

Abstract
Gastric cancer is common among the world, but genetic mechanisms of gastric carcinogenesis are not well understood. Gastric polypoid adenomas and flat dysplasias are regarded as precursor lesions. However, a detailed molecular study of these lesions has not been done to determine their role as precancerous lesions. We investigated mutations of the APC, β-catenin, and K-ras genes, and microsatellite instability (MSI) status in 35 adenomas and 47 flat dysplasias without adenocarcinoma, 35 adenomas/dysplasias associated with adenocarcinomas, and 39 adenocarcinomas (20 diffuse type and 19 intestinal type). Somatic APC gene mutations were identified in 76% (59 of 78) of adenomas or flat dysplasias without associated adenocarcinoma, but in only 3% (1 of 30) of adenomas/dysplasias associated with adenocarcinoma, and in only 4% (3 of 69) of adenocarcinomas (P < 0.000001). No mutations of β-catenin were found in adenocarcinomas, or adenomas/dysplasia without APC mutation. K-ras mutations were detected in 5% (4 of 82) of gastric adenomas/dysplasia without carcinoma, 3% (1 of 39) of adenocarcinomas without associated adenoma/dysplasia, and not in 32 adenocarcinomas with associated adenoma/dysplasia. High level of MSI (MSI-H) was more frequent in gastric adenoma/dysplasia associated with carcinoma (17%, 6 of 35) than in adenomas/dysplasia without carcinoma (3%, 2 of 75; P = 0.01). MSI-H was also more frequent in intestinal type adenocarcinoma (20%, 11 of 54) than in diffuse type (0%, 0 of 20; P = 0.03). APC gene mutations were present in six of nine (67%) of gastric adenomas/dysplasias with low level of MSI, but in none of the eight adenomas/dysplasia with MSI-H phenotype (P = 0.009). Our results indicate that somatic mutation of the APC gene plays an important role in the pathogenesis of gastric adenoma and dysplasia but has a limited role in neoplastic progression to adenocarcinoma. Gastric adenomas or dysplasias without APC mutations but with or without MSI may have a different biological behavior, and are precursors of intestinal-type of gastric adenocarcinomas.
Gastric cancer is one of the most common malignant neoplasms among the world. Genetic alterations have been shown to play roles in gastric carcinogenesis including abnormalities in proto-oncogenes (K-ras and β-catenin), tumor suppressor genes (p53 and APC), cell-cycle regulator genes (E-cadherin), tissue invasion-related genes (CD44), and mismatch repair genes (hMLH1). 1-13 Gastric adenomas and dysplasias are considered precursor lesion of invasive gastric cancer, but the genetic mechanisms of early gastric carcinogenesis are not well understood.
There are two major histological types of gastric adenocarcinoma (intestinal and diffuse) according to the Lauren classification. 14 The pathogenesis and genetic alterations for these two distinct types of adenocarcinoma are also different. 15-17 The most frequent gastric malignancy is the intestinal type, which is often preceded by sequential steps of precancerous changes, including atrophic gastritis, intestinal metaplasia, and either dysplasia or adenoma. In contrast, the diffuse type of gastric carcinoma tends to arise de novo and is infrequently associated with dysplasia or adenoma. 18-21 Abnormal expression and amplification of the Met gene, inactivation of the p53 tumor suppressor gene, abnormal transcription of CD44, and loss of telomerase are common events in both types. 15-17,22,23 Reduced or absent expression of E-cadherin and K-sam gene amplification are unique to the diffuse type gastric adenocarcinoma. 16,17,22 By contrast, K-ras mutations, c-erb2 gene amplification, mutations of the APC gene, allelic loss of BclII gene and DCC locus, and microsatellite instability (MSI, replication error) are preferentially associated with the intestinal-type. 16,17,22 Gastric adenoma (polypoid dysplastic mucosal lesion) and flat dysplasia have been regarded as precancerous lesions for intestinal type adenocarcinoma. The sequential accumulation of alternations of APC and K-ras genes, characteristic of the colorectal adenoma-carcinoma sequence, however, does not frequently occur between adenoma and intestinal-type adenocarcinoma of the stomach. 3-7,24-26
There are two lines of evidence indicating that not all gastric dysplastic lesions are precursor lesions for gastric carcinoma. First, gastric dysplasia can undergo spontaneous regression clinically, especially low-grade dysplastic lesions. 27,28 Only 11 to 40% of adenoma/dysplasia progress to carcinoma. 29-31 Secondly, APC mutations have been reported to occur more frequently in gastric adenomas than in gastric adenocarcinomas. 24-26 However, the APC mutation status of gastric adenoma/dysplasia lesions associated with adenocarcinomas has not been studied in detail. It is not clear whether the APC gene plays any role in the pathogenesis of adenocarcinomas arising from pre-existing adenoma/dysplasia. Furthermore, genetic alterations separating these two distinct morphological precancerous lesions (flat dysplasia and polypoid adenoma) remain unclear. In this study, we investigated genetic alterations in gastric adenomas, flat dysplasias, adenocarcinoma with associated adenoma/dysplasia, and adenocarcinoma without associated adenoma/dysplasia to determine their potential roles in gastric carcinogenesis.
Materials and Methods
Case Selection
This study included 35 adenomas and 47 flat dysplasias from endoscopic biopsies of patients without gastric carcinoma, 39 adenocarcinomas (20 diffuse type and 19 intestinal type) without an associated adenoma/dysplasia, and 35 adenocarcinomas associated with adenoma/dysplasia (29 adenomas/dysplasias were immediately adjacent to the carcinoma, and 6 were distant) from surgical resection specimens (approved by University of Texas MD Anderson Cancer Center Institutional Review Board). All of the cases were retrospectively identified from the surgical pathology files of Chonnam National University Hospital, Kwangiu, South Korea between 1996 to 2000. No patient had familial adenomatous polyposis (FAP). The distinction between adenoma and dysplasia was based on endoscopic findings. Gastric adenoma was defined as a polypoid, elevated or exophytic lesion, and flat dysplasia as a flat or depressed lesion. Gastric adenomas and dysplasias were subclassified in hematoxylin and eosin (H&E)-stained slides into low-grade and high-grade dysplasia according to published criteria. 32 The carcinomas were classified histologically according to Lauren 14 and staged according to the criteria of the International Union Against Cancer. 33 Location and size of tumor were also recorded. Gastric nonneoplastic mucosa was evaluated for the presence of intestinal metaplasia, mucosal atrophy by Sydney criteria, and Helicobacter pylori by Giemsa stain and Campylobacter-like organism (CLO) test. 34 The clinicopathogical features of patients are summarized in Table 1 ▶ . Most of adenocarcinomas associated with adenoma/dysplasia were stage 1 tumors and intestinal type carcinomas.
Table 1.
Clinicopathological Characteristics of Gastric Adenoma, Dysplasia, and Adenocarcinoma
| Adenocarcinoma (−) | Adenocarcinoma (+) | |||
|---|---|---|---|---|
| Adenoma | Dysplasia | with Adenoma/dysplasia | without Adenoma/dysplasia | |
| Case no. | 35 | 47 | 35 | 39 |
| Age (mean± SD) | 67 ± 9.5 | 61 ± 8.3 | 63 ± 8.1 | 59 ± 11.3 |
| Sex | ||||
| Male | 28 | 37 | 27 | 22 |
| Female | 7 | 10 | 8 | 17 |
| Location | ||||
| Body | 22 | 19 | 17 | 22 |
| Antrum | 13 | 28 | 18 | 17 |
| Size (mm) | ||||
| ≤10 | 10 | 21 | 9 | 8 |
| 11–20 | 14 | 16 | 12 | 8 |
| ≥21 | 11 | 10 | 14 | 23 |
| Tumor size (mm) | ||||
| Mean± SD | 20.7 ± 15.8 | 17.8 ± 11.4 | 21.3 ± 11.1* | 35.4 ± 24.8* |
| Stage | ||||
| I | 34† | 13† | ||
| II | 1† | 4† | ||
| III | 0† | 15† | ||
| IV | 0† | 7† | ||
| Histologic grade | ||||
| Low grade | 24 | 24 | ||
| High grade | 11 | 23 | ||
| Histologic subtype | ||||
| Intestinal | 35‡ | 19‡ | ||
| Diffuse | 0‡ | 20‡ | ||
| Intestinal metaplasia | ||||
| Present | 27 | 36 | 31 | 28 |
| Absent | 8 | 11 | 4 | 11 |
| Mucosal atrophy | ||||
| Present | 11§ | 28§ | 28 | 35 |
| Absent | 24§ | 19§ | 7 | 4 |
| H. pylori infection | ||||
| Present | 23 | 30 | 32 | 31 |
| Absent | 12 | 17 | 3 | 8 |
*P = 0.003;
†P <0.001;
‡P <0.001;
§P = 0.01.
DNA Extraction
Microdissection from formalin-fixed and paraffin-embedded tissue was performed on H&E-stained slides for both tumor and normal mucosa. A 271/2-gauge needle was used for microdissection of H&E-stained slides under a low-power (×4) objective. In cases of adenocarcinoma associated with adenoma/dysplasia, carcinoma and adenoma/dysplasia components were separately microdissected and analyzed. Genomic DNA was extracted from microdissected tissue as described previously. 35
APC Gene Mutation Analysis
Four sets of oligonucleotide primers (5′-CAGACTTATTGTGTAGAAGA-3′ and 5′-CTCCTGAAGAAAATTCAACA-3′ for codons 1260 to 1350; 5′-AGGGTTCTAGTTTATCTTCA-3′ and 5′-TCTGCTTGGTGGCATGGTTT-3′ for codons 1339 to 1436; 5′-GGCATTATAAGCCCCAGTGA-3′ and 5′-AAATGGCTCATCGAGGCTCA-3′ for codons 1417 to 1516; 5′-ACTCCAGATGGATTTTCTTG-3′ and 5′-GGCTGGCTTTTTTGCTTTAC-3′ for codons 1497 to 1596) were used to amplify the mutation cluster region of the APC gene for gastrointestinal tumors. Polymerase chain reaction (PCR) was performed under standard conditions in a 50-μl volume using PCR Master (Boehringer Mannheim, Mannheim, Germany) and 1 μmol/L of both 5′ and 3′ oligonucleotides with 40 cycles (94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes). PCR products were purified using shrimp alkaline phosphatase and exonuclease I (Amersham, Buckinghamshire, UK). Purified PCR products were sequenced directly with SequiTherm Excel II DNA Sequencing Kit (Epicentre, Madison, WI) with the same primers used for DNA amplification. Oligonucleotides were end-labeled with [γ-32P]-ATP (DuPont-New England Nuclear Research Products, Boston, MA) using T4 polynucleotide kinase (New England Biolabs, Beverly, MA). All mutations were verified in both the sense and anti-sense directions.
β-Catenin Gene Mutation Analysis
Mutation analysis of the β-catenin gene was performed only on cases that did not show detectable APC mutations. Genomic DNA from each sample was amplified by PCR using the forward primer 5′-ATGGAACCAGACAGAAAAGC-3′ and reverse primer 5′-GCTACTTGTTCTTGAGTGAAG-3′. These amplified a 200-bp fragment of exon 3 of the β-catenin gene encompassing the region for GSK-3β phosphorylation. PCR reaction was performed under standard conditions in a 50 μl volume using PCR Master (Boehringer Mannheim) and 1 μmol/L of both 5′ and 3′ oligonucleotides with 40 cycles (94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes). PCR products were purified and sequenced directly using internal primers (forward, 5′-AAAGCGGCTGTTAGTCACTGG-3′; reverse, 5′-CCTGTTCCCACTCATACAGG-3′) as described above. All mutations were verified in both the sense and anti-sense directions.
K-ras Oncogene Mutation Analysis
The first exon of K-ras was amplified in a 50-μl volume using the reaction mixture described above with oligonucleotide primers (5′-GGCCGGTAGTGTATTAACCTTATGTGTGACAT-3′ and 5′-CCGCGGCCGGCGGCCAAAACAAGATTTACCTCTATTGTTGG-3′). PCR products were purified as described. The purified PCR products were sequenced using ABI PRISM BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) with an internal primer 5′-ATTCGTCCACAAAATGAT-3′. The sequencing reactions were run on an ABI PRISM 3700 DNA Analyzer (Applied Biosystems). The data were collected and analyzed using Applied Biosystems sequencing software, according to the manufacturer’s protocols.
Microsatellite Instability Analysis
MSI status was determined by five fluorescently labeled PCR amplifications using fluorescent dye-labeled forward primer and unlabeled reverse primer (BAT-25, BAT-26, D2S123, D5S346, and D17S250). The forward oligonucleotide was end-labeled with 6-FAM (Applied Biosystems). PCR was performed in 15-μl volumes containing 40 ng of DNA, 9 μl ABI Prism True Allele PCR Premix (Applied Biosystems), 5 pmol of 6-FAM-labeled forward primer, and 10 pmol of unlabeled reverse primer. PCR was performed using the following cycling conditions: denaturation at 95°C for 6 minutes, 45 cycles of 94°C for 45 seconds, 55°C for 45 seconds, 72°C for 1 minute, and extension at 72°C for 30 minutes. The PCR product was diluted further with 30 μl of H2O, and a 1.0 μl aliquot of each diluted fluorescent-labeled PCR product was combined with 12 μl of formamide and 0.5 μl of Genescan 400HD (ROX) size standard (Applied Biosystems). The samples were then capillary electrophoresed on an ABI 3700 DNA Analyzer using GeneScan Analysis software (Applied Biosystems). Allelic shift (MSI) of a microsatellite marker was defined by the presence of at least one additional band in the DNA from tumor or invasive carcinoma that was not present in the control normal DNA. A specimen was considered as MSI-high (MSI-H) when at least two markers showed allelic shift or MSI-low (MSI-L) when only one marker was shifted.
Statistical Analysis
Fisher’s exact test was used to compare differences in clinical or pathological characteristics, gene mutation, and MSI phenotype. A P value of <0.05 was considered statistically significant.
Results
Somatic Mutations of APC, β-Catenin, and K-ras Genes
Somatic APC mutations were more frequent in gastric adenomas and flat dysplasia without carcinoma (76%, 59 of 78) than in adenocarcinomas (4%, 3 of 69; P < 0.000001) or in adenomas/dysplasias associated with adenocarcinoma (3%, 1 of 30; P < 0.000001) (Table 2) ▶ . There was no difference in the APC mutation rate between polypoid adenomas (77%, 24 of 31) and flat dysplasias (74%, 35 of 47). Similarly, there was no difference in the APC mutation rate between adenocarcinomas associated with adenoma/dysplasia (7%, 2 of 30) and adenocarcinomas without associated adenoma/dysplasia (3%, 1 of 39). Two adenocarcinomas associated with adjacent adenoma/dysplasia had APC mutations. One was a frameshift mutation (4-bp deletion of TCTC spanning codons 1464 to 1465) observed in both the carcinoma and adenoma/dysplasia components, and the other was a frame shift mutation (1-bp insertion of A in a poly(A) tract spanning codons 1554 to 1556) detected only in the carcinoma component not in the adenoma/dysplasia component. There was no correlation between the presence of APC mutation and grade of dysplasia in flat dysplasia or adenoma (88% and 76% in low grade versus 61% and 80% in high grade, respectively, P = 0.4). The distribution of APC mutations is summarized in Figure 1 ▶ . Seventy-nine percent (50 of 63) of APC mutations were frameshifts (26 deletions and 24 insertions), and 21% (13 of 63) were nonsense point mutations resulting in truncation of the APC gene product. An insertion of A into the poly(A) tract at codons 1554 to 1556 was the most common (42%, 21 of 50) of the frameshift mutations. There were no significant associations between APC mutations and other clinicopathological parameters. No β-catenin mutations were detected in adenocarcinomas (n = 74), adenomas (n = 10), or dysplasia (n = 14) without APC mutation.
Table 2.
APC, β-catenin, and K-ras Mutations in Gastric Adenomas, Flat Dysplasias, and Adenocarcinomas
| APC | β-catenin | K-ras | |
|---|---|---|---|
| Adenoma/dysplasia without carcinoma (n = 82) | 76% (59/78)*† | 0% (0/19) | 5% (4/82) |
| Polypoid adenoma (n = 35) | 77% (24/31) | 0% (0/7) | 9% (3/35) |
| Flat dysplasia (n = 47) | 74% (35/47) | 0% (0/12) | 2% (1/47) |
| Adenoma/dysplasia associated with carcinoma (n = 35) | 3% (1/30)* | 0% (0/35) | 0% (0/32) |
| Adenocarcinoma (n = 74) | 4% (3/69)† | 0% (0/74) | 1% (1/71) |
| Intestinal type (n = 54) | 4% (2/49) | 0% (0/54) | 2% (1/51) |
| with adenoma/dysplasia (n = 35) | 7% (2/30) | 0% (0/35) | 0% (0/32) |
| without adenoma/dysplasia (n = 19) | 0% (0/19) | 0% (0/19) | 5% (1/19) |
| Diffuse type (n = 20) | 5% (1/20) | 0% (0/20) | 0% (0/20) |
*P <0.000001;
†P < 0.000001.
Figure 1.
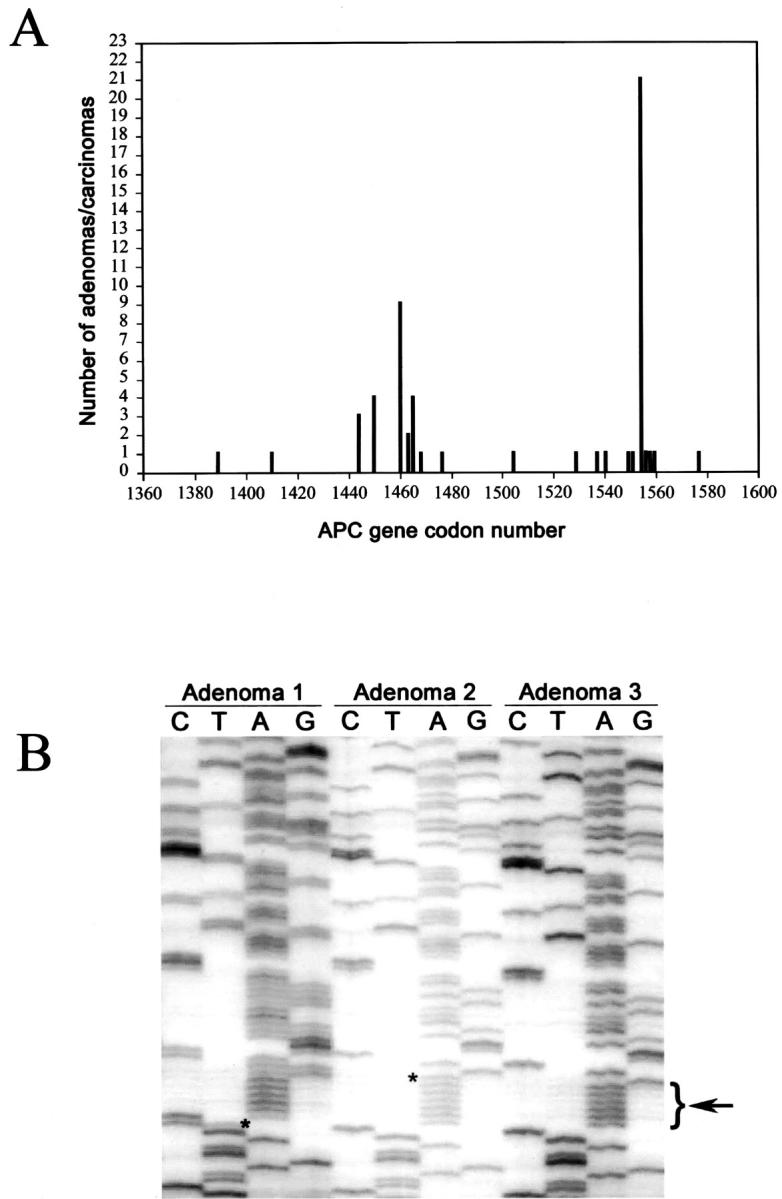
Somatic APC gene mutations in gastric adenomas/dysplasias and adenocarcinomas. A: Summary of the APC gene mutations in the mutation cluster region. An insertion of A into the poly(A) tract at codons 1554 to 1556 is the most common mutation. B: An insertion of T (asterisk) at codon 1556 (adenoma 1), and an insertion of A (asterisk) into the poly(A) tract (bracketwith arrow) at codons 1554 to 1556 (adenoma 2). In adenoma 2, the absence of normal sequence also indicates loss of the nonmutated APC allele. Adenoma 3 has normal APC sequence.
K-ras mutations were detected in 5% (4 of 82) of gastric adenomas/dysplasia without carcinoma, and 3% (1 of 39) of adenocarcinoma without associated adenoma/dysplasia, but were not found in adenocarcinoma with associated adenoma/dysplasia (n = 32). Only one gastric adenoma had both an activating codon 12 K-ras mutation and a frame shift mutation of the APC gene (Table 3) ▶ .
Table 3.
APC Mutation and Microsatellite Instability (MSI) Status in Gastric Tumors with K-ras Mutation
| No. | Histologic grade/type | K-ras mutation | APC mutation | MSI status | ||
|---|---|---|---|---|---|---|
| Codon | Nucleotide change | Amino acid change | ||||
| 1 | Dysplasia, HG | 12 | GGT→GAT | Gly→Asp | − | MSI-S |
| 2 | Adenoma, LG | 12 | GGT→GCT | Gly→Ala | ND | MSI-L |
| 3 | Adenoma, HG | 12 | GGT→GAT | Gly→Asp | + | MSI-L |
| 4 | Adenoma, HG | 12 | GGT→GCT | Gly→Ala | ND | MSI-S |
| 5 | Adenocarcinoma intestinal-type | 12 | GGT→GAT | Gly→Asp | − | MSI-H |
HG, high grade; LG, low grade; ND, not done; MSI-S, MSI stable phenotype; MSI-L, MSI-low phenotype; MSI-H, MSI-high phenotype.
Microsatellite Instability
MSI-H phenotype was more frequent in gastric adenoma/dysplasia associated with carcinoma (17%, 6 of 35) than in adenoma/dysplasia without carcinoma (3%, 2 of 75; P = 0.01) (Table 4) ▶ . Two of 30 adenomas, but none of 45 flat dysplasias had MSI-H phenotype. MSI-H phenotype was also more frequent in intestinal-type adenocarcinoma (20%, 11 of 54) than in diffuse-type (0%, 0 of 20; P = 0.03). There was no difference in the frequency of MSI-L phenotype between gastric adenoma/dysplasia associated with carcinoma (9%, 3 of 35) and adenomas/dysplasia without carcinoma (9%, 7 of 75); or between intestinal type adenocarcinoma and diffuse type [6% (3 of 54) versus 5% (1 of 20), respectively].
Table 4.
Microsatellite Instability (MSI) in Gastric Adenoma/Dysplasia and Adenocarcinoma
| MSI (+) | MSI-H | MSI-L | |
|---|---|---|---|
| Adenoma/dysplasia without carcinoma (n = 82) | 12% (9/75) | 3% (2/75)* | 9% (7/75) |
| Polypoid adenoma (n = 35) | 23% (7/30) | 7% (2/30) | 17% (5/30) |
| Flat dysplasias (n = 47) | 4% (2/45) | 0% (0/45) | 4% (2/45) |
| Adenoma/dysplasia associated with carcinoma (n = 35) | 26% (9/35) | 17% (6/35)* | 9% (3/35) |
| Adenocarcinoma (n = 74) | |||
| Intestinal type (n = 54) | 26% (14/54) | 20% (11/54)† | 6% (3/54) |
| with adenoma/dysplasia (n = 35) | 26% (9/35) | 17% (6/35) | 9% (3/35) |
| without adenoma/dysplasia (n = 19) | 26% (5/19) | 26% (5/19) | 0% (0/19) |
| Diffuse type (n = 20) | 5% (1/20) | 0% (0/20)† | 5% (1/20) |
*P = 0.01;
†P = 0.03.
MSI (+) includes both MSI-H and MSI-L phenotypes; MSI-H, MSI high phenotype; MSI-L, MSI low phenotype.
MSI phenotype was not correlated with grade of dysplasia. Among two MSI-L flat gastric dysplasias, one was high-grade dysplasia and one was low-grade dysplasia. Similarly, among two MSI-H and five MSI-L adenomas, three were high-grade dysplasia and four were low-grade dysplasia. Among adenocarcinomas associated with adenoma/dysplasia, MSI+ phenotype was present in both the carcinoma and adenoma/dysplasia components in five cases (four MSI-H and one MSI-L), in carcinoma alone in four cases (two MSI-H and two MSI-L), and in adenoma/dysplasia alone in four cases (two MSI-H and two MSI-L). Three of five MSI+ cases had a different allelic shift pattern between the carcinoma and adjacent adenoma/dysplasia components (Figure 2) ▶ .
Figure 2.

Microsatellite instability in gastric adenocarcinoma and the associated adenoma/dysplasia. The adenoma/dysplasia component shows a different allelic shift pattern in all five markers (D2S123, D5S346, D17S250, BAT25, and BAT26) as compared to the adenocarcinoma component.
Correlation between APC, K-ras, and Microsatellite Instability Status
There was an inverse correlation between the presence of MSI-H and the presence of APC gene mutations. APC gene mutations were present in six of nine (67%) gastric adenomas/dysplasias with MSI-L phenotype, but in none of the eight adenomas/dysplasia with MSI-H phenotype (P = 0.009) (Table 5) ▶ . The three MSI-L adenomas/dysplasias without APC mutations were associated with adenocarcinomas. No APC mutations were detected in any of the 15 MSI+ (11 MSI-H and 4 MSI-L) gastric adenocarcinomas.
Table 5.
APC Gene Mutation Status in MSI+ Gastric Adenoma/Dysplasia
| MSI-H | MSI-L | |
|---|---|---|
| APC Gene Mutation | ||
| Present | 0% (0/8)* | 67% (6/9)* |
| Absent | 100% (8/8)* | 33% (3/9)* |
*P = 0.009.
MSI-H, microsatellite instability high phenotype; MSI-L, microsatellite instability low phenotype.
There was no association between K-ras mutation and MSI phenotype. Among five tumors with K-ras mutations, two were MSI-S, two were MSI-L, and one was MSI-H (Table 3) ▶ .
Discussion
Gastric dysplastic lesions such as polypoid adenomas or flat dysplasia are frequently seen in patients with chronic atrophic gastritis and intestinal metaplasia. Intestinal-type gastric carcinomas also typically arise in a background of atrophic gastritis with intestinal metaplasia. 18 Although gastric carcinomas can arise from pre-existing dysplastic lesions, only a subset of gastric carcinoma has identifiable preneoplastic lesions. 29-31 The adenoma-carcinoma sequence typically seen in colorectal carcinoma does not seem to be a major pathway in gastric carcinogenesis. This issue is further complicated by the facts that only a small subset of gastric adenoma or dysplastic lesions eventually progress to carcinoma.
The reason for lack of neoplastic progression in the majority of gastric adenomas and dysplastic lesions is not clear. APC gene mutations have been previously reported in up to 40% of gastric adenomas but only rarely in gastric adenocarcinomas. 3,4,24-26,36,37 The presence of frequent somatic APC mutations in gastric adenoma but not in gastric carcinoma suggests that there is a different genetic pathway for the pathogenesis of adenoma and carcinoma. However, in previous studies only isolated adenomas and gastric carcinomas have been studied. Gastric adenoma/dysplasia lesions associated with adenocarcinomas have not been studied in detail for the presence of APC mutation. 3,4,25,26,36 In most cases, the adenocarcinoma can be assumed to have arisen within the dysplasia or adenoma. If the APC gene plays any role in the pathogenesis of adenocarcinomas arising from pre-existing adenoma/dysplasia, APC gene mutations should be detected in both adenoma/dysplasia and adenocarcinoma components, similar to the frequency of APC gene mutations in adenomas or dysplasias without adenocarcinoma. Our study demonstrates that APC mutations are frequent in sporadic gastric adenomas and dysplasias, but only rarely in adenocarcinomas. This rare presence of APC mutations was true for both adenocarcinomas with or without associated adenoma/dysplasia. The findings in this study therefore strongly indicate that APC gene mutations play an important role in the pathogenesis of gastric adenoma and dysplasia, but have only a limited role in the pathogenesis of gastric adenocarcinomas. β-catenin gene mutations have previously been reported in 5% (4 of 77) to 16% (7 of 43) of gastric adenocarcinomas from the Korean population. 8,38 In contrast, we found no β-catenin gene mutations in 54 intestinal and 20 diffuse type adenocarcinomas. The reason for this discrepancy is not clear.
Our results also suggest that APC gene mutation status could predict the biological behavior of individual gastric adenoma/dysplasia lesions (Figure 3) ▶ . Gastric adenomas/dysplasias with APC gene mutations only rarely progress to adenocarcinoma. This corroborates the clinical observation that only 11 to 40% of adenomas/dysplasias can progress to carcinoma. 29-31 This is contradictory to the role of the APC gene in colorectal carcinogenesis, in which it is regarded as a gatekeeper gene and is involved in the vast majority of colorectal carcinomas. 39 Because we only sequenced the mutation cluster region of the APC gene, we cannot exclude APC mutations outside this region. It is possible that APC gene mutations in adenocarcinomas either associated or not associated with adenoma/dysplasia are different from sporadic gastric adenomas or dysplasias, and are not present within the mutation cluster region we have sequenced. However, the presence of only rare APC gene mutations in gastric carcinomas in previously published studies further corroborates our results. 40 In addition, we have previously shown frequent somatic second-hit of APC genes in FAP-associated gastric fundic gland polyps. 41 Foveolar dysplasia can present in up to 25% of FAP-associated fundic gland polyps, but rare occurrence of adenocarcinomas in FAP-associated fundic gland polyps. Therefore, APC gene mutations seem to have a different biological behavior in the stomach as compared to the colorectum, and gastric adenomas/dysplasia with APC mutations only rarely progress to adenocarcinomas.
Figure 3.

Role of APC gene and MSI in gastric adenoma/dysplasia-carcinoma sequence. Adenocarcinoma can arise de novo or from pre-existing adenoma/dysplasia. Adenomas or dysplasias with APC gene mutations rarely progress to adenocarcinoma, and have a different biological behavior as compared to adenomas or dysplasias with MSI.
Gastric dysplastic lesions can be divided into adenomas (with a polypoid configuration) or flat dysplasias based on growth pattern. 32 However, it is difficult to differentiate adenoma from flat dysplasia once adenocarcinoma has arisen from these pre-existing dysplastic lesions. Furthermore, genetic alterations of these two distinct morphological gastric dysplastic lesions (polypoid adenoma and flat dysplasia) have not been studied in detail. We found no difference in the frequency of APC mutations in gastric adenomas versus flat dysplasias. In the colorectum, K-ras mutations are associated with a polypoid growth pattern in colorectal adenomas and adenocarcinomas, and are more frequent in polypoid adenomas as compared to flat adenomas. 42-44 In contrast, in our study the prevalence of K-ras mutations was not different between polypoid adenomas and flat dysplasias in the stomach.
Microsatellite instability (MSI) because of DNA replication errors has been widely observed in a variety of sporadic tumors in addition to tumors associated with hereditary nonpolyposis colorectal cancer syndrome because of germ line mutations in mismatch repair genes. 45-48 MSI has been identified in 7 to 50% of gastric carcinomas with geographic variation in prevalence. 49 We found MSI-H phenotype in 11% (8 of 75) of gastric adenomas/dysplasias and in 20% (11 of 54) of gastric intestinal type adenocarcinomas, which are similar to previously published results; 21% (13 of 63) in gastric adenomas and 30% (19 of 63) in gastric carcinomas from the Korean population. 46 Similar to colorectal carcinomas, MSI-positive gastric carcinomas have distinct clinicopathological features including better prognosis, and prominent lymphoid infiltration. MSI-positivity is more common in intestinal type gastric carcinoma located in the antrum than in diffuse type carcinoma. 50-53 In the present study, we found a higher prevalence of MSI-H phenotype in adenoma/dysplasia associated with adenocarcinoma than adenomas not associated with adenocarcinoma, as previously reported. 54 Three of five MSI+ adenocarcinomas associated with adenoma/dysplasia components had a different allelic shift pattern between carcinoma and adjacent adenoma/dysplasia components. One explanation for this finding is that an adenocarcinoma acquires a different allelic size during progression of tumor from adenoma/dysplasia. This phenomenon has been described in MSI+ tumor cells. 55 However, the possibility of adenocarcinoma arising de novo, and not from the pre-existing adenoma/dysplasia cannot be completely excluded. MSI+ colorectal and pancreatic carcinomas have a distinct genetic profile with wild-type K-ras and lack of p53 gene mutations. 55-57 Similarly, MSI-H gastric adenocarcinomas or adenomas/dysplasias also correlated with the absence of APC gene mutations in our study. In contrast, MSI-L gastric adenomas/dysplasias had frequent APC mutations.
In conclusion, our results demonstrate that somatic mutation of the APC gene plays an important role in the pathogenesis of gastric adenoma and dysplasia but has limited role in neoplastic progression to adenocarcinoma. In contrast, gastric adenomas or dysplasias without APC mutations may have a different biological behavior, and are precursors of intestinal type of gastric adenocarcinomas. MSI-H can play an important role in a subset of adenocarcinomas arising from gastric adenomas/dysplasias without APC mutations.
Footnotes
Address reprint requests to Tsung-Teh Wu, M.D., Ph.D., UT M.D. Anderson Cancer Center, Department of Pathology G1.3595C, Box 85, 1515 Holcombe Blvd., Houston, TX 77030. E-mail: twu@mdanderson.org.
References
- 1.Rhyu MG, Park WS, Jung YJ, Choi SW, Meltzer SJ: Allelic deletions of MCC/APC and p53 are frequent late events in human gastric carcinogenesis. Gastroenterology 1994, 106:1584-1588 [DOI] [PubMed] [Google Scholar]
- 2.Kang GH, Kim CJ, Kim WH, Kang YK, Kim HO, Kim YI: Genetic evidence for the multicentric origin of synchronous multiple gastric carcinoma. Lab Invest 1997, 76:407-417 [PubMed] [Google Scholar]
- 3.Horii A, Nakatsuru S, Miyoshi Y, Ichii S, Nagase H, Kato Y, Yanagisawa A, Nakamura Y: The APC gene, responsible for familial adenomatous polyposis, is mutated in human gastric cancer. Cancer Res 1992, 52:3231-3233 [PubMed] [Google Scholar]
- 4.Nishimura K, Yokozaki H, Jaruma K, Kajiyama G, Tahara E: Alternations of the APC gene in carcinoma cell lines and precancerous lesions of the stomach. Int J Oncol 1995, 7:587-592 [DOI] [PubMed] [Google Scholar]
- 5.Arber N, Shapira I, Ratan J, Stern B, Hibshoosh H, Moshkowitz M, Gammon M, Fabian I, Halpern Z: Activation of c-K-ras mutations in human gastrointestinal tumors. Gastroenterology 2000, 118:1045-1050 [DOI] [PubMed] [Google Scholar]
- 6.Kihana T, Tsuda H, Hirota T, Shimosato Y, Sakamoto H, Terada M, Hirohashi S: Point mutation of c-Ki-ras oncogene in gastric adenoma and adenocarcinoma with tubular differentiation. Jpn J Cancer Res 1991, 82:308-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KH, Lee JS, Suh C, Kim SW, Kim SB, Lee JH, Lee MS, Park MY, Sun HS, Kim SH: Clinicopathologic significance of the K-ras gene codon 12 point mutation in stomach cancer. An analysis of 140 cases. Cancer 1995, 75:2794-2801 [DOI] [PubMed] [Google Scholar]
- 8.Park WS, Oh RR, Park JY, Lee SH, Shin MS, Kim YS, Kim SY, Lee HK, Kim PJ, Oh ST, Yoo NJ, Lee JY: Frequent somatic mutations of the beta-catenin gene in intestinal-type gastric cancer. Cancer Res 1999, 59:4257-4260 [PubMed] [Google Scholar]
- 9.Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, Hofler H: E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 1994, 54:3845-3852 [PubMed] [Google Scholar]
- 10.Becker KF, Kremmer E, Eulitz M, Becker I, Handschuh G, Schuhmacher C, Muller W, Gabbert HE, Ochiai A, Hirohashi S, Hofler H: Analysis of E-cadherin in diffuse-type gastric cancer using a mutation-specific monoclonal antibody. Am J Pathol 1999, 155:1803-1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gayther SA, Gorringe KL, Ramus SJ, Huntsman D, Roviello F, Grehan N, Machado JC, Pinto E, Seruca R, Halling K, MacLeod P, Powell SM, Jackson CE, Ponder BA, Caldas C: Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res 1998, 58:4086-4089 [PubMed] [Google Scholar]
- 12.Yokozaki H, Ito R, Nakamura H, Kuniyasu H, Taniyama K, Tahara E: Expression of CD44 abnormal transcripts in human gastric carcinomas. Cancer Lett 1994, 83:229-234 [DOI] [PubMed] [Google Scholar]
- 13.Kang GH, Shim YH, Ro JY: Correlation of methylation of the hMLH1 promoter with lack of expression of hMLH1 in sporadic gastric carcinomas with replication error. Lab Invest 1999, 79:903-909 [PubMed] [Google Scholar]
- 14.Lauren P: The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. Acta Pathol Microbiol Scand 1965, 64:31-49 [DOI] [PubMed] [Google Scholar]
- 15.Stadtlander CT, Waterbor JW: Molecular epidemiology, pathogenesis and prevention of gastric cancer. Carcinogenesis 1999, 20:2195-2208 [DOI] [PubMed] [Google Scholar]
- 16.Tahara E, Semba S, Tahara H: Molecular biological observations in gastric cancer. Semin Oncol 1996, 23:307-315 [PubMed] [Google Scholar]
- 17.Chan AO, Luk JM, Hui WM, Lam SK: Molecular biology of gastric carcinoma: from laboratory to bedside. J Gastroenterol Hepatol 1999, 14:150-160 [DOI] [PubMed] [Google Scholar]
- 18.Correa P: Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res 1992, 52:6735-6740 [PubMed] [Google Scholar]
- 19.Correa P, Shiao YH: Phenotypic and genotypic events in gastric carcinogenesis. Cancer Res 1994, 54(Suppl):1941s-1943s [PubMed] [Google Scholar]
- 20.Correa P: A human model of gastric carcinogenesis. Cancer Res 1988, 48:3554-3560 [PubMed] [Google Scholar]
- 21.Solcia E, Fiocca R, Luinetti O, Villani L, Padovan L, Calistri D, Ranzani GN, Chiaravalli A, Capella C: Intestinal and diffuse gastric cancers arise in a different background of Helicobacter pylori gastritis through different gene involvement. Am J Surg Pathol 1996, 20(Suppl 1):S8-S22 [DOI] [PubMed] [Google Scholar]
- 22.Tahara E: Molecular mechanism of stomach carcinogenesis. J Cancer Res Clin Oncol 1993, 119:265-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiyama E, Yokoyama T, Tatsumoto N, Hiyama K, Imamura Y, Murakami Y, Kodama T, Piatyszek MA, Shay JW, Matsuura Y: Telomerase activity in gastric cancer. Cancer Res 1995, 55:3258-3262 [PubMed] [Google Scholar]
- 24.Maesawa C, Tamura G, Suzuki Y, Ogasawara S, Sakata K, Kashiwaba M, Satodate R: The sequential accumulation of genetic alterations characteristic of the colorectal adenoma-carcinoma sequence does not occur between gastric adenoma and adenocarcinoma. J Pathol 1995, 176:249-258 [DOI] [PubMed] [Google Scholar]
- 25.Nakatsuru S, Yanagisawa A, Furukawa Y, Ichii S, Kato Y, Nakamura Y, Horii A: Somatic mutations of the APC gene in precancerous lesion of the stomach. Hum Mol Genet 1993, 2:1463-1465 [DOI] [PubMed] [Google Scholar]
- 26.Tamura G, Maesawa C, Suzuki Y, Tamada H, Satoh M, Ogasawara S, Kashiwaba M, Satodate R: Mutations of the APC gene occur during early stages of gastric adenoma development. Cancer Res 1994, 54:1149-1151 [PubMed] [Google Scholar]
- 27.Rugge M, Farinati F, Di Mario F, Baffa R, Valiante F, Cardin F: Gastric epithelial dysplasia: a prospective multicenter follow-up study from the Interdisciplinary Group on Gastric Epithelial Dysplasia Multicenter Study. Hum Pathol 1991, 22:1002-1008 [DOI] [PubMed] [Google Scholar]
- 28.Rugge M, Farinati F, Baffa R, Sonego F, Di Mario F, Leandro G, Valiante F: Gastric epithelial dysplasia in the natural history of gastric cancer: a multicenter prospective follow-up study. Interdisciplinary Group on Gastric Epithelial Dysplasia. Gastroenterology 1994, 107:288-296 [DOI] [PubMed] [Google Scholar]
- 29.Kolodziejczyk P, Yao T, Oya M, Nakamura S, Utsunomiya T, Ishikawa T, Tsuneyoshi M: Long-term follow-up study of patients with gastric adenomas with malignant transformation. An immunohistochemical and histochemical analysis Cancer 1994, 74:2896-2907 [DOI] [PubMed] [Google Scholar]
- 30.Orlowska J, Jarosz D, Pachlewski J, Butruk E: Malignant transformation of benign epithelial gastric polyps. Am J Gastroenterol 1995, 90:2152-2159 [PubMed] [Google Scholar]
- 31.Kamiya T, Morishita T, Asakura H, Miura S, Munakata Y, Tsuchiya M: Long-term follow-up study on gastric adenoma and its relation to gastric protruded carcinoma. Cancer 1982, 50:2496-2503 [DOI] [PubMed] [Google Scholar]
- 32.Genta RM, Rugge M: Gastric precancerous lesions; heading for an international consensus. Gut 1999, 45(Suppl):I5-I8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobin LH: International Union Against Cancer. Sobin LH Wittekind C eds. Stomach. TNM Classification of Malignant Tumours, ed 5 1997:pp 59-62 Wiley-Liss, New York
- 34.Stolte M, Meining A: The updated Sydney system: classification and grading of gastritis as the basis of diagnosis and treatment. Can J Gastroenterol 2001, 15:591-598 [DOI] [PubMed] [Google Scholar]
- 35.Goelz SE, Hamilton SR, Vogelstein B: Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochem Biophys Res Commun 1985, 130:118-126 [DOI] [PubMed] [Google Scholar]
- 36.Nakatsuru S, Yanagisawa A, Ichii S, Tahara E, Kato Y, Nakamura Y, Horii A: Somatic mutation of the APC gene in gastric cancer: frequent mutations in very well differentiated adenocarcinoma and signet-ring cell carcinoma. Hum Mol Genet 1992, 1:559-563 [DOI] [PubMed] [Google Scholar]
- 37.Powell SM, Cummings OW, Mullen JA, Asghar A, Fuga G, Piva P, Minacci C, Megha T, Tosi P, Jackson CE: Characterization of the APC gene in sporadic gastric adenocarcinomas. Oncogene 1996, 12:1953-1959 [PubMed] [Google Scholar]
- 38.Woo DK, Kim HS, Lee HS, Kang YH, Yang HK, Kim WH: Alterated expression and mutation of β-catenin gene in gastric carcinomas and cell lines. Int J Cancer 2001, 95:108-113 [DOI] [PubMed] [Google Scholar]
- 39.Peifer M, Polakis P: Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science 2000, 287:1606-1609 [DOI] [PubMed] [Google Scholar]
- 40.Beroud C, Soussi T: APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 1996, 24:121-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT: Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am J Pathol 2000, 157:747-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang JM, Chou YH, Chou TB: K-ras codon 12 mutation determines the polypoid growth of colorectal cancer. Cancer Res 1998, 58:3289-3293 [PubMed] [Google Scholar]
- 43.Yashiro M, Carethers JM, Laghi L, Saito K, Slezak P, Jaramillo E, Rubio C, Koizumi K, Hirakawa K, Boland CR: Genetic pathways in the evolution of morphologically distinct colorectal neoplasms. Cancer Res 2001, 61:2676-2683 [PubMed] [Google Scholar]
- 44.van Wyk R, Slezak P, Hayes VM, Buys CH, Kotze MJ, de Jong G, Rubio C, Dolk A, Jaramillo E, Koizumi K, Grobbelaar JJ: Somatic mutations of the APC, KRAS, and TP53 genes in nonpolypoid colorectal adenomas Genes Chromosom Cancer 2000, 27:202-208 [PubMed] [Google Scholar]
- 45.Keller G, Rudelius M, Vogelsang H, Grimm V, Wilhelm MG, Mueller J, Siewert JR, Hofler H: Microsatellite instability and loss of heterozygosity in gastric carcinoma in comparison to family history. Am J Pathol 1998, 152:1281-1289 [PMC free article] [PubMed] [Google Scholar]
- 46.Kim HS, Woo DK, Bae SI, Kim YI, Kim WH: Microsatellite instability in the adenoma-carcinoma sequence of the stomach. Lab Invest 2000, 80:57-64 [DOI] [PubMed] [Google Scholar]
- 47.Leung WK, Kim JJ, Kim JG, Graham DY, Sepulveda AR: Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. Am J Pathol 2000, 156:537-543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ottini L, Palli D, Falchetti M, D’Amico C, Amorosi A, Saieva C, Calzolari A, Cimoli F, Tatarelli C, De Marchis L, Masala G, Mariani-Costantini R, Cama A: Microsatellite instability in gastric cancer is associated with tumor location and family history in a high-risk population from Tuscany. Cancer Res 1997, 57:4523-4529 [PubMed] [Google Scholar]
- 49.Sepulveda AR, Santos AC, Yamaoka Y, Wu L, Gutierrez O, Kim JG, Graham DY: Marked differences in the frequency of microsatellite instability in gastric cancer from different countries. Am J Gastroenterol 1999, 94:3034-3038 [DOI] [PubMed] [Google Scholar]
- 50.Wu MS, Lee CW, Shun CT, Wang HP, Lee WJ, Chang MC, Sheu JC, Lin JT: Distinct clinicopathologic and genetic profiles in sporadic gastric cancer with different mutator phenotypes. Genes Chromosom Cancer 2000, 27:403-411 [PubMed] [Google Scholar]
- 51.Wu MS, Lee CW, Shun CT, Wang HP, Lee WJ, Sheu JC, Lin JT: Clinicopathological significance of altered loci of replication error and microsatellite instability-associated mutations in gastric cancer. Cancer Res 1998, 58:1494-1497 [PubMed] [Google Scholar]
- 52.Ohmura K, Tamura G, Endoh Y, Sakata K, Takahashi T, Motoyama T: Microsatellite alterations in differentiated-type adenocarcinomas and precancerous lesions of the stomach with special reference to cellular phenotype. Hum Pathol 2000, 31:1031-1035 [DOI] [PubMed] [Google Scholar]
- 53.Chong JM, Fukayama M, Hayashi Y, Takizawa T, Koike M, Konishi M, Kikuchi-Yanoshita R, Miyaki M: Microsatellite instability in the progression of gastric carcinoma. Cancer Res 1994, 54:4595-4597 [PubMed] [Google Scholar]
- 54.Isogaki J, Shinmura K, Yin W, Arai T, Koda K, Kimura T, Kino I, Sugimura H: Microsatellite instability and K-ras mutations in gastric adenomas, with reference to associated gastric cancers. Cancer Detect Prev 1999, 23:204-214 [DOI] [PubMed] [Google Scholar]
- 55.Shibata D: Molecular tumor clocks and dynamic phenotype. Am J Pathol 1997, 151:643-646 [PMC free article] [PubMed] [Google Scholar]
- 56.Ruschoff J, Dietmaier W, Luttges J, Seitz G, Bocker T, Zirngibi H, Schlegel J, Schackert HK, Jauch KW, Hofstaedter F: Poorly differentiated colonic adenocarcinoma, medullary type: clinical, phenotypic, and molecular characteristics. Am J Pathol 1997, 150:1815-1825 [PMC free article] [PubMed] [Google Scholar]
- 57.Goggins M, Offerhaus GJA, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology: poor differentiation, a syncytial growth pattern, and pushing borders suggests RER+. Am J Pathol 1998, 152:1501-1507 [PMC free article] [PubMed] [Google Scholar]