

Abstract
The outbreak of new variant Creutzfeldt-Jakob disease has raised the specter of a potentially large population being at risk to develop this prionosis. None of the prionoses currently have an effective treatment. Recently, vaccination has been shown to be effective in mouse models of another neurodegenerative condition, namely Alzheimer’s disease. Here we report that vaccination with recombinant mouse prion protein delays the onset of prion disease in mice. Vaccination was performed both before peripheral prion exposure and after exposure. A delay in disease onset was seen in both groups, but was more prolonged in animals immunized before exposure. The increase in the incubation period closely correlated with the anti-prion protein antibody titer. This promising finding suggests that a similar approach may work in humans or other mammalian species at risk for prion disease.
Prions are very unusual infectious agents. Current evidence suggests that they lack nucleic acid and their pathogenic potential is dependent on the conformation of prion protein (PrPSc). 1 The normal mammalian prion protein is known as PrPC. The disease-associated protein, PrPSc, has the same amino acid sequence but the conformation is altered, having a higher β-sheet content. Experimental treatment approaches that have been reported for prion diseases include the use of amphotericin B, 2 Congo Red, 3 sulfated polyanions, 4 anthracyclines, 5 β-sheet breaker peptides, 6 porphyrin, and phthalocyanine compounds. 7 Some of these compounds delay the incubation time of animals infected with PrPSc but all have limitations in terms of toxicity and/or pharmacokinetics.
Prion infections do not elicit a classical immune response; however, transport of prions from the periphery to the central nervous system is critically dependent on the lymphoreticular system. 8-11 The immune system appears to assist rather than impair the propagation of prions and their access to the central nervous system, which is required for pathogenicity. 10 In the current study, we sought to determine how overcoming the natural immunological tolerance to PrP by active immunization would influence progression of the disease.
Materials and Methods
Prophylactic Treatment Group
Twenty female CD-1 mice, 2 to 3 months of age, were immunized with mouse recombinant prion protein (recPrP). The recPrP was prepared as previously described. 12 For the first injection, the recPrP (1 mg/ml in 0.5 mol/L urea) was mixed with an equal volume of complete Freund’s adjuvant immediately before subcutaneous administration (50 μg recPrP/100 μl). Twenty control mice received the adjuvant plus vehicle. Subsequent immunizations were performed at 2-week intervals in incomplete Freund’s adjuvant. Fourteen weeks after the first vaccination, the mice were bled and the anti-recPrP antibody titer was determined by enzyme-linked immunosorbent assay (see below). The mice were subsequently divided into two groups matched for their titer to recPrP and were inoculated intraperitoneally with a brain homogenate of the mouse-adapted scrapie strain 139A at a 10-fold or a 1000-fold dilution. The control mice were also divided into two groups that received either the 10-fold or 1000-fold dilution of the same 139A inoculum. This represents a well-established model of prion disease in mice, which leads to central nervous system scrapie infection and death in all cases, if the disease is allowed to progress. 6,11,13-15 The immunization was continued thereafter at monthly intervals until the first mice showed clinical symptoms of scrapie. The mice were bled again at 14 weeks after the intraperitoneal PrPSc inoculation, a few weeks before they were expected to show clinical signs of the disease. Final bleeding was performed at the time of sacrifice, which occurred when the mice scored positive for clinical signs of prion infection using a test of motor coordination for 3 consecutive weeks by observers blinded to the experimental status of the animals, as per an established protocol. 6,11,15 The analysis of clinical symptoms consists of observing the activity level and competency of the mice on an apparatus containing a series of parallel bars (3 mm in diameter) placed 7 mm apart. The initial clinical findings are a reduction in activity and/or the ability of the mice to traverse the parallel bars. This clinical endpoint correlates with the pathological development of central nervous system scrapie infection. 6,11,15 The plasma was tested for reactivity against recPrP and PrPC by enzyme-linked immunosorbent assay (see below). The brains from ketamine/xylazine-anesthetized mice were removed, and the right hemisphere was immersion-fixed overnight in periodate-lysine-paraformaldehyde, whereas the left hemisphere was snap-frozen for Western blots. The fixed brain hemispheres were subsequently transferred to a solution containing 20% glycerol and 2% dimethyl sulfoxide in 0.1 mol/L of sodium phosphate buffer, and stored at 4°C until sectioned. Serial coronal sections (40 μm) were cut and series of sections at 0.2-mm intervals were obtained for histological analysis. The diagnosis of prion disease was confirmed by staining brain sections with cresyl violet, immunostaining with a polyclonal anti-PrP antibody (anti-PrP 78295), and by the detection of proteinase K-resistant PrP on Western blots as previously described. 16
Rescue Treatment Group
This experiment was performed similarly to the prophylactic treatment mouse group, except immunization with recPrP was started 24 hours after intraperitoneal inoculation with mouse-adapted scrapie strain 139A. As before, there were two groups of 10 female CD-1 mice each at 2 to 3 months of age, which received either a 10-fold or 1000-fold dilution of the 139A brain homogenate inoculum intraperitoneally, followed in this case by the first recPrP injection in complete Freund’s adjuvant 24 hours later. There were also two control groups of 10 CD-1 mice, which received either the 10-fold or 1000-fold dilution of the same 139A brain homogenate inoculum intraperitoneally, but followed by injection of adjuvant plus vehicle (0.5 mol/L urea). The rest of the protocol was then identical to the prophylactic treatment mouse group.
