

Abstract
Human papillomaviruses (HPVs) infect cervical epithelial cells and induce both benign and precancerous lesions. High-risk HPVs promote the development of cervical cancer in vivo and can immortalize cervical epithelial cells in vitro, whereas low-risk HPVs cannot. We used cDNA microarrays and quantitative polymerase chain reaction to compare cellular gene expression in primary cervical epithelial cells during a time course after retroviral transduction with either low-risk or high-risk E6/E7 genes. At early passages, cervical cells transduced with high-risk E6/E7 genes demonstrated increased expression of the cell cycle-regulated genes CDC2 and ubiquitin carrier E2-C. At later passages, these same cells exhibited dramatic increases in insulin-like growth factor-binding protein-3 (IGFBP-3) mRNA and both secreted an intracellular protein, with mRNA levels increasing ∼85-fold. Corroborating these in vitro studies, in situ hybridization of cervical biopsies with an IGFBP-3 riboprobe revealed high levels of expression in high-grade squamous intraepithelial neoplasia but not in normal cervical epithelium. Our in vitro results indicate that overexpression of IGFBP-3 is a late event after E6/E7 expression, and analysis of cervical lesions indicates that overexpression of this gene is also seen in vivo.
Human papillomaviruses (HPVs) are the major cause of cervical cancer and precancerous cervical lesions. 1 Epidemiological and molecular studies have identified high-risk types of HPV, such as HPV-16 and HPV-18, which are present in >90% of high-grade cervical lesions and cervical cancers. In contrast, low-risk types of HPV such as HPV-6 and HPV-11 are found in genital warts and low-grade cervical lesions but rarely in high-grade lesions or cancer. The difference between high-risk and low-risk HPV types is also apparent in cultured cells. The E6 and E7 oncogenes of high-risk HPV are sufficient to immortalize human foreskin keratinocytes and cervical epithelial cells in vitro. 2,3 In contrast, the E6 and E7 genes of low-risk HPV can prolong life span in culture but cannot immortalize cells. 4 This difference between viral types can be explained by the different interactions of high-risk and low-risk E6 and E7 with the key human cell-cycle regulatory proteins p53 and pRb. 5,6 Destabilization of p53 and pRb proteins by high-risk but not low-risk HPV oncogenes results in altered levels of other cell-cycle regulatory proteins. For example, levels of cyclin A, cyclin B, cyclin E, cdc2, cdk2, and cdk4 proteins are elevated in HPV-16 expressing keratinocytes 7,8 when compared to controls. Recent results from cDNA microarray experiments that surveyed several thousand genes indicate that mRNA levels of these cell cycle genes are also elevated in HPV-16 E6/E7 expressing keratinocytes. 9,10 We used cDNA microarrays containing 25,000 clones to identify additional genes that are differentially expressed in HPV-immortalized cervical keratinocytes when compared with vector control and early passage HPV-16 E6/E7-transduced cells. Furthermore, we aimed to identify molecular differences between cervical keratinocytes expressing high-risk and low-risk HPV E6/E7. We were particularly interested in evaluating chronological changes in cellular gene expression of proliferating cervical cells. Cellular genes expressed only at late passage might offer insight into critical regulatory pathways that are perturbed during cell immortalization.
We found two cell-cycle regulated genes, CDC2 and ubiquitin carrier protein E2-C, that are rapidly overexpressed in cervical cells after transduction with high-risk E6/E7 oncogenes when compared with cells transduced with low-risk E6/E7. Most importantly, we found one gene, insulin-like growth factor binding protein 3 (IGFBP-3), which showed a dramatic increase in expression at a time point in cell passaging experiments that correlated with the immortalization of cervical keratinocytes in vitro. Expression of this gene was also elevated in clinical samples of high-grade cervical lesions but not of normal ectocervix.
Materials and Methods
Cell Culture
Primary human ectocervical keratinocytes were derived from fresh cervical tissue obtained after hysterectomy for benign uterine diseases. Standard trypsinization procedures were used to isolate the keratinocytes, 4 after which they were cultured in serum-free keratinocyte medium supplemented with 50 μg/ml of bovine pituitary extract and 26 ng/ml of recombinant epidermal growth factor (Invitrogen, Carlsbad, CA). Primary cultures of human ectocervical keratinocytes were infected with high-titer (∼106 colony-forming units/ml) LXSN retroviruses 11 expressing either the HPV-6 E6/E7 genes or the HPV-16 E6/E7 genes. 7 Control LXSN retroviruses contained no inserted genes and expressed only the neomycin resistance gene. After infection, keratinocytes were selected with 50 μg/ml of G418 for 5 days and were subcultured once before extraction of RNA. All subsequent passages were performed at a split ratio of 1:4.
Transcriptional Profiling
cDNA microarrays contained 25,000 sequence-verified Unigene clones from Research Genetics (Hunstville, AL). Arrays were prepared at Millennium Pharmaceuticals, Inc. (Cambridge, MA) by polymerase chain reaction (PCR) amplifying each clone with M13 primers, ethanol precipitating the PCR products, and spotting the products on Biodyne B nylon membranes (Pall, Ann Arbor MI) using a customized Yaskawa robot (Motoman, West Carrolton, OH). Total RNA was prepared from cell pellets using RNeasy Midi Columns (Qiagen, Valencia CA). Fifteen μg of RNA was radioactively labeled by reverse transcription with 33P-dCTP, oligo dT-30 primer and Superscript II reverse transcriptase (Invitrogen). Labeled first-strand cDNA was preannealed with cot-1 DNA (Invitrogen) and polydA 40-60 (Pharmacia, Peapack, NJ) to reduce nonspecific hybridization. Each labeled cDNA was hybridized in duplicate to a set of five nylon membranes. Microarrays were exposed to Fuji Phosphoimager screens and scanned using a Fuji scanner BAS 2500 (Fujifilm Medical Systems, Stamford, CT). Spots were quantitated using Array Vision software (Imaging Research, St. Catherines, Ontario, Canada) and data were analyzed using custom software developed at Millennium Pharmaceuticals. Data were normalized to the median intensity of all spots on each filter and data from duplicate filters were averaged after normalization.
Quantitative PCR
One μg of DNase-treated total RNA was reverse-transcribed using random hexamers and oligo dT from the Superscript First-Strand Synthesis system for reverse transcriptase-PCR (Invitrogen). A control reaction with no reverse transcriptase was performed to check for contamination by genomic DNA. Multiplex PCR reactions contained first-strand cDNA, recommended amounts of 18S rRNA Predeveloped Taqman Assay Reagent and Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA), 900 nmol/L forward primer, 900 nmol/L reverse primer, and 250 nmol/L FAM-labeled probe. PCR reactions were run on the ABI Prism 7700 sequence detector with the following conditions: 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute. All reactions were done in duplicate, and data were analyzed with Sequence Detector v1.6.3 software (Applied Biosystems). 18S rRNA was used as a housekeeping gene to normalize amounts of RNA. For each reaction, expression was calculated as 2−ΔCt, where ΔCt is the difference between the Ct for the gene of interest and the Ct for 18S RNA. The average expression for the duplicates is reported with the SD. The following primers (f and r) and probes (p) were designed using Primer Express 1.0 (Applied Biosystems): HPV-16 E6f GCACAGAGCTGCAAACAACTATACA, HPV-16 E6r ATCCCGAAAAGCAAAGTCATATACC, HPV-16 E6p TGTGTGTACTGCAAGCAACAGTTACTGCGA; HPV-16 E7f AAGTGTGACTCTACGCTTCGGTT, HPV-16 E7r GCCCATTAACAGGTCTTCCAAA, HPV-16 E7p TGCGTACAAAGCACACACGTAGACATTCG; CDC2f AATCTATGATCCAGCCAAACGAA, CDC2r TCTTAATCTGATT GTCCAAATCATTAAAA, CDC2p TTCTGGCAAAATGGCACTGAATCATCC; E2-Cf TTCCCCAGTGGCTACCCTTAC, E2-Cr TGGAGAGCAGAATGGTCCTGA, E2-Cp TCCTCACGCCCTGCTATCACCCC; IGFBP-3f AGCACAGCACCCAGACTTCA, IGFBP-3r TTCTGCATAAAGCCTGTGCG, IGFBP-3p CGCCCGTGGAATGCTCACCA.
Western Blots
For p53, endogenous IGFBP-3, and β-actin detection, cell cultures were harvested at 80% confluent growth on 100-mm plastic dishes. The cells were lysed in sodium dodecyl sulfate-extraction buffer (0.8% sodium dodecyl sulfate, 20% glycerol, 0.125 mol/L Tris-HCl, pH 6.8), reduced with 10% β-mercaptoethanol and heated for 7 minutes at 95°C. Fifty μg of the extracted protein was electrophoretically separated on a 10% Tris-Glycine-Gel (Invitrogen, Carlsbad, CA), transferred to a polyvinylidene difluoride membrane, and either incubated overnight at 4°C with anti-p53 antibody at 0.4 μg/ml (Santa Cruz Biotechnology, Santa Cruz, CA) or with anti IGFBP-3 antibody (BD Transduction Laboratories, San Diego, CA) at 1.0 μg/ml. After reaction with secondary goat anti-mouse IgG antibody at 0.3 μg/ml (Tropix, Foster City, CA), p53 and IGFBP-3 were visualized with a chemiluminescent substrate for alkaline phosphatase (Tropix). The p53 immunoblot was then stripped and reprobed with β-actin antibody (1:1000 dilution, Amersham). To verify that equal amounts of protein were used for the IGFBP-3 Western blot analysis, 10 μg of protein from the same lysates were separated in polyacrylamide Tris-glycine minigels, transferred to polyvinylidene difluoride membranes and incubated for 90 minutes at room temperature with anti β-actin antibody clone AC-15 (Sigma Chemical Co., St. Louis, MO) at 0.27 μg/ml and secondary goat anti-mouse IgG antibody at 0.3 μg/ml (Tropix). β-actin was visualized with a chemiluminescent substrate for alkaline phosphatase (Tropix).
For analysis of secreted IGFBP-3, 35 μl of conditioned cell culture medium (1 × 105 cells plated in 12-well-plates, incubated for 6 days in 300 μl of supplemented keratinocyte medium) was treated with 15 μl of sodium dodecyl sulfate-extraction buffer containing 10% 2-mercaptoethanol and heated for 7 minutes at 95°C. Samples were electrophoretically separated, blotted with anti IGFBP-3 antibody (1 μg/ml), and visualized by chemiluminescence as above.
In Situ Hybridization
Tissue microarrays were obtained from Clinomics (Pittsfield, MA). Arrays were constructed by collecting 0.6-mm cores of paraffin-embedded archival cervical samples and embedding these samples into a new paraffin block. 12 Sections from this block were then cut and used for in situ hybridization. Samples included triplicate cores from normal ectocervix samples, normal endocervix samples, and high-grade squamous intraepithelial neoplasia (HGSIL). In situ hybridization with IGFBP-3 riboprobe was performed as described. 13 IMAGE clone 898218, which encodes a 1.1-kb fragment of IGFBP-3, was linearized and labeled with 35S-UTP. A β-actin riboprobe was used as a positive control for RNA preservation.
Results
Transduction of E6/E7 Oncogenes and Effects on Cervical Cell Life Span and p53 Levels
We infected primary ectocervical keratinocytes at passage 4 with three different LXSN retroviruses: an empty LXSN virus as control, virus containing low-risk HPV-6 E6/E7, and virus containing high-risk HPV-16 E6/E7. As expected, control LXSN-transduced cells had a very limited life span and proliferated for only four to five passages after infection, whereas HPV-6 E6/E7 induced an extended life span in which the cells proliferated for six passages after infection. HPV-16 E6/E7-infected cervical cells continued proliferating for more than 200 passages after infection without undergoing a detectable crisis period. We repeated these transductions on a second set of the same cervical cells and obtained similar results.
HPV-16 E6/E7 and HPV-6 E6/E7 transcripts were detectable by Northern blot at the first passage after infection (data not shown). Quantitative PCR (Figure 1A) ▶ using primers specific for HPV-16 E6 and E7 shows that these genes were already expressed by passage 1 after infection and remained at relatively constant levels from passage 6 through passage 20. Expression was further increased at the latest time point examined (passage 30). As expected, p53 protein levels were greatly reduced by HPV-16 E6/E7 at the earliest time point examined, passage 4 after infection (Figure 1B) ▶ . Although very low levels of p53 reappeared at late passages, the levels remained much lower than those in the LXSN or HPV-6E6/E7 cells, indicating that HPV-16 E6 was expressed and functional during the entire time course.
Figure 1.
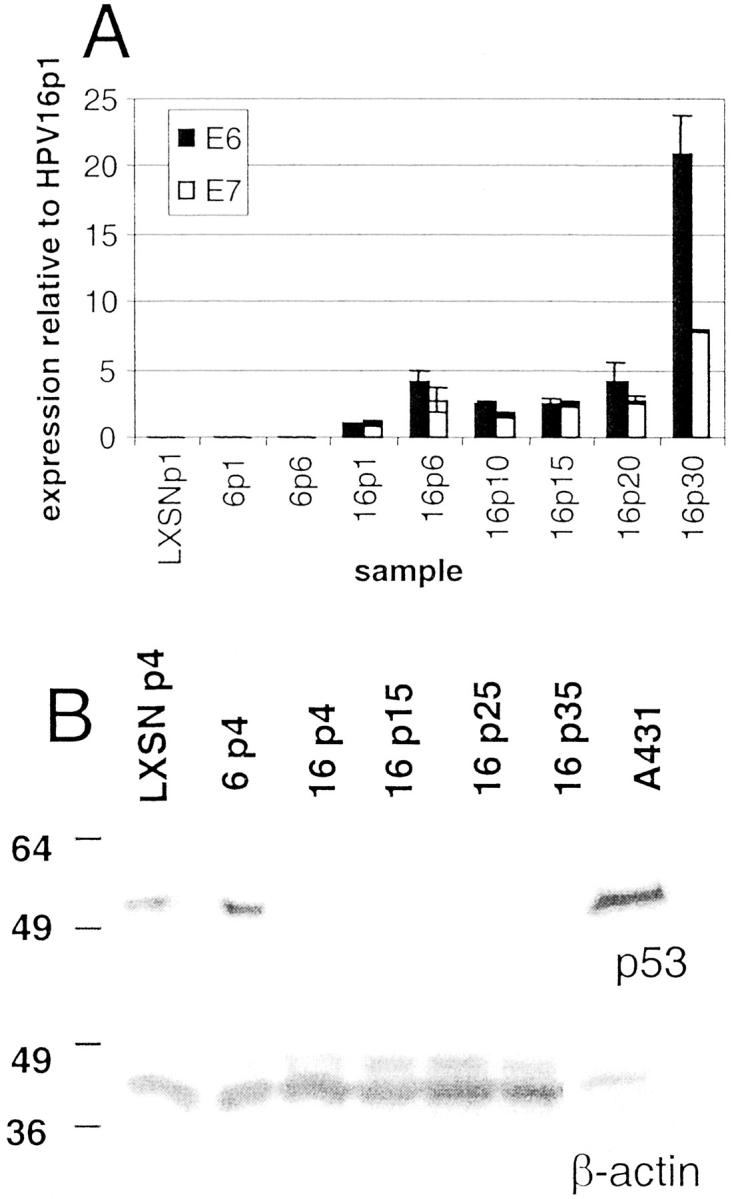
E6/E7 and p53 expression in transduced human ectocervical epithelial cells. Expression of the HPV-16 E6 and E7 oncogenes was detected by quantitative PCR (A) and p53 protein levels by Western blotting (B) of cervical keratinocytes infected with control retrovirus LXSN or retrovirus containing the HPV-6 E6/E7 or HPV-16 E6/E7 genes. Samples from infection 1 are shown. Abbreviations indicate viral type and passage number after infection, ie, 6p1 refers to HPV-6 E6/E7 cells one passage after infection. A: Relative expression levels are calculated based on the expression of each gene in HPV-16 E6/E7-infected cells at passage 1 after infection. Error bars represent the SD for duplicate PCR reactions. B: Western blot analysis of total cellular p53 protein was performed as described in Materials and Methods for each of the indicated samples. A431 cells are epidermoid carcinoma cells used as a positive control for p53. Western blotting of these samples for β-actin levels was used to verify equal loading of cellular protein.
Transcriptional Profiling
For each set of infections, we profiled RNA from six samples: acutely infected LXSN, HPV-6, and HPV-16 cells, extended life span HPV-6 and HPV-16 cells, and immortalized HPV-16 cells. Pairwise comparisons between samples showed surprisingly few changes in gene expression between any two samples. For example, using samples from the first infection, we found ∼50 genes of 25,000 (0.2%) on the cDNA arrays that were expressed at least threefold higher in immortalized HPV-16 cells when compared with extended life span HPV-16 cells. Likewise, we found ∼50 genes that were expressed at least threefold higher in HPV-16-extended life span cells when compared with HPV-6-extended life span cells in the first infection (data not shown). To eliminate false-positive results, we required that genes show threefold or greater expression in the same comparison in both infection 1 and infection 2. Using these criteria, we identified three genes of interest: the cell cycle kinase CDC2, the ubiquitin carrier protein E2-C, and insulin-like growth factor binding protein 3 (IGFBP-3). CDC-2 and E2-C show higher expression in HPV-16-extended life span cells than in HPV-6-extended life span cells in both infections, whereas IGFBP-3 was chosen because it shows higher expression in HPV-16-immortalized cells compared to HPV-16-extended life span cells in both infections. We also observed a set of cellular genes that showed significant decreases in expression; however, these were not characterized in the current study.
Figure 2 ▶ shows the chronological changes in the expression of CDC2, E2-C, and IGFBP-3 in the six samples from infection 2. At passage 4 after infection, CDC2 and E2-C displayed approximately threefold higher expression in HPV-16-infected cells when compared with HPV-6-infected cells. In contrast, IGFBP-3 was not elevated in HPV-16 cells at passage 4 but showed more than a sixfold higher expression in HPV-16-immortalized cells (passage 20) when compared with earlier passages of HPV-16, HPV-6, or LXSN. Similar results were seen with samples from infection 1 (data not shown). In all cases the genes showed at least threefold changes between the relevant samples in both infections.
Figure 2.
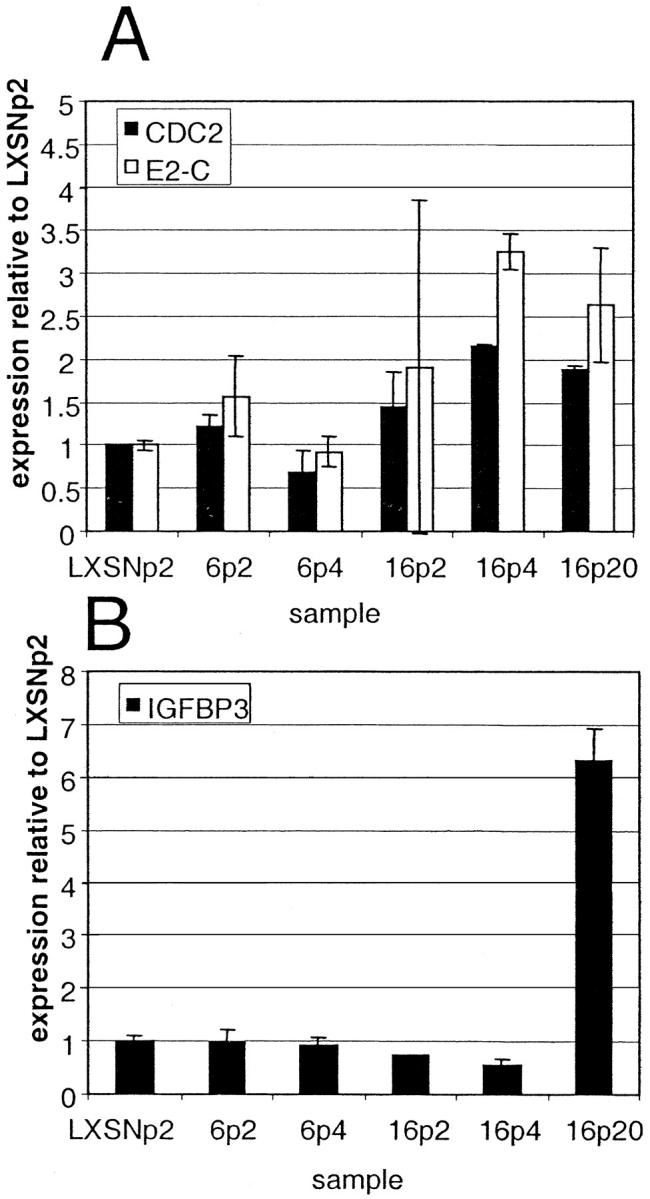
Temporal changes in CDC2, E2-C, and IGFBP-3 expression in transduced cervical cells as determined by transcriptional profiling of samples from infection 2. Expression of CDC2, E2-C, and IGFBP-3 was determined by transcriptional profiling of cervical keratinocytes infected with control retrovirus LXSN or retrovirus containing the HPV-6 E6/E7 or HPV-16 E6/E7 genes. Abbreviations indicate viral type and passage number after infection, ie, 6p2 refers to HPV-6 E6/E7 cells two passages after infection. Relative expression levels were calculated based on the expression of each gene in the control LXSN-infected cells at passage 2. Error bars represent the SD for duplicate spots on cDNA microarrays.
Confirmation by Quantitative PCR
To confirm these expression differences with another method and to assay gene expression in additional samples in the time course, we performed quantitative PCR using the Taqman system. Figure 3 ▶ shows data from infection 1; we performed Taqman assays on infection 2 samples with equivalent results (data not shown). Figure 3A ▶ shows that CDC2 and E2-C were up-regulated in all passages of HPV-16 cells when compared with LXSN or HPV-6 cells. In HPV-16 cells, CDC2 and E2-C were up-regulated as early as passage 1 after infection and maintained their high levels through immortalization (passage 20). In addition, these genes showed even higher up-regulation at passage 30, the point at which E6/E7 expression reached the highest levels. The expression pattern of CDC2 and E2-C appeared to parallel that of HPV-16 E6/E7 (Figure 1A) ▶ .
Figure 3.
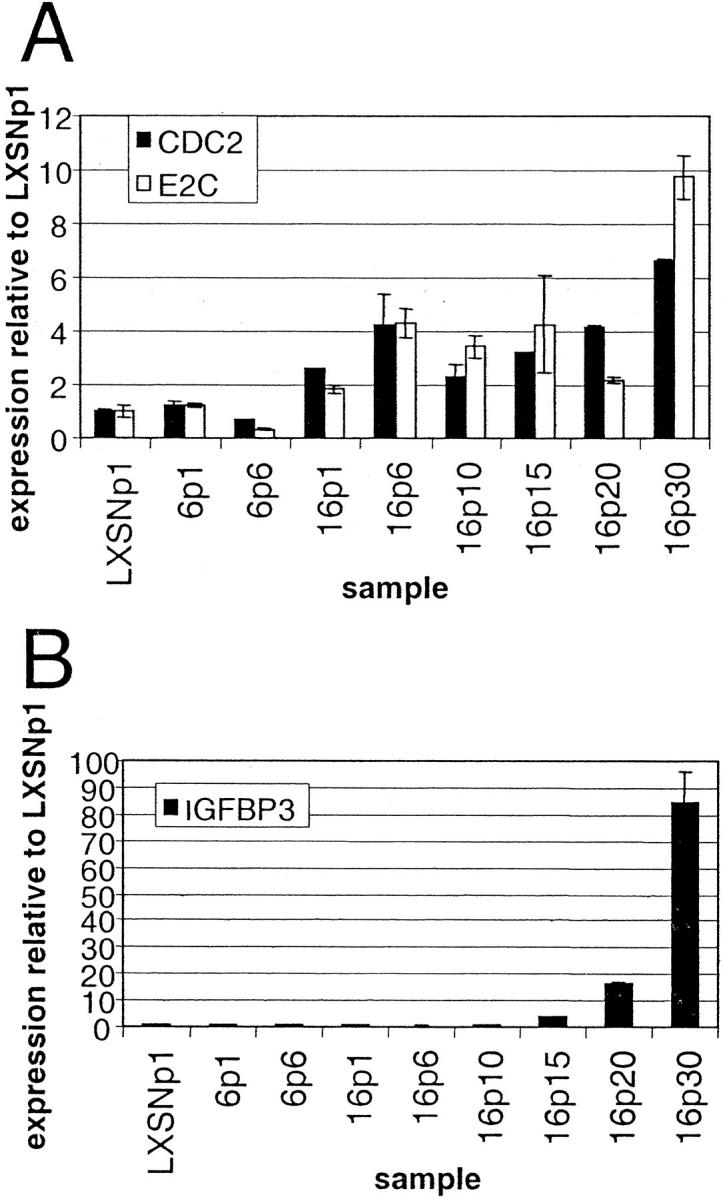
Temporal changes in expression of CDC2, E2-C, and IGFBP-3 mRNA as determined by quantitative PCR using samples from infection 1. The level of mRNA expression in the indicated cervical cell samples was determined by quantitative PCR as described in Materials and Methods for CDC2 and E2-C (A) and for IGFBP-3 (B). Abbreviations indicate viral type and passage number after infection, ie, 6p1 refers to HPV-6 E6/E7 cells one passage after infection. Error bars in A and B indicate the SD between expression values from duplicate PCR reactions. Expression levels are normalized to those of LXSNp1.
Taqman results for IGFBP-3 (Figure 3B) ▶ show that this gene was induced much later than HPV-16 E6/E7, CDC2, and E2-C. Instead of showing an immediate induction in HPV-16 passage 1 cells, IGFBP-3 did not begin to show increased expression until passage 15. Levels continued to rise at later passages, showing an ∼85-fold increase by passage 30. The expression of IGFBP-3 was not directly correlated with that of HPV-16 E6/E7, because a 16-fold induction of IGFBP-3 occurred between passages 10 and 20 while HPV-16 gene expression remained constant. However, the spike in IGFBP-3 levels at passage 30 may be related to the increased expression of HPV-16 E6/E7 at this time point.
Confirmation by Western Blot
To determine whether the levels of IGFBP-3 protein increase coordinately with the levels of mRNA in HPV-16 E6/E7-infected cells, we assayed conditioned media from cells of various passages from infection 1 (Figure 4A) ▶ . Secreted IGFBP-3 was undetectable in media from LXSN controls at passages 1 and 5, HPV-6-infected cells at passages 1 and 6, and HPV-16-infected cells at passages 1 and 6. Secreted IGFBP-3 was detectable in media from HPV-16-infected cells by passage 20 and increased further at passage 30. IGFBP-3 protein secretion is therefore directly correlated with levels of IGFBP-3 mRNA in these cells. Levels of intracellular IGFBP-3 protein increased in HPV-16-infected cells in a similar manner (Figure 4B) ▶ . IGFBP-3 was undetectable at passage 4 but was present in increasing amounts at passages 22 and 54. High levels of IGFBP-3 protein were also seen in CaSki cells that were derived from a human cervical cancer specimen (Figure 4B) ▶ .
Figure 4.
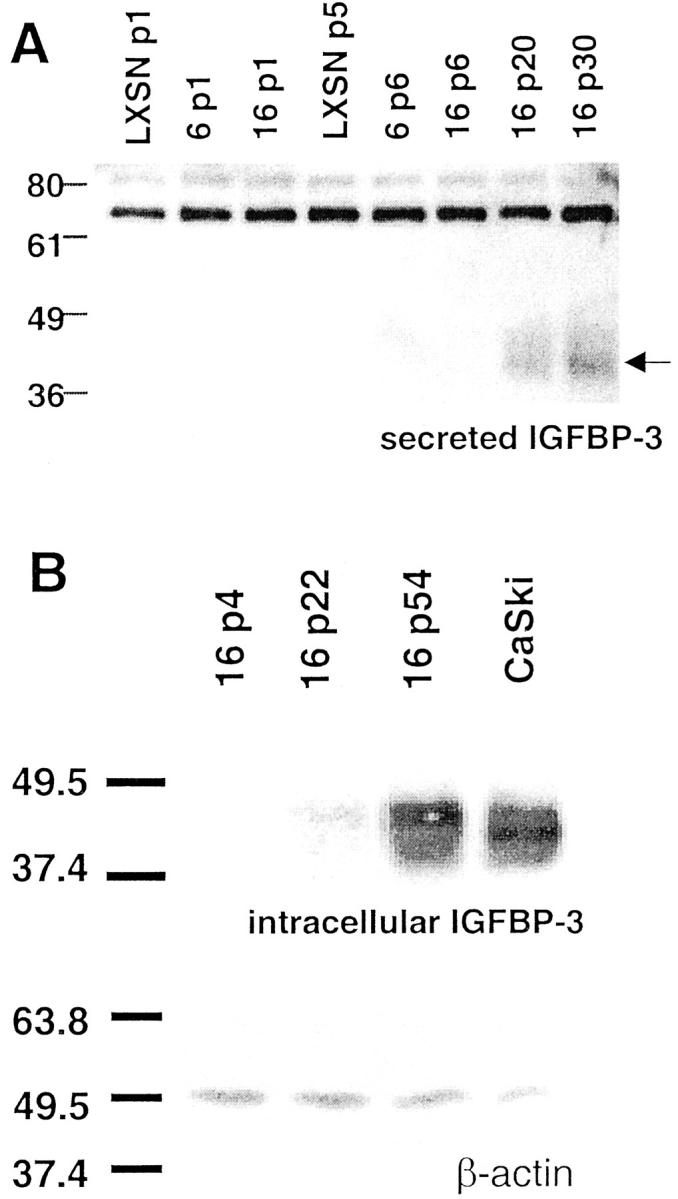
Intracellular and extracellular IGFBP-3 protein expression in E6/E7-transduced cervical epithelial cultures. A: Extracellular levels of secreted IGFBP-3 protein were detected by Western blotting of conditioned medium as described in Materials and Methods. Arrowhead indicates the position of IGFBP-3 protein. B: Intracellular levels of IGFBP-3 were determined by Western blotting of cellular lysates as described in Materials and Methods. IGFBP-3 was only detectable in late passage HPV-16-expressing cervical cells and in the CaSki cervical carcinoma cell line (top). Equal loading of proteins was verified by Western blot analysis of β-actin (bottom).
IGFBP-3 Expression in Human Clinical Samples
Because levels of IGFBP-3 mRNA and protein increased dramatically in HPV-16 E6/E7-infected cervical keratinocytes in late-passage culture, we examined whether this gene was overexpressed in precancerous human cervical lesions. Cervical cells immortalized with HPV-16 and HPV-18 grown in vitro form poorly differentiated stratified epithelial rafts that morphologically resemble HGSILs, 14,15 suggesting a parallel between immortalized cells in culture and high-grade cervical lesions. At least 80 to 95% of HGSILs contain high-risk types of HPV. 16,17 We analyzed a tissue microarray containing normal ectocervix sections and sections containing HGSIL. In situ hybridization with an IGFBP-3 riboprobe showed that this gene was not expressed in any of four samples of normal ectocervical epithelium but was expressed in five of five HGSIL samples, indicating that overexpression of IGFBP-3 is not just a cell culture phenomenon, but is also seen in relevant clinical samples. A representative set of samples is shown in Figure 5 ▶ . Expression of IGFBP-3 was also observed in nonepithelial cells including scattered stromal cells underlying the epithelium, normal and precancerous endocervical glands, endothelial cells, and fibroblasts surrounding the blood vessels of the cervix (not shown).
Figure 5.
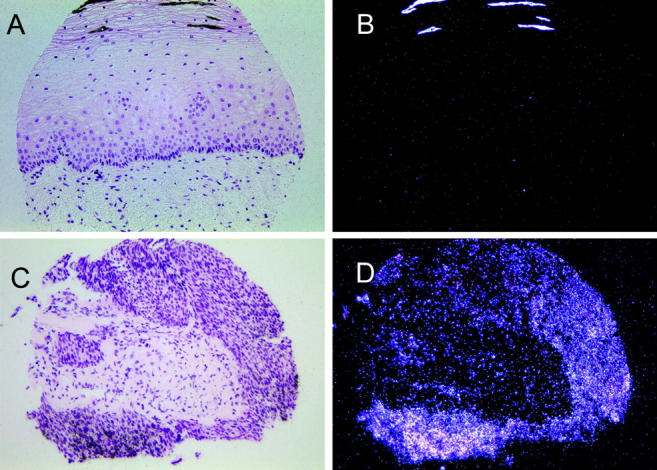
In situ hybridization detection of IGFBP-3 mRNA in samples from a cervical tissue microarray stained with H&E. A and B: Bright-field and dark-field views of normal ectocervix. Bright-field view shows the normal histology of the ectocervix: (from bottom) stromal cells, basal cells, parabasal and intermediate cells, and stratified squamous cells. C and D: Bright-field and dark-field views of a HGSIL sample. White grains in the dark-field panel indicate IGFBP-3 mRNA expression in the HGSIL sample (D) but not in the normal cervix (B).
Discussion
The high-risk HPV oncogenes are retained and expressed in HGSIL and cervical carcinomas and are required for the maintenance of the transformed phenotype. 1,6 The E6 and E7 proteins interact with cellular tumor suppressor proteins, p53 and Rb, respectively. In addition, the E6 gene has been shown to increase hTERT expression, thereby increasing telomerase activity and contributing to continued cellular proliferation. 18,19 It is likely that E6 and E7 also alter the expression of other cellular genes either directly or indirectly. The experiments outlined in the