

To the Editor-in-Chief:
Many gastrointestinal stromal tumors (GISTs) contain oncogenic mutations of the KIT receptor tyrosine kinase gene. These genomic mutations encode structurally abnormal KIT proteins with constitutive enzymatic activity. In GISTs, most KIT mutations are within the juxtamembrane region (encoded by exon 11). However, smaller numbers of mutations are found in the KIT extracellular region (exon 9), the first part of the split kinase domain (exon 13), or the catalytic activation loop in the second part of the kinase domain (exon 17). 1–8
Recently, Andersson and colleagues 9 reported a possible novel recurrent KIT mutation involving exon 15 and resulting in deletions of SER715. They demonstrated Ser715− KIT transcripts in GISTs from 6 of the 11 patients where exon 15 cDNA was evaluated, and 5 of the Ser715− GISTs also had mutations involving the KIT juxtamembrane region. Because the authors regard the Ser715− transcript as mutant, they state that multiple KIT mutations in the same tumor, although not previously described in the GIST literature, were quite common in their series. We do not agree with this conclusion, and we are concerned that the authors did not discuss the substantial evidence for Ser715− KIT as a normal variant. We summarize this evidence below.
The Ser715− “mutations” reported by Andersson and co-workers 9 were demonstrated by cDNA sequencing, but no evidence of genomic mutation was provided. Absent a convincing genomic mutation, we see no reason to view these as anything other than normal KIT Ser715− isoforms. 10,11 Such isoforms were reported by Crosier and colleagues in 1993, 10 who found expression of Ser715− and Ser715+ KIT in nonneoplastic and neoplastic tissues. These isoforms apparently result from use of an alternate splice acceptor site at the start of exon 15 (Figure 1a) ▶ . Interestingly, as discussed by Crosier et al, 10 the more 5′ splice acceptor option is not present in murine KIT (Figure 1) ▶ . Therefore, the residue corresponding to human KIT Ser715 is not expressed in the mouse (GenBank NM 021099, Y00864, AW762164, BF136330, BF124268, BI134457, BG800962, BE281825, BE949406, BI696285), nor, for similar reasons, in the rat (eg, GenBank NM 022264). KIT Ser715 is poorly conserved generally in mammals, and there is an asparagine at this position in the cow, horse, goat, and cat (eg, GenBank AF263827, AJ224645, D45168, S76596). Given that murine KIT lacks SER715 and that human Ser715− transcript is found in nonneoplastic cells, we do not believe Ser715− KIT should be viewed as an intrinsic oncoprotein.
Figure 1.
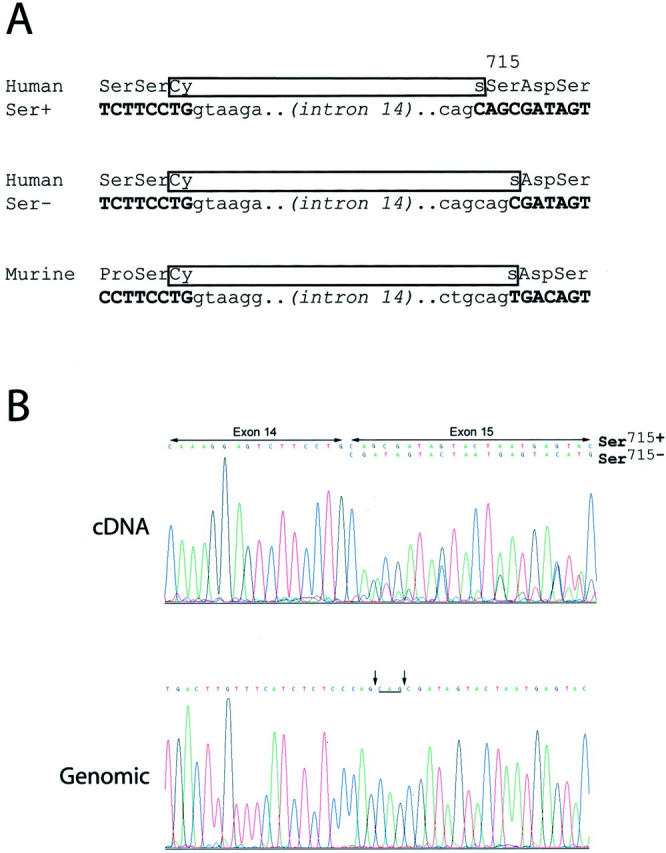
We have compared KIT cDNA and genomic sequence for the exon 14 and exon 15 splice junctions in seven human primary GISTs. KIT Ser715− and Ser715+ transcripts were coexpressed at approximately equal levels in five of the GISTs, whereas the splice junction genomic sequences were normal in all (Figure 1b) ▶ . No GIST had a genomic mutation affecting the Ser715 codon. The genomic and cDNA sequence comparisons show that the mechanism for the Ser715− transcript is not as proposed by Andersson and co-workers, 9 ie, a deletion after the start of exon 15. Rather, this transcript results from an alternate splicing event, exactly as one would surmise from the work of Crosier et al, 10 resulting in omission of the first three nucleotides in exon 15 [163 C, 2164 A, 2165G – GenBank X06182].