Antibody Titers
Antibody titers to recPrP were determined by serial dilutions of plasma, in which mouse recPrP at 100 ng/well is coated overnight onto microtiter wells. The titer, defined as the dilution yielding 50% of the maximum signal, was detected by a goat anti-mouse IgG linked to horseradish peroxidase (Amersham Pharmacia Biotech, Piscataway, NJ) and tetramethyl benzene (Pierce, Rockford, IL) was the substrate.
To determine the correlation between PrPC antibody recognition and disease incubation time, enzyme-linked immunosorbent assay plates were coated overnight with PrPC at 50 ng/well. The PrPC was prepared by the purification of ME7 strain PrPSc and reverting the conformation and proteinase K sensitivity to a protease-sensitive PrPC-like state by solubilization in 99% formic acid, as previously described. 16 Mouse plasma was used at a 1000-fold dilution in duplicate and the signal was detected by goat anti-mouse alkaline phosphatase (Biosource International, Camarillo, CA) and p-nitrophenyl phosphate (Sigma Diagnostics, St. Louis, MO) was the substrate.
Results
Prophylactic Treatment Group
The anti-recPrP titer in the mice just before receiving the 1:10 and 1:1000 dilution of the mouse-adapted scrapie strain 139A intraperitoneally was 20,135 ± 17,916 (±SEM) and 9645 ± 8063, respectively. When the mice were bled at 14 weeks after the intraperitoneal PrPSc 139A inoculation the group inoculated with the higher dose (1:10 dilution) had a titer of 6618 ± 2481 and the group inoculated with the lower dose had a titer of 1562 ± 621 (P = 0.09 by unpaired t-test). As depicted in Figure 1A ▶ , the mice immunized with the recPrP had a statistically significant delay in the onset of scrapie symptoms. The treatment effect was more pronounced at the 10-fold dilution [days to sacrifice, 173 ± 2 days (control) versus 189 ± 4 days; P = 0.002, two-tailed t-test], than at the 1000-fold dilution [days to sacrifice, 197 ± 3 (control) versus 205 ± 3; P = 0.040, one-tailed t-test]. This may be related to the trend for a higher titer of anti-PrP antibodies in the 10-fold PrPSc dilution group (see above), although titer alone may not be the sole determining factor. The epitope specificity of this response is also likely to influence progression of disease. A higher anti-PrPC antibody level in vaccinated animals correlated with a longer incubation time (Figure 1, B and C) ▶ in both PrPSc-inoculated mouse groups (lower dilution group: r2 = 0.453, P < 0.005; higher dilution group: r2 = 0.744, P < 0.0001).
Figure 1.

A: Prophylactic vaccination with recPrP delays disease onset in mice inoculated intraperitoneally both at the 10-fold (P = 0.002) and 1000-fold (P = 0.040) dilution of PrPSc with day 0 being the first day an animal scored positive for disease. Group 1 mice were controls receiving brain inoculum at a 10-fold dilution, while group 2 was inoculated at the same dilution but also received recPrP vaccination. Group 3 mice were controls inoculated with brain homogenate at a 1000-fold dilution, while group 4 received the same dilution of inoculum along with recPrP vaccination. The two control groups received adjuvant and vehicle injections. B and C: Levels of serum antibody against PrPC (PrPSc treated with formic acid rendering it protease-sensitive) correlate with incubation time of the disease both at 10-fold (B; r2 = 0.453, P < 0.005) and 1000-fold (C; r2 = 0.744, P < 0.0001) dilution of brain inoculum. Each data point represents the mean ± SEM of duplicate plasma samples.
Rescue Treatment Group
As expected, the effects of the treatment (Figure 2A) ▶ were not as pronounced in the rescue mouse group, compared to the prophylactically treated mice (Figure 1A) ▶ . No significant group difference was observed in disease onset in mice receiving the 10-fold dilution of the brain inoculum [days to sacrifice, 192 ± 5 days (control) versus 190 ± 5 days], although the levels of antibodies against PrPC correlated with disease onset (r2 = 0.279, P < 0.017, data not shown). However, at the 1000-fold dilution, a delay in symptoms was observed in the vaccinated mice [days to sacrifice, 210 ± 3 days (control) versus 222 ± 4 days; P = 0.018, t-test, one-tailed]. As with the prophylactic treatment, the anti-PrPC antibody levels in the immunized mice correlated with a longer incubation time (Figure 2B ▶ ; r2 = 0.772, P < 0.0001).
Figure 2.

A: Vaccination with recPrP initiated 24 hours after inoculation with PrPSc delays disease onset only at the lower dilution (P = 0.021). Group assignments were the same as depicted in Figure 1A ▶ . B: Levels of serum antibodies against PrPC (PrPSc treated with formic acid rendering it protease-sensitive) correlate with incubation time of the disease at the 1000-fold dilution of brain inoculum (r2 = 0.772, P < 0.0001). Each data point represents the mean ± SEM of duplicate plasma samples.
Histological and Western blot evaluations of all of the brains of the treated and control groups did not reveal any apparent differences in the degree of spongiform change or PrPSc levels at the time of sacrifice (Figure 3) ▶ in either the prophylactic or rescue treatment mouse experiments. Hence, immunization delayed PrPSc propagation, but ultimately similar pathology and levels of PrPSc were obtained in treatment and control groups.
Figure 3.
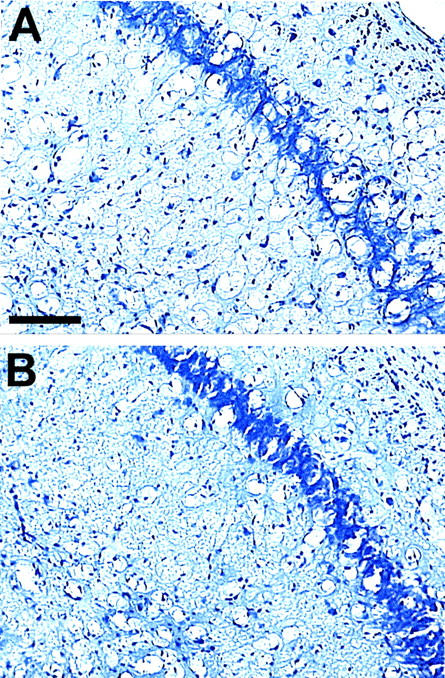
Coronal representative sections stained with cresyl violet through the pyramidal layer of the hippocampus in a control (A) and in a prophylactically vaccinated (B) mouse, showing extensive spongiform change. Both mice were infected intraperitoneally with a 10-fold dilution of a 139A PrPSc brain homogenate. All mice were examined histologically and had similar spongiform change. Even though the vaccinated animals had a longer incubation period, the extent of pathology was not apparently different in the two groups. This is expected because all of the mice were sacrificed at an equivalent time point after developing disease (each mouse had been rated clinically ill for 3 consecutive weeks using an established protocol). 6,11,15 Original magnifications, ×100. Scale bar, 100 μm.
Discussion
The prionoses belong to the category of conformational disorders, which are all characterized by the accumulation of a constitutively expressed protein in an abnormal, toxic conformation. 17 Alzheimer’s disease also falls into this category, in which the disease state is characterized by the conformational change of normal soluble amyloid β (Aβ) peptide to aggregated/fibrillar Aβ. A number of recent reports have shown that immunization with Aβ peptides is highly successful at reducing cerebral amyloid accumulation, a key neuropathological feature of Alzheimer’s disease, in transgenic mouse models of this disease. 18-21 Passive immunization studies in the Alzheimer’s disease mouse model suggest that an antibody-mediated clearance of Aβ is critical for a therapeutic response. 22 In this report we extend this immunological approach to prion disease and suggest that it may be applied to all members of this extended category of conformational diseases. Although neither of our treatment paradigms prevented prion disease, the close correlation between antibody levels and incubation time shows the promise of vaccination therapy for this untreatable and fatal neurodegenerative disease. Overall, the vaccination-mediated delay in the onset of prion disease is highly reproducible, correlates well with antibody titer, with the best therapeutic effect being obtained in mice preimmunized before infection.
It is not clear how the immunization delays the onset of prion disease in these mice. Our preliminary studies of passive immunization using anti-PrP antibodies suggest that humoral immunity is critical for a therapeutic response. It is possible that antibody binding to PrPC and/or PrPSc may interfere with PrPSc-mediated conversion of PrPC to PrPSc and thereby delay the onset of clinical symptoms. Recent in vitro studies support this view, 23,24 and immunization with prion peptides of 20 amino acids has been shown to reduce the levels of PrPSc in scrapie-infected mouse tumors 25 without affecting PrPC levels. Hence, epitope mapping of the anti-PrP antibodies produced by immunization may provide insights on which portions of the prion molecule are important for prion replication. The ultimate goal of such immunological approaches is for human testing; although the recently reported problems with the Aβ1-42 vaccine for Alzheimer’s disease highlight the difficulties of translating successful approaches in mice to humans. There are a number of potential toxic side effects of vaccine-based approaches in humans that will require further animal and in vitro experimentation. One source of toxicity is from the immunogen that is used. In our Alzheimer’s disease vaccine development studies we altered the Aβ sequence making it nonfibrillogenic and nontoxic, while maintaining or increasing its immunogenicity, reducing the potential of this toxicity. 26 Similar types of alterations are underway to limit any potential toxicity from using the native PrP sequence as an immunogen. Our in vivo findings serve as a starting point for the development of vaccine-based approaches for the prion diseases and suggest that prion-based immunization is promising as a potential therapy.
Footnotes
Address reprint requests to Thomas Wisniewski, M.D., New York University School of Medicine, Millhauser Laboratory, Room HN419, 550 First Ave., New York, NY 10016. E-mail: thomas.wisniewski@med.nyu.edu.
Supported by National Institutes of Health grants AG15408, AR02594, AG17617, AG05891, and AG20197.
References
- 1.Prusiner SB: Neurodegenerative diseases and prions. N Engl J Med 2001, 344:1516-1526 [DOI] [PubMed] [Google Scholar]
- 2.Pocchiari M, Schmittinger S, Masullo C: Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J Gen Virol 1987, 68:219-223 [DOI] [PubMed] [Google Scholar]
- 3.Caughey B, Race RE: Potent inhibition of scrapie-associated PrP accumulation by Congo Red. J Neurochem 1992, 59:768-771 [DOI] [PubMed] [Google Scholar]
- 4.Ladogana A, Casaccia P, Ingrosso L, Cibati M, Salvatore M, Xi YG, Masullo C, Pocchiari M: Sulphate polyanions prolong the incubation period of scrapie-infected hamsters. J Gen Virol 1992, 73:661-665 [DOI] [PubMed] [Google Scholar]
- 5.Tagliavini F, McArthur RA, Canciani B, Giaccone G, Porro M, Bugiani M, Lievens PM, Bugiani O, Peri E, Dall’Ara P, Rocchi M, Poli G, Forloni G, Bandiera T, Varasi M, Suarato A, Cassutti P, Cervini MA, Lansen J, Salmona M, Post C: Effectiveness of anthracycline against experimental prion disease in Syrian hamsters. Science 1997, 276:1119-1122 [DOI] [PubMed] [Google Scholar]
- 6.Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, Tagliavini F, Carp RI, Frangione B: Reversion of prion protein conformational changes by synthetic β-sheet breaker peptides. Lancet 2000, 355:192-197 [DOI] [PubMed] [Google Scholar]
- 7.Priola SA, Raines A, Caughey WS: Porphyrin and phthalocyanine antiscrapie compounds. Science 2000, 287:1503-1506 [DOI] [PubMed] [Google Scholar]
- 8.Weissmann C, Raeber AJ, Montrasio F, Hegyi I, Frigg R, Klein MA, Aguzzi A: Prions and the lymphoreticular system. Philos Trans R Soc Lond B Biol Sci 2001, 356:177-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mabbott NA, Bruce ME: The immunobiology of TSE diseases. J Gen Virol 2001, 82:2307-2318 [DOI] [PubMed] [Google Scholar]
- 10.Aucouturier P, Carp RI, Carnaud C, Wisniewski T: Prion diseases and the immune system. Clin Immunol 2000, 96:79-85 [DOI] [PubMed] [Google Scholar]
- 11.Aucouturier P, Geissmann F, Damotte D, Saborio GP, Meeker HC, Kascsak R, Kascsak R, Carp RI, Wisniewski T: Infected dendritic cells are sufficient for prion transmission to the CNS in mouse scrapie. J Clin Invest 2001, 108:703-708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown DR, Wong B-S, Hafiz F, Clive C, Haswell SJ, Jones IM: Normal prion protein has an activity like that of superoxide dismutase. Biochem J 2001, 344:1-5 [PMC free article] [PubMed] [Google Scholar]
- 13.Kimberlin RH, Walker CA: Pathogenesis of mouse scrapie: patterns of agent replication in different parts of the CNS following intraperitoneal infection. J Royal Soc Med 1982, 75:618-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimberlin RH, Walker CA: The role of the spleen in the neuroinvasion of scrapie in mice. Virus Res 1989, 12:201-212 [DOI] [PubMed] [Google Scholar]
- 15.Carp RI, Callahan SM, Sersen EA, Moretz RC: Preclinical changes in weight of scrapie-infected mice as a function of scrapie agent-mouse strain combination. Intervirology 1984, 27:61-69 [DOI] [PubMed] [Google Scholar]
- 16.Kascsak RJ, Fersko R, Pulgiano D, Rubenstein R, Carp RI: Immunodiagnosis of prion disease. Immunol Invest 1997, 26:259-268 [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski T, Sigurdsson EM, Aucouturier P, Frangione B: Conformation as a therapeutic target in the prionoses and other neurodegenerative conditions. Baker HF eds. Molecular and Cellular Pathology in Prion Disease. 2001:pp 223-236 Humana Press, Totowa [DOI] [PubMed]
- 18.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P: Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mice. Nature 1999, 400:173-177 [DOI] [PubMed] [Google Scholar]
- 19.Weiner HL, Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, Issazadeh S, Hancock WW, Selkoe D: Nasal administration of amyloid-β peptide decreases cerebral amyloid burden in a mouse model of Alzheimer’s disease. Ann Neurol 2000, 48:567-579 [PubMed] [Google Scholar]
- 20.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW: Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 2001, 408:982-985 [DOI] [PubMed] [Google Scholar]
- 21.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D: Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408:979-982 [DOI] [PubMed] [Google Scholar]
- 22.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T: Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 2000, 6:916-919 [DOI] [PubMed] [Google Scholar]
- 23.Enari M, Flechsig E, Weissmann C: Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA 2001, 98:9295-9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB: Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001, 412:739-743 [DOI] [PubMed] [Google Scholar]
- 25.Souan L, Tal Y, Felling Y, Cohen IR, Taraboulos A, Mor F: Modulation of proteinase-K resistant prion protein by prion peptide immunization. Eur J Immunol 2001, 31:2338-2346 [DOI] [PubMed] [Google Scholar]
- 26.Sigurdsson EM, Scholtzova H, Mehta P, Frangione B, Wisniewski T: Immunization with a non-toxic/non-fibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. Am J Pathol 2001, 159:439-447 [DOI] [PMC free article] [PubMed] [Google Scholar]