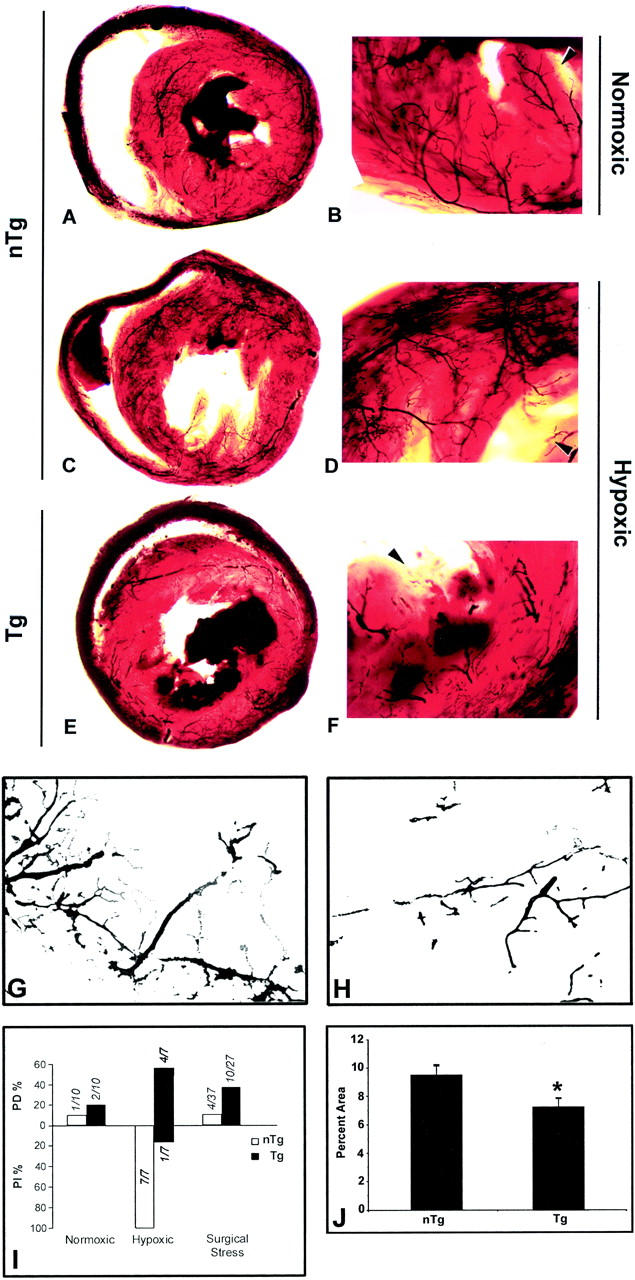
Perfusion defects of coronary arterial vessels under hypoxia in CD4C/HIV Tg mice. Arterial perfusion was performed on Tg mice bred for at least 10 generations on C3H background. Microfil-perfused coronary vasculature is seen by transillumination of cleared heart sections. Under normal oxygen tension larger coronary arteries of non-Tg mice taper gently down to smaller ones without constriction or dilation (A and B). Under hypoxia, general vessel dilatation was observed in non-Tg mice (C and D). In contrast, under hypoxia, Tg mouse vasculature showed sparseness of perfused vessels, areas of constriction with areas of pre- or postnarrowing dilatation and discontinuity, mainly in the intramuscular and endocardial zone (E and F). Arrowheads indicate endocardial side. G and H: Images of Microfil perfused non-Tg (G) and Tg (H) hearts electronically processed to remove background staining (see Materials and Methods). Note the sparseness, narrowing, and discontinuity of the Tg vasculature. I: Quantitation of the incidence of hypoxic stress-induced perfusion defects (PD) or perfusion increase (PI) in Tg and non-Tg mice. J: Quantitation of the extent of perfusion difference between Tg and non-Tg mice exposed to hypoxic stress was based on images as those shown in G and H, as described in Materials and Methods. Error bars indicate the SEM for seven mice assessed in each group. Significance was assessed by a two-tailed Student’s t-test. *, (P < 0.05). Original magnifications: ×10 (A, C, and E); ×30 (B, D, and F); ×50 (G and H).
Together, these results indicated that Nef expression was sufficient to generate cardiac lesions in these Tg mice.
Cell-Type Specificity of Tg Expression
We have previously reported that transgene expression with the CD4C regulatory sequences was primarily restricted to the immature CD4+CD8+ and mature CD4+ T cells as well as cells of the macrophage/dendritic lineage. 31,32,47,48 No evidence of expression could be documented in smooth, skeletal muscle, endothelial, or in various epithelial cells. To further confirm that Tg expression was restricted in the heart to cells of hematopoietic origin, Tg expression was assessed by in situ hybridization using HIV-1-specific riboprobes, 31 both in CD4C/HIVMutA and CD4C/HIVMutG Tg hearts. No Tg expression was observed in cardiomyocytes or in heart vessels (n = 15 Tg hearts, two sections each) (Figure 3, A and B) ▶ . Expression was only observed in mononuclear interstitial cells (Figure 3, A and C) ▶ and, in some animals, in rare single cells dispersed throughout the myocardium (Figure 3B) ▶ . Therefore, cardiac lesions were observed most often in the absence of detectable transgene expression, even in mononuclear cells. These results suggest that the cardiac pathology observed in these Tg mice develops by an indirect mechanism and is likely immune-mediated.
Figure 3.
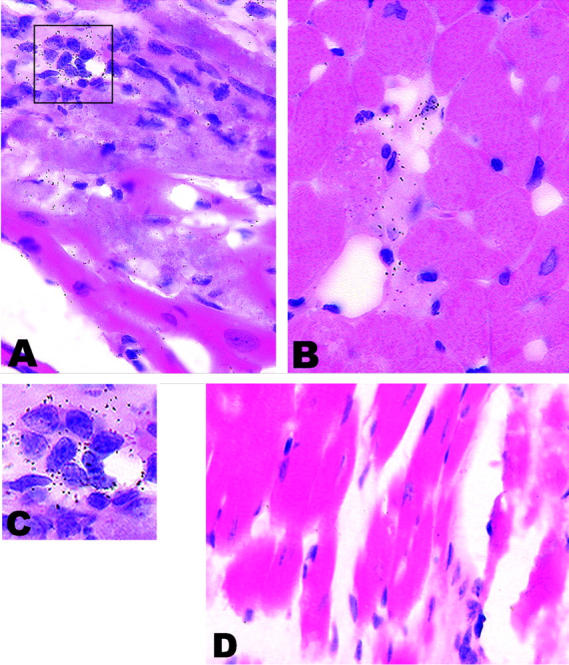
Rare HIV-1 transgene expression detected by in situ hybridization in CD4C/HIVMutA Tg mice. Anti-sense (A–C) or control sense (D) 35S-labeled riboprobes were used. A: Transgene expression was rarely detected in mononuclear cells infiltrating the myocardium. Note the myocytolysis and vacuolation of adjacent cardiomyocytes. B: Tg expression is also rarely seen in isolated mononuclear cells of the myocardium. Note the small focus of myocytolysis adjacent to Tg-expressing cells. Also note that adjacent healthy cardiomyocytes do not express detectable levels of the transgene. Similar results were obtained in CD4C/HIVMutG Tg mice. C: Enlargement of boxed region of A demonstrating the nuclear morphology of the transgene expressing cells. D: Tg heart hybridized with a sense probe. Note the absence of signal. Original magnifications: ×550 (A, B, and D); ×1100 (C). Counterstain, H&E.
Apoptosis of Cardiomyocytes in Diseased CD4C/HIVMutA Tg Mice
Apoptosis has been postulated to be one mechanism by which cardiomyocytes are lost in other cardiac diseases. 49,50 To determine whether apoptosis of cardiomyocytes was an important phenotype in these Tg mice, the TUNEL method and IHC with antibodies against the activated form of caspase-3 were used to detect apoptotic cardiomyocytes. Only rare single TUNEL- or caspase-3-positive cardiomyocytes were observed in non-Tg mice (n = 4 and n = 3, respectively). Evidence of apoptosis in cells having the morphology of cardiomyocytes was seen in only 2 Tg hearts each of 12 (TUNEL) or of 9 (caspase-3) CD4C/HIVMutA or CD4C/HIVMutG Tg mice assessed, despite the fact that several hearts had extensive pathology. Cardiomyocyte apoptosis as assessed by TUNEL (Figure 4 ▶ ; A to D) or caspase-3 (Figure 4, E and F) ▶ staining was not seen in undiseased regions, but was only discernable in areas of very mild pathology in the form of clusters of several apoptotic cardiomyocytes and in association with a diffuse mild myocarditis. In two Tg mice, 10 and 14 TUNEL-positive cardiomyocytes, respectively, were clustered around regions of mild pathology. In contrast, in non-Tg hearts, between zero to two TUNEL-positive cardiomyocytes were seen to occur sporadically. A similar distribution of apoptotic cardiomyocytes, associated with areas of myocarditis but not areas exhibiting chronic fibrotic lesions, has recently been reported to occur in hearts of SIV-infected Rhesus monkeys. 51,52 These results suggest that apoptosis may be one mechanism by which cardiomyocytes are lost in this disease.
Figure 4.
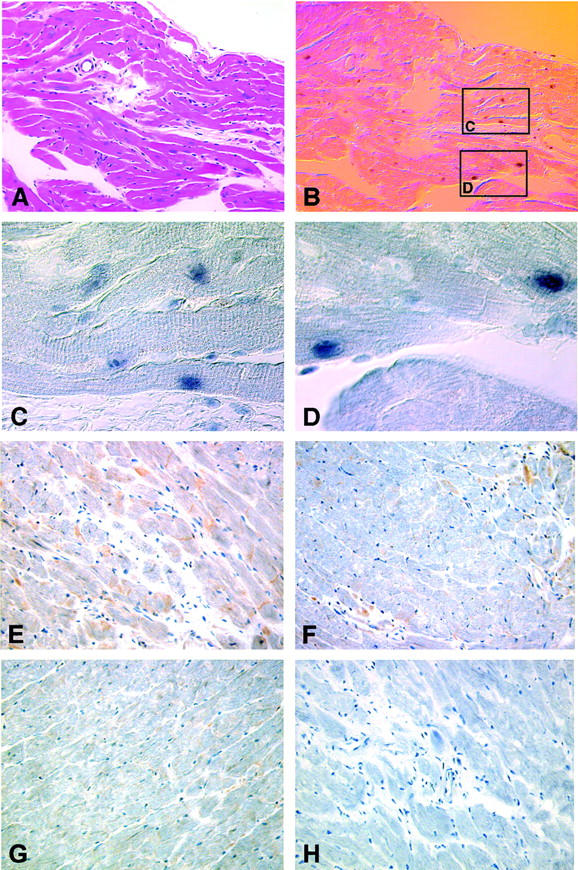
Detection of cardiomyocyte apoptosis in CD4C/HIV Tg mice. A–D: TUNEL. Cardiomyocytes adjacent to a region of minimal pathology, representing borderline myocarditis (A; H&E) are TUNEL-positive (B–D). Alkaline phosphatase staining of TUNEL-positive nuclei, no counterstain, background color derives from Namarsky optics. Boxed regions of B refer to regions shown in C and D. E, F, and G: Caspase-3 IHC. IHC to detect the active form of caspase-3 in a CD4C/HIVMutG Tg mouse. Active caspase-3 is detected in cardiomyocytes, in areas of diffuse mild myocarditis (E and F), but not in a morphologically normal-appearing region of the same heart (G). H: Same field as shown in E in a serial section exposed to only secondary antibody. Original magnifications: ×230 (A and B); ×1200 (C and D); ×250 (E–H). Counterstain: H&E (A); hematoxylin (E–H).
Decoration of Hearts of CD4C/HIV Tg Mice by Endogenous Immunoglobulin (Ig)
Autoimmunity is a well-known cause of myocarditis and cardiomyopathy. 53 Anti-heart antibodies have been detected in a significant percentage of HIV-infected patients with heart muscle diseases. 54 Moreover, we recently found that autoantibody levels (anti-DNA) were elevated in the sera of the CD4C/HIVMutA Tg mice. 33 Given these findings, we looked for the presence of signs of anti-heart antibodies in these Tg mice. Heart sections probed with anti-mouse Ig antibodies revealed decoration of the cytoplasm of cardiomyocytes, either isolated or in bundles, in areas exhibiting minimal or no histological evidence of disease in a significant proportion (5 of 30) of CD4C/HIVMutA and CD4C/HIVMutG Tg hearts. These were both associated with (Figure 5D) ▶ or distinct (Figure 5B) ▶ from areas of frank pathology in Tg mice and were not found in non-Tg mice (0 of 22) (Figure 5F) ▶ or in undiseased hearts from CD4C/HIVWT Tg mice (0 of 9) (Figure 5E) ▶ .
Figure 5.
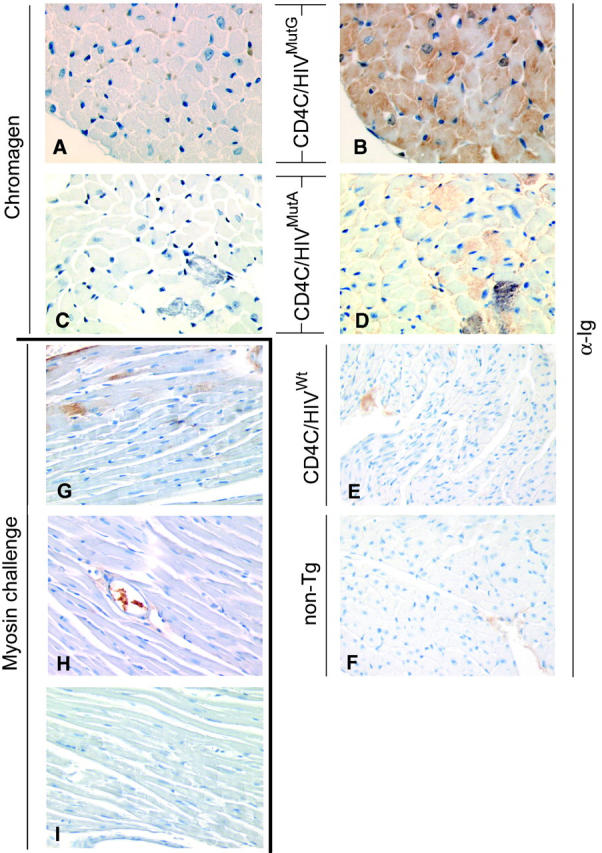
Deposition of endogenous Ig in myocardium of CD4C/HIVMutG and CD4C/HIVMutA Tg mice. Myocardium of control non-Tg (F), CD4C/HIVMutG (B), CD4C/HIVMutA (D), or CD4C/HIVWt (E) Tg mice were probed with α-mouse Ig antibodies. A and C: Adjacent sections to B and D, respectively, reacted with chromagen alone. Note that deposition of endogenous Ig was observed in CD4C/HIVMutG and CD4C/HIVMutA Tg myocardium, but not in CD4C/HIVWT Tg or non-Tg myocardium. G: As a positive control, sections of myocardium of a BALB/c mouse developing experimental autoimmune myocarditis after injection of cardiac myosin was probed with α-mouse Ig antibodies. Note the deposition of Ig. H: No Ig deposition was apparent in the myocardium of the vehicle-challenged mouse probed similarly. I: Myocardium of a myosin-challenged mouse probed with α-rat Ig antibodies as a negative control. Immunoreaction products in E, F, and H are a blood component inside the vasculature. Original magnifications: ×300 (A–I). Counterstain, hematoxylin.
As a comparator, heart sections from BALB/c mice in whom an experimental autoimmune myocarditis had been induced by challenge with cardiac myosin 42 were also probed with anti-mouse Ig antibodies. Heart sections from the cardiac myosin-challenged mice (three of three) (Figure 5G) ▶ but not from the vehicle-injected mice (zero of two) (Figure 5H) ▶ exhibited cytoplasmic decoration of cardiomyocytes by endogenous Ig very similar to that observed in the CD4C/HIV Tg mice. Because the pattern of endogenous Ig staining in CD4C/HIV Tg mice is similar as that of a well-established model of experimental autoimmune myocarditis, this suggests that these Ig deposits in Tg hearts may represent anti-heart antibodies. In such an event, these data would indicate that the CD4C/HIV Tg mice have the ability to produce autoantibodies of different specificities, most likely as a consequence of the dysregulation of T- and B-cell functions previously documented. 33
Cardiac Pathology Occurs in the Absence of Detectable Disruption of the Dystrophin-Associated Glycoprotein Complex
In rodent cardiomyopathies resulting in nearly identical histopathology, the dystrophin-associated glycoprotein complex of cardiomyocytes, or/and of smooth muscle cells, is disrupted. 43,55,56 We used antibodies against α, γ, ε sarcoglycans and against dystobrevin to determine the integrity of the dystrophin-associated glycoprotein complex and against smooth muscle actin to assess smooth muscle cell abnormalities. No differences in staining patterns could be observed between Tg and non-Tg myocardium or vasculature with any of these antibodies (data not shown).
Coronary Vascular Function Is Compromised in CD4C/HIV Tg Mice
One possible etiological condition that could give rise to the multifocal nature of the cardiac lesions of these HIV Tg mice is an altered vascular function. 43,55 Therefore, we studied the heart vasculature in two groups of Tg mice, more extensively bred in C3H (>10 generations) by perfusing the heart with Microfil, a liquid silicon rubber. In the first group, mice enrolled in several other protocols, and subjected to both anesthesia and surgical procedures (to remove lymph nodes, thymus, and spleen; followed by thoracotomy) immediately before Microfil perfusion were used. In this group, 37% (10 of 27) of Tg versus 11% (4 of 37)of non-Tg mice exhibited coronary perfusion defects. Among these, a higher percentage of younger (2 to 5 months) Tg [58% (7 of 12)] or non-Tg [17% (3 of 17)] mice showed perfusion defects when compared to older mice (>6 months old) [Tg, 20% (3 of 15)] and non-Tg, 5% (1 of 20)]. A second group of mice (2 to 5 months old) were anesthetized and either exposed or not to hypoxic stress before thoracotomy and perfusion (Figure 6) ▶ . Without hypoxic stress, 2 of 10 Tg mice versus 1 of 10 non-Tg mice demonstrated coronary microvascular perfusion defects. However, under hypoxia, four of seven Tg mice demonstrated perfusion defects, and one mouse showed evidence of increased perfusion. In contrast all seven non-Tg mice showed evidence of increased perfusion, which likely represents vascular dilatation in response to decreased oxygen supply. 57
In all cases, the perfusion defects observed consisted of a decreased abundance of perfused vessels, with patchy vessel constriction, vascular discontinuities and irregularities, vascular dilatation pre- and postnarrowing (Figure 6 ▶ ; E, F, and H). This apparent arteriospasm mainly affected medium and small coronary vessels in intramuscular and endocardial regions. Quantitation of the tissue area occupied by Microfil-filled vessels revealed a statistically significant decrease (25%) in the perfused area of Tg as compared to non-Tg heart vessels (Figure 6J) ▶ . These results suggest that abnormal coronary vascular function may be an important etiological factor of the multifocal cardiac lesions seen in these mice.
Cardiac Function Is Depressed in CD4C/HIV Tg Mice
In view of the multiple focal lesions, the more diffuse interstitial fibrosis and the apparent coronary vasospasm observed in the hearts of these CD4C/HIV Tg mice, it is possible that their cardiac function may be compromised, as reported in human AIDS. Left ventricular dimensions, wall thickness, and cardiac function were assessed by echocardiography in 15 CD4C/HIVMutA Tg and in 12 age- and sex-matched control mice (3 to 6 months old) (Figure 7 ▶ , Table 2 ▶ ). There was no significant difference between Tg and non-Tg mice in left ventricular dimensions and wall thickness. However, a significant (P < 0.01) increased systolic left ventricular internal dimension was observed in Tg mice, a finding that implies depressed contractility and that is consistent with the increased collagen deposition seen in these Tg mice. In addition, the fractional shortening (FS%), the ejection fraction (EF%), the stroke volume (SV), and the cardiac output (CO) were all significantly decreased (P < 0.01) in Tg mice as compared to non-Tg mice, indicating a compromised left ventricular function.
Figure 7.

Echocardiographic demonstration of compromised cardiac function in a CD4C/HIV Tg mouse. Echocardiography was performed on Tg mice bred for at least 10 generations on C3H background. Typical M-mode and aortic Doppler echocardiograms from a CD4C/HIVMutA Tg mouse (Tg) and an age, sex-matched non-Tg control mouse (non-Tg) are shown. Note the decreased contractility and increased systolic left ventricular internal dimensions of the Tg heart (arrowheads, bottom left). Decreased aortic flow rate is seen in the Tg heart in Doppler mode (bottom right). Note the difference in scales between non-Tg and Tg Doppler images. IVS, interventricular septal thickness; LVPW, left ventricular posterior wall thickness; LVIDd, end-diastolic left ventricular dimension; LVIDs, end-systolic left ventricular dimension.
Table 2.
Echocardiographic Indices in CD4C/HIVMutA Tg Mice
| Parameters | Non-Tg | Tg |
|---|---|---|
| HR (bpm) | 624 ± 53 | 572 ± 78 |
| IVS (mm) | 0.105 ± 0.021 | 0.097 ± 0.021 |
| LVPW (mm) | 0.103 ± 0.019 | 0.092 ± 0.023 |
| LVIDd (mm) | 0.270 ± 0.027 | 0.283 ± 0.060 |
| LVIDs (mm) | 0.121 ± 0.018 | 0.183 ± 0.060* |
| FS (%) | 55.2 ± 5.0 | 36.7 ± 8.6* |
| EF (%) | 80.6 ± 7.9 | 60.3 ± 16.1* |
| SV (μl) | 98 ± 18 | 74 ± 23* |
| CO (ml/min) | 61.1 ± 12.1 | 42.4 ± 14.6* |
Values are means ± SD;
*P < 0.01 Tg (n = 15) versus non-Tg (n = 12) mice. HR, (bpm); heart rate, beats per minute. IVS, interventricular septal thickness; LVPW, left ventricular posterior wall thickness; LVIDd, end-diastolic left ventricular dimension; LVIDs, end-systolic left ventricular dimension; FS, fractional shortening; EF, ejection fraction; SV, stroke volume; and CO, cardiac output.
Discussion
Novel Observations in the CD4C/HIV Tg Model of AIDS: Cardiovascular Findings
We show here that CD4C/HIV Tg mice develop, in addition to the previously reported 31,32 AIDS-like disease, a global cardiac dysfunction (vascular defects and decreased left ventricular function) as well as various focal cardiac pathological lesions (myocytolysis, myocarditis, replacement fibrosis, and interstitial fibrosis), similar to those found in individuals with AIDS. 13-18,58 We observed that the decreased left ventricular function phenotype seems to segregate independently of the focal lesions in these Tg mice, suggesting that they may be induced by different mechanisms. However, the fact that the global vasospasm may give rise stochastically to focal lesions may also indicate the presence of a single underlying defect. Our present data do not allow us to determine whether the focal and global lesions are related or not. Remarkably, only one HIV-1 gene product (Nef) is sufficient to elicit these cardiac lesions, consistent with our previous results showing that Nef is sufficient to elicit the whole AIDS-like disease in CD4C/HIVMutG Tg mice. 32 We have also recently determined that expression of the functionally similar SIV Nef in Tg mice under the control of the same CD4C promoter results in very similar cardiac lesions. 59
It is unlikely that the cardiac pathology in these mice was simply a result of a generalized severe illness, because the CD4C/HIVWt Tg mice that exhibited a virtually indistinguishable AIDS-like disease to that of the CD4C/HIVMutA and CD4C/HIVMutG Tg mice, except for an earlier death, did not develop grossly or histologically detectable heart disease. Also, very ill and moribund C3H mice infected with the paralytogenic/leukemogenic variant of Moloney MuLV (ts-1) 60 and showing a very severe clinical phenotype (kyphosis, wasting, partial to complete hind limb paralysis, and in some animals advanced lung disease) at the time of sacrifice exhibited no histopathological heart lesions (Kay GD, Massé G, Jolicoeur P, unpublished).
The compromised left ventricular function detected by echocardiography in these CD4C/HIV Tg mice, which in some mice was severe, is reminiscent of the finding in human AIDS. Indeed, left ventricular dysfunction, seen even in asymptomatic HIV-1 carriers, 61 is among the most frequently described cardiac disorders in AIDS. 20,62 The gross and the histological cardiac abnormalities noted in these CD4C/HIV Tg mice are also similar to those observed in HIV-1 infection. Myofibrillar necrosis sometimes accompanied by a myocarditis, or myocarditis alone are commonly observed in human AIDS. 14,21,22 It should be pointed out that such histological lesions are not specific to AIDS cardiac lesions and are found in other heart diseases, of idiopathic or of various known etiologies. Myocardial calcification seen in this model is not however a feature of AIDS cardiomyopathy and may simply reflect the susceptibility of the mouse strain used (C3H), and not of other strains of mice, to develop muscle calcifications after trauma. 63 Together, these results suggest that the AIDS cardiomyopathy in humans may be caused by expression of HIV-1 itself.
Surprisingly, in contrast to all of the other phenotypes of these Tg mice, the extent of the cardiac lesions did not correlate with the levels of expression of the HIV-1 transgene in the thymus or spleen. Indeed, cardiac lesions were not observed in high expressor Tg mice that died within an average of 5 weeks of age, perhaps because they were exposed to the effects of transgene expression for an insufficient length of time. This feature of the Tg model reflects the heart disease in humans that has been found to develop at a higher incidence in patients infected for a longer time or with a more advanced disease. 18,64
Recently, dilated cardiomyopathy exhibiting several features of the human disease, except for more severe arteriopathy, has been reported in SIV-infected monkeys. 46 Also, generalized muscle degeneration, both skeletal and heart, and arteriopathy of several organs, including of the heart, have been observed in other HIV Tg mice, especially in the d1443 Tg line. 65 But heart disease was observed only sporadically in these older Tg mice. More recently, functional cardiac abnormalities, in the absence of detectable histological lesions, were described in these mice. 66 In other Tg mice in which expression of HIV-1 outside lymphoid organs was restricted to smooth muscle cells, a severe generalized vasculopathy developed. 67
Our new findings of a cardiac disease in the CD4C/HIV-1 Tg model adds to this list and presents specific distinguishing characteristics: 1) HIV-1 and not SIV is being studied; 2) the cardiac disease develops in the specific context of animals expressing HIV-1 in the same CD4+ target cells as those infected in humans, 31,32 and developing a severe immunodeficiency, associated with an autoantibody response similar to human AIDS; 33 3) the HIV-1 gene product harboring the major determinant for the cardiac lesions (Nef) has been identified; 4) an enhanced response of the coronary vasculature (arteriospasm) to stimuli has been documented; 5) both functional defects and histological lesions of the heart are observed at high frequency; 6) functional defects of the coronary vasculature occur in the absence of detectable transgene expression in this structure; 7) and most importantly, expression of Nef in immune cells, and not in cardiomyocytes or in smooth muscle cells, triggers the disease (see below).
Development of Cardiac Lesions in Tg Mice Occurs in the Absence of Detectable Tg Expression in Cardiomyocytes and Seems to Be Caused by Tg Expression in Immune Cells
As expected from the specificity of the hCD4 promoter in these Tg mice, we found that neither cardiomyocytes nor cells of the cardiac vasculature express detectable levels of the HIV-1 RNA. These results suggest strongly that the cardiac lesions resulted from expression of HIV-1 Nef in specific immune cells, and therefore is immune-mediated. The literature regarding the cardiac cell-type specificity of infection by HIV-1 is currently unclear, with both mononuclear cells and cardiomyocytes having been reported to be infected. 23-30 However, our present results suggest that cardiomyocyte expression of HIV-1 gene products may not be necessary for the development of HIV-1-associated cardiomyopathy in humans.
Consistent with the absence of a correlation between the levels of transgene expression in lymphoid organs and the extent of the cardiac lesions, we observed that the number of HIV-1-expressing cells in the heart was low or zero and did not correlate with the extent of the cardiac lesions. In human AIDS a similar paradox has been observed, in which very low numbers of HIV-1-expressing cells are detectable in the heart. 26-29 This animal model seems to replicate this intriguing cardiac phenotype faithfully, as it does for many other phenotypes in other organs, suggesting similar underlying mechanisms for the development of both the mouse and human heart diseases.
Pathogenesis of Cardiac Lesions in CD4C/HIV Tg Mice
Because these CD4C/HIV Tg mice exhibit a multiorgan disease, it is at present impossible to definitively determine whether their heart disease is primary or secondary. The situation is also similar in human AIDS, which is a syndrome with multiple phenotypes. 1,2 However, we sometimes observe pathological lesions in the CD4C/HIV Tg hearts, where only minimal lung or kidney pathology exists (Table 1) ▶ . Conversely and perhaps more importantly, in CD4C/HIV Tg mice bred for several generations on the C3H background, kidney disease is still present at high frequency, but is less frequently associated with histological lesions of the heart. These observations indicate that the heart disease can segregate independently of the kidney and lung diseases and suggest that the heart disease may not be secondary to lung or kidney disease.
Our results suggest that in these Tg mice, cardiomyocytes die, at least in part, by apoptosis. Although a low number of apoptosing cardiomyocytes was detected, this may throughout time have a significant impact on cardiomyocyte attrition.
The heart disease of these Tg mice is unlikely to be caused by the encephalomyocarditis virus or the coxsackievirus 68 because in addition to heart disease, these viruses induce widespread lesions. Furthermore, coxsackievirus virus-encoded protease 2A cleaves dystrophin resulting in the morphological disruption of α-sarcoglycan, 69 a phenotype not seen in the CD4C/HIV Tg mice. The cardiac lesions could also arise because of their close proximity to HIV-1-expressing immune cells that could disappear once the lesions are initiated. The observation of increased numbers of TUNEL- and caspase-3-positive cells associated with mild diffuse myocarditis is consistent with this possibility.
Alternatively, development of cardiac disease may involve an indirect humoral factor, such as the anti-heart autoantibodies that may be present in these mice, because decoration of cardiomyocytes by endogenous Ig was documented. Heart diseases induced by anti-heart autoantibodies have been described both in humans 53 and in animal models. 70 Also anti-heart antibodies have been detected in patients with HIV-1-associated heart muscle disease. 54 Moreover, we have already shown that the CD4C/HIV Tg mice produced elevated levels of anti-DNA autoantibodies, indicating that the presence of autoantibodies is another feature of this model similar to the human disease. Although our experiment does not establish that the endogenous Ig deposits on cardiomyocytes are pathogenic, they may contribute to the development of cardiac lesions. Further work such as deletion of Ig-producing B cells from these Tg mice will be needed to assess the pathogenic role of these putative anti-heart antibodies.
Another attractive hypothesis to explain the development of the multifocal cardiac lesions of these mice is the global functional defect of the vasculature. This defect appears to manifest itself as an arteriospasm affecting mainly the medium and small cardiac vessels, and is enhanced under hypoxia or stress. Such widespread arteriospasm, if sustained at some restricted sites, would be expected to result in microinfarction and in the appearance of multifocal lesions as the ones we detected. In fact, identical histopathology resulting from arteriospasm arises in mice and hamsters in which the δ-sarcoglycan has been mutated. 43,55,56 Altered endothelial cell function leading to vasospasm and consequent focal myofibrolysis may represent one component of the pathogenic cascade leading to the cardiac pathology observed in AIDS patients. This would be consistent with the endothelial cell dysfunction observed in HIV-1 infection. 71 Indeed, focal and multifocal hypoperfusion with alterations of endothelial cell structure and membrane composition has been reported in the central nervous system of AIDS patients. 72 In light of these observations, further investigation of endothelial cell-immune cell interactions may prove fruitful.
Therefore, it seems that, in these CD4C/HIV Tg mice, Nef is able to induce in one or more than one immune cell types in which it is expressed (CD4+ T cells, macrophages, and dendritic cells) the production of factors [possibly cytokine(s), chemokine(s) and/or receptors] leading to decreased cardiac function and facilitating cardiac arteriospasm. This arteriospasm possibly associated with other insults, such as heart autoantibodies, may be the underlying cause of the multifocal cardiac lesions. The cardiac disease in CD4/HIV Tg mice is a novel feature of this mouse model of AIDS and seems to faithfully reflect many of the AIDS-related human cardiac lesions. It thus should prove to be a valuable tool in understanding their pathogenesis. In addition, with the data presented here, it is tempting to hypothesize that some other noninfectious human heart diseases may also be mediated by some yet unrecognized dysfunctions of the same type(s) of immune system cells.
Acknowledgments
We thank Karina Lamarre, Chunyan Hu, Benoît Laganière, Martin Demers, Claire Crevier, Annie Vallée, Viorica Lascau, and Francine Poulin for excellent technical assistance; Christian Charbonneau and Hélène Lienard for image processing; Drs. D. Nicholson (Merck Frost, Canada) and J. Sanes (Washington University School of Medicine) for their gift of anti-caspase-3 and α- and ε- sarcoglycan Ab, respectively; our colleagues Drs. Mona Nemer, Pierre Paradis, and Dr. Marc Martin (Hôtel-Dieu Hospital), Drs. Adel Giaid and Takayoki Saito (Montreal General Hospital), and Dr. Jean-Claude Tardif (Montreal Heart Institute) for helpful discussions; and Rita Gingras for preparing the manuscript.
Footnotes
Address reprint requests to Dr. Paul Jolicoeur, M.D., Ph.D., Clinical Research Institute of Montreal, 110 Pine Ave. West, Montreal, Québec, Canada H2W 1R7. E-mail: jolicop@ircm.qc.ca.
Supported by grants from the Medical Research Council (MRC) (Canada) (no. MT-10313) and from the National Heart, Lung, and Blood Institute, National Institutes of Health (no. HL59846 to P. J.).
D. G. K. and P. Y. contributed equally to this work.
References
- 1.Levy JA: Pathogenesis of human immunodeficiency virus infection. Microbiol Rev 1993, 57:183-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pantaleo G, Graziosi C, Fauci AS: New concepts in the immunopathogenesis of human immunodeficiency virus infection. N Engl J Med 1993, 328:327-335 [DOI] [PubMed] [Google Scholar]
- 3.Pantaleo G, Graziosi C, Demarest JF, Cohen OJ, Vaccarezza M, Gantt KMCC, Fauci AS: Role of lymphoid organs in the pathogenesis of human immunodeficiency virus (HIV) infection. Immunol Rev 1994, 140:105-130 [DOI] [PubMed] [Google Scholar]
- 4.McSherry GD: Human immunodeficiency-virus-related pulmonary infections in children. Semin Respir Infect 1996, 11:173-183 [PubMed] [Google Scholar]
- 5.Clarke JR, Robinson DS, Coker RJ, Miller RF, Mitchell DM: Role of the human immunodeficiency virus within the lung. Thorax 1995, 50:567-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rappaport J, Kopp JB, Klotman PE: Host virus interactions and the molecular regulation of HIV-1: role in the pathogenesis of HIV-associated nephropathy. Kidney Int 1994, 46:16-27 [DOI] [PubMed] [Google Scholar]
- 7.Seney FD, Jr, Burns DK, Silva FG: Acquired immunodeficiency syndrome and the kidney. Am J Kidney Dis 1990, 16:1-13 [DOI] [PubMed] [Google Scholar]
- 8.Moses A, Nelson J, Bagby GC, Jr: The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998, 91:1479-1495 [PubMed] [Google Scholar]
- 9.Grunfeld C: What causes wasting in AIDS? N Engl J Med 1995, 333:123-124 [DOI] [PubMed] [Google Scholar]
- 10.Simpson DM, Olney RK: Peripheral neuropathies associated with human immunodeficiency virus infection. Neurol Clin 1992, 10:685-711 [PubMed] [Google Scholar]
- 11.de Gans J, Portegies P: Neurological complications of infection with human immunodeficiency virus type 1. A review of literature and 241 cases. Clin Neurol Neurosurg 1989, 91:199-219 [DOI] [PubMed] [Google Scholar]
- 12.Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P: The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 1988, 239:586-592 [DOI] [PubMed] [Google Scholar]
- 13.Acierno LJ: Cardiac complications in acquired immunodeficiency syndrome (AIDS): a review. J Am Coll Cardiol 1989, 13:1144-1154 [DOI] [PubMed] [Google Scholar]
- 14.Ferguson DW, Volpp BD: Cardiovascular complications of AIDS. Heart Dis Stroke 1994, 3:388-394 [PubMed] [Google Scholar]
- 15.Kaul S, Fishbein MC, Siegel RJ: Cardiac manifestations of acquired immune deficiency syndrome: a 1991 update. Am Heart J 1991, 122:535-544 [DOI] [PubMed] [Google Scholar]
- 16.Anderson DW, Virmani R: Emerging patterns of heart disease in human immunodeficiency virus infection. Hum Pathol 1990, 21:253-259 [DOI] [PubMed] [Google Scholar]
- 17.Herskowitz A: Cardiomyopathy and other symptomatic heart diseases associated with HIV infection. Curr Opin Cardiol 1996, 11:325-331 [DOI] [PubMed] [Google Scholar]
- 18.Lipshultz SE: Dilated cardiomyopathy in HIV-infected patients. N Engl J Med 1998, 339:1153-1155 [DOI] [PubMed] [Google Scholar]
- 19. Lipshultz SE eds. Cardiology in AIDS. 1998. Chapman & Hall, New York
- 20.Cohen IS, Anderson DW, Virmani R, Reen BM, Macher AM, Sennesh J, DiLorenzo P, Redfield RR: Congestive cardiomyopathy in association with the acquired immunodeficiency syndrome. N Engl J Med 1986, 315:628-630 [DOI] [PubMed] [Google Scholar]
- 21.Grenier MA, Lipshultz SE: Left ventricular hypertrophy in HIV disease. Cardiology in AIDS. 1998:pp 111-139 Chapman & Hall, New York
- 22.Currie PF, Boon NA: Prospective adult cardiovascular morbidity and mortality studies: the world. Cardiology in AIDS. 1998:pp 59-75 Chapman & Hall, New York
- 23.Rodriguez ER, Nasim S, Hsia J, Sandin RL, Ferreira A, Hilliard BA, Ross AM, Garrett CT: Cardiac myocytes and dendritic cells harbor human immunodeficiency virus in infected patients with and without cardiac dysfunction: detection by multiplex, nested, polymerase chain reaction in individually microdissected cells from right ventricular endomyocardial biopsy tissue. Am J Cardiol 1991, 68:1511-1520 [DOI] [PubMed] [Google Scholar]
- 24.Kovacs A, Hinton DR, Wright D, Xu J, Li XL, Rasheed S, Hofman F: Human immunodeficiency virus type 1 infection of the heart in three infants with acquired immunodeficiency syndrome and sudden death. Pediatric Infect Dis J 1996, 15:819-824 [DOI] [PubMed] [Google Scholar]
- 25.Wu AY, Forouhar F, Cartun RW, Berman MM, Shiue ST, Louie AT, Grunnet M: Identification of human immunodeficiency virus in the heart of a patient with acquired immunodeficiency syndrome. Mod Pathol 1990, 3:625-630 [PubMed] [Google Scholar]
- 26.Grody WW, Cheng L, Lewis W: Infection of the heart by the human immunodeficiency virus. Am J Cardiol 1990, 66:203-206 [DOI] [PubMed] [Google Scholar]
- 27.Lipshultz SE, Fox CH, Perez-Atayde AR, Sanders SP, Colan SD, McIntosh K, Winter HS: Identification of human immunodeficiency virus-1 RNA and DNA in the heart of a child with cardiovascular abnormalities and congenital acquired immune deficiency syndrome. Am J Cardiol 1990, 66:246-250 [DOI] [PubMed] [Google Scholar]
- 28.Herskowitz A, Wu TC, Willoughby SB, Vlahov D, Ansari AA, Beschorner WE, Baughman KL: Myocarditis and cardiotropic viral infection associated with severe left ventricular dysfunction in late-stage infection with human immunodeficiency virus. J Am Coll Cardiol 1994, 24:1025-1032 [DOI] [PubMed] [Google Scholar]
- 29.Barbaro G, Di Lorenzo G, Grisorio B, Barbarini G: Incidence of dilated cardiomyopathy and detection of HIV in myocardial cells of HIV-positive patients. Gruppo Italiano per lo Studio Cardiologico dei Pazienti Affetti da AIDS. N Engl J Med 1998, 339:1093-1099 [DOI] [PubMed] [Google Scholar]
- 30.Beschorner WE, Baughman K, Turnicky RP, Hutchins GM, Rowe SA, Kavanaugh-McHugh AL, Suresch DL, Herskowitz A: HIV-associated myocarditis. Pathology and immunopathology Am J Pathol 1990, 137:1365-1371 [PMC free article] [PubMed] [Google Scholar]
- 31.Hanna Z, Kay DG, Cool M, Jothy S, Rebai N, Jolicoeur P: Transgenic mice expressing human immunodeficiency virus type 1 in immune cells develop a severe AIDS-like disease. J Virol 1998, 72:121-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P: Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell 1998, 95:163-175 [DOI] [PubMed] [Google Scholar]
- 33.Poudrier J, Weng X, Kay DG, Paré G, Calvo EL, Hanna Z, Kosco-Vilbois MH, Jolicoeur P: The AIDS disease of CD4C/HIV transgenic mice shows impaired germinal centers and autoantibodies and develops in the absence of IFN-γ and IL-6. Immunity 2001, 15:173-185 [DOI] [PubMed] [Google Scholar]
- 34.Cullen BR: The role of Nef in the replication cycle of the human and simian immunodeficiency viruses. Virology 1994, 205:1-6 [DOI] [PubMed] [Google Scholar]
- 35.Harris M: From negative factor to a critical role in virus pathogenesis: the changing fortunes of Nef. J Gen Virol 1996, 77:2379-2392 [DOI] [PubMed] [Google Scholar]
- 36.Peter F: HIV nef: the mother of all evil? Immunity 1998, 9:433-437 [DOI] [PubMed] [Google Scholar]
- 37.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC: Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med 1995, 332:228-232 [DOI] [PubMed] [Google Scholar]
- 38.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J: Genomic structure of an attenuated quasi species of hiv-1 from a blood transfusion donor and recipients. Science 1995, 270:988-991 [DOI] [PubMed] [Google Scholar]
- 39.Kestler HW, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC: Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991, 65:651-662 [DOI] [PubMed] [Google Scholar]
- 40.McLean IW, Nakane PK: Periodate-lysine-paraformaldehyde fixative. a new fixation for immunoelectron microscopy. J Histochem Cytochem 1974, 22:1077-1083 [DOI] [PubMed] [Google Scholar]
- 41.Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJJ, Olsen EGJ, Schoen FJ: Myocarditis: a histopathologic definition and classification. Am J Cardiovascular Pathol 1986, 1:3-14 [PubMed] [Google Scholar]
- 42. Coligah JE eds. Animal models for autoimmune and inflammatory disease: autoimmune myocarditis. Current Protocols in Immunology, chapter 15.14. 2001. John Wiley & Sons, New York
- 43.Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP: Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell 1999, 98:465-474 [DOI] [PubMed] [Google Scholar]
- 44.Ammarguellat F, Larouche I, Schiffrin EL: Myocardial fibrosis in DOCA-salt hypertensive rats effect of endothelin ETA receptor antagonism. Circulation 2001, 103:319-324 [DOI] [PubMed] [Google Scholar]
- 45.Sahn DJ, DeMaria A, Kisslo J, Weyman A: Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation 1978, 58:1072-1083 [DOI] [PubMed] [Google Scholar]
- 46.Shannon RP, Simon MA, Mathier MA, Geng YJ, Mankad S, Lackner AA: Dilated cardiomyopathy associated with simian AIDS in nonhuman primates. Circulation 2000, 101:185-193 [DOI] [PubMed] [Google Scholar]
- 47.Hanna Z, Simard C, Jolicoeur P: Specific expression of the human CD4 gene in mature CD4+CD8− and immature CD4+CD8+ T cells, and in macrophages of transgenic mice. Mol Cell Biol 1994, 14:1084-1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanna Z, Rebai N, Poudrier J, Jolicoeur P: Distinct regulatory elements are required for faithful expression of human CD4 in T-cells, macrophages and dendritic cells of transgenic mice. Blood 2001, 98:2275-2278 [DOI] [PubMed] [Google Scholar]
- 49.Colucci WS: Apoptosis in the heart. N Engl J Med 1996, 335:1224-1226 [DOI] [PubMed] [Google Scholar]
- 50.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P: Apoptosis in the failing human heart. N Engl J Med 1997, 336:1131-1141 [DOI] [PubMed] [Google Scholar]
- 51.Lewis W: Cardiomyopathy in AIDS: a pathophysiological perspective. Prog Cardiovasc Dis 2000, 43:151-170 [DOI] [PubMed] [Google Scholar]
- 52.Shannon RP, Lackner AA: Acute myocarditis associated with SIV infection causes myocardial injury through T cell induced apoptosis. Circulation 2000, 102(18)Suppl II(Abstract 3803)
- 53.Cetta F, Michels VV: The autoimmune basis of dilated cardiomyopathy. Ann Med 1995, 27:169-173 [DOI] [PubMed] [Google Scholar]
- 54.Currie PF, Goldman JH, Caforio AL, Jacob AJ, Baig MK, Brettle RP, Haven AJ, Boon NA, McKenna WJ: Cardiac autoimmunity in HIV related heart muscle disease. Heart 1998, 79:599-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Factor SM, Minase T, Cho S, Dominitz R, Sonnenblick EH: Microvascular spasm in the cardiomyopathic Syrian hamster: a preventable cause of focal myocardial necrosis. Circulation 1982, 66:342-354 [DOI] [PubMed] [Google Scholar]
- 56.Sakamoto A, Abe M, Masaki T: Delineation of genomic deletion in cardiomyopathic hamster. FEBS Lett 1999, 447:124-128 [DOI] [PubMed] [Google Scholar]
- 57.Bache RJ, Dymek DJ: Local and regional regulation of coronary vascular tone. Prog Cardiovasc Dis 1981, 24:191-212 [DOI] [PubMed] [Google Scholar]
- 58.Michaels AD, Lederman RJ, MacGregor JS, Cheitlin MD: Cardiovascular involvement in AIDS. Curr Probl Cardiol 1997, 22:109-148 [DOI] [PubMed] [Google Scholar]
- 59.Simard M-C, Chrobak P, Kay DG, Hanna Z, Jothy S, Jolicoeur P: Expression of simian immunodeficiency virus nef in immune cells of transgenic mice leads to a severe AIDS-like disease. J Virol 2002, 76:3981-3995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong PK, Prasad G, Hansen J, Yuen PH: ts1, a mutant of Moloney murine leukemia virus-TB, causes both immunodeficiency and neurologic disorders in BALB/c mice. Virology 1989, 170:450-459 [DOI] [PubMed] [Google Scholar]
- 61.Coudray N, Dezuttere D, Force G, Deribes DC, Pourny JC, Antony I, Lecarpentier Y, Chemla D: Left ventricular diastolic function in asymptomatic and symptomatic human immunodeficiency virus carriers—an echocardiographic study. Eur Heart J 1995, 16:61-67 [DOI] [PubMed] [Google Scholar]
- 62.Rekpattanapipat P, Wongpraparut N, Jacobs LE, Kotler MN: Cardiac manifestations of acquired immunodeficiency syndrome. Arch Intern Med 2000, 160:602-608 [DOI] [PubMed] [Google Scholar]
- 63.Brunnert SR: Morphologic response of myocardium to freeze-thaw injury in mouse strains with dystrophic cardiac calcification. Lab Animal Sci 1997, 47:11-18 [PubMed] [Google Scholar]
- 64.Barbaro G, Fisher SD, Pelllicelli A, Lipshultz SE: The expanding role of the cardiologist in the care of HIV infected patients. Heart 2001, 86:365-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, Notkins AL: HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology 1991, 185:109-119 [DOI] [PubMed] [Google Scholar]
- 66.Lewis W, Grupp IL, Grupp G, Hoit B, Morris R, Samarel AM, Bruggeman L, Klotman P: Cardiac dysfunction occurs in the HIV-1 transgenic mouse treated with zidovudine. Lab Invest 2000, 80:187-197 [DOI] [PubMed] [Google Scholar]
- 67.Tinkle BT, Ngo L, Luciw PA, Maciag T, Jay G: Human immunodeficiency virus-associated vasculopathy in transgenic mice. J Virol 1997, 71:4809-4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matsumori A, Kawai C: An animal model of congestive (dilated) cardiomyopathy: dilatation and hypertrophy of the heart in the chronic stage in DBA/2 mice with myocarditis caused by encephalomyocarditis virus. Circulation 1982, 66:355-360 [DOI] [PubMed] [Google Scholar]
- 69.Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU: Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med 1999, 5:320-326 [DOI] [PubMed] [Google Scholar]
- 70.Huber SA: Autoimmunity in myocarditis: relevance of animal models. Clin Immunol Immunopathol 1997, 83:93-102 [DOI] [PubMed] [Google Scholar]
- 71.Lafeuillade A, Alessi MC, Poizot-Martin I, Boyer-Neumann C, Zandotti C, Quilichini R, Aubert L, Tamalet C, Juhan-Vague I, Gastaut JA: Endothelial cell dysfunction in HIV infection. J AIDS 1992, 5:127-131 [PubMed] [Google Scholar]
- 72.Buttner A, Mehraein P, Weis S: Vascular changes in the cerebral cortex in HIV-1 infection. 2. An immunohistochemical and lectin histochemical investigation. Acta Neuropathol 1996, 92:35-41 [DOI] [PubMed] [Google Scholar]