In several large published series, each including sequence analyses of more than 40 GISTs, the incidence of KIT V560D mutations has been less than 5%. 3,4,7 We have recently completed analysis of GISTs in an additional group of 220 patients, and found KIT V560D in only 5.9% of these tumors, whereas the overall KIT mutation rate was in excess of 80% (M.C. Heinrich and C.L. Corless, unpublished data). By contrast, 5 of 12 patients (42%) in the series from Andersson and co-workers 9 had GISTs with apparent V560D substitution. There is no clear explanation for the apparent overrepresentation of V560D mutations in this study, but we believe PCR cross-contamination must be considered as a potential confounding factor. We raise this possibility because there is no mention that any of the cDNA mutations were confirmed by PCR amplification of genomic DNA, or by repeating the cDNA sequencing studies from an independent RNA isolate. We believe the interpretation of differing mutations in two metastases from one patient (case 5), only one of these metastases having the V560D mutation, is clouded in the absence of such confirmatory studies. Likewise, the report of differing KIT mutations in primary versus metastatic GIST (case 4) is unconvincing, because the assumed mutation in this GIST was the exon 15 alternative splicing variant, and no genomic mutation was demonstrated. We have analyzed GIST specimen pairs, either primary and metastasis (N = 8), or two metastases (N = 2) in 10 patients, and in each patient the identical KIT mutation was found in both biopsies (Heinrich, Corless, and Fletcher, unpublished data). The KIT mutations in those patients involved exon 11 (N = 9) or exon 9 (N = 1). Therefore, we disagree with the suggestion that activating KIT mutations, in GISTs, are substantially more complex than previously appreciated.
Johanna Andersson, Helene Sjögren, Jeanne Meis-Kindblom, Göran Stenman, Pierre Åman, Lars-Gunnar Kindblom
Author’s Reply:
After the completion of our study we became aware of the true nature of the Ser715− KIT transcript in GIST. As pointed out by Dr. Jonathan Fletcher and co-workers, the Ser715− KIT transcript that was detected in 7 of 13 gastrointestinal stromal tumors (GIST) in our series 1 are indeed a normal splice variant. The main reason we interpreted this as a mutation was the fact that this isoform was not recorded in any available public database despite its original description in 1993 by Crosier et al. 2 This Ser715− splice variant has now been submitted to EMBL (Accession number AJ438313). We have subsequently analyzed the distribution of the Ser715+/− isoforms in 18 GIST as well as in normal human bone marrow and fetal liver by RT-PCR and restriction enzyme analyses. Both isoforms were detected and most tumors showed similar expression levels of the two transcripts. In contrast, normal bone marrow and fetal liver showed a clear predominance of the shorter Ser715− isoform (Figure 2) ▶ . Our misinterpretation of the Ser715− isoform as a mutation could have been avoided by analysis of genomic DNA. However, cDNA-based analyses generally detect what is truly expressed in a tumor and are therefore particularly useful in lesions with an admixture of normal cells. The finding of a predominant GNNK-isoform in GIST would not have been possible if the mutation analysis had been performed on genomic DNA alone.
Figure 2.

Expression of the Ser715+ and Ser715− isoforms of the KIT transcript. cDNA was amplified with primers designed for a region of the KIT gene including the exons 14–15 border. The PCR products were digested with PstI, a restriction enzyme that only cuts the Ser715+ isoform, and separated by gel electrophoresis. Fifteen of the 18 tested tumors are indicated by case numbers. A and B represent clonal control fragments, A: Ser715+; B: Ser715−; BM, human bone marrow; FL, human fetal liver
Dr. Fletcher and co-workers suggest that the detection of the V560D mutation of exon 11 in 5 of 14 GIST in our series may be due to PCR cross-contamination, since they have observed this particular mutation in only 5.9% of 220 GIST (M.C. Heinrich and C.L. Corless, unpublished data). We have now repeated the sequence analyses of genomic DNA from the five tumors in which we originally detected the V560D mutation. In three cases (cases 1, 5a, and 9), the mutation was confirmed and in two cases (cases 2 and 10), a normal KIT sequence was detected. We were thus able to confirm the existence of a differing mutation pattern in two metachronous metastases (cases 5a and 5b), one of the findings questioned by Dr. Fletcher and co-workers. Our inability to confirm the V560D mutation using genomic DNA in cases 2 and 10 could be due to tumor cell heterogeneity or that the mutation was obscured due to an admixture of non-neoplastic cells in the sample used for extraction of genomic DNA. Although a number of steps were taken to avoid the well known risks of PCR cross-contamination, we cannot entirely exclude this possibility. The reasons for the more frequent occurrence of this particular mutation of exon 11 in our series are not clear. The relatively small size of our series as well as geographic differences could account for the discrepancy. In a subsequent analysis of six additional GIST in which three tumors had mutations in exon 11, we did not detect the V560D mutation in any of the tumors (unpublished data).
Previous studies correlating KIT mutation pattern with phenotypic and biological behavior in GIST have been limited to analysis of exon 11 mutations. Several studies have indicated a strong correlation between the existence of exon 11 mutations, a malignant phenotype and aggressive clinical behavior. 3–9 Our recent study demonstrated the occurrence of clinically highly malignant GIST lacking any KIT gene mutations as well as KIT mutation in a small incidentally detected GIST with an entirely benign histology and clinical course. These findings suggest that the relationship between KIT mutations and biological behavior is more complex than previously recognized. In addition, we did not detect any clear correlation between chromosomal aberrations, KIT mutations, and phenotypic and biological characteristics in GIST. The recent study of Dr. Rubin and co-workers 10 supports our conclusions regarding the complex relationship between KIT mutations and the biological behavior of GIST. Dr. Rubin et al 10 detected KIT gene mutations of exons 9, 11, 13 and/or 17 in 44 of 48 GIST, including 10 of 10 benign GIST, 8 of 10 borderline tumors and 26 of 28 malignant GIST. KIT mutational status did not correlate with histological subtype or with morphologically determined biological potential. Their findings of KIT gene activation without the presence of activating mutations further indicate the complexity of this issue. In view of new therapeutic approaches, 11 further analyses attempting to correlate KIT gene function and mutations, biological behavior, and treatment response are essential.
- 1.Andersson J, Sjögren H, Meis-Kindblom JM, Stenman G, Åman P, Kindblom LG: The complexity of KIT gene mutations and chromosome rearrangements and their clinical correlation in gastrointestinal stromal (pacemaker cell) tumors. Am J Pathol 2002, 160:15-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crosier PS, Ricciardi ST, Hall LR, Vitas MR, Clark SC, Crosier KE: Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood 1993, 82:1151-1158 [PubMed] [Google Scholar]
- 3.Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O′Leary TJ: KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998, 78:1633-1636 [PubMed] [Google Scholar]
- 4.Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M: Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999, 154:53-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF, Jr: Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18:1897-1902 [DOI] [PubMed] [Google Scholar]
- 6.Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, Matsuda H, Kitamura Y: Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999, 59:4297-4300 [PubMed] [Google Scholar]
- 7.Nishida T, Hirota S: Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol 2000, 15:1293-1301 [DOI] [PubMed] [Google Scholar]
- 8.Nishida T, Nakamura J, Taniguchi M, Hirota S, Ito T, Kitamura Y, Matsuda H: Clinicopathological features of gastric stromal tumors. J Exp Clin Cancer Res 2000, 19:417-425 [PubMed] [Google Scholar]
- 9.Pidhorecky I, Cheney RT, Kraybill WG, Gibbs JF: Gastrointestinal stromal tumors: current diagnosis, biologic behavior, and management. Ann Surg Oncol 2000, 7:705-712 [DOI] [PubMed] [Google Scholar]
- 10.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA: KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001, 61:8118-8121 [PubMed] [Google Scholar]
- 11.Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman SL, Capdeville R, Dimitrijevic S, Druker B, Demetri GD: Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001, 344:1052-1056 [DOI] [PubMed] [Google Scholar]
References
- 1.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279:577-580 [DOI] [PubMed] [Google Scholar]
- 2.Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O’Leary TJ: KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998, 78:1633-1636 [PubMed] [Google Scholar]
- 3.Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M: Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999, 154:53-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, Matsuda H, Kitamura Y: Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999, 59:4297-4300 [PubMed] [Google Scholar]
- 5.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA: KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000, 156:791-795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, Sobin LH, Miettinen M: Mutations in exon 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors: a study of 200 cases. Am J Pathol 2000, 157:1091-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA: KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001, 61:8118-8121 [PubMed] [Google Scholar]
- 8.Hirota S, Nishita T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, Kitamura Y: Gain-of-function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol 2001, 193:505-510 [DOI] [PubMed] [Google Scholar]
- 9.Andersson J, Sjögren H, Meis-Kindblom JM, Stenman G, Åman P, Kindblom LG: The complexity of KIT gene mutations and chromosome rearrangements and their clinical correlation in gastrointestinal stromal (pacemaker cell) tumors. Am J Pathol 2002, 160:15-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crosier PS, Ricciardi ST, Hall LR, Vitas MR, Clark SC, Crosier KE: Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood 1993, 82:1151-1158 [PubMed] [Google Scholar]
- 11.Ashman LK: The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol 1999, 31:1037-1051 [DOI] [PubMed] [Google Scholar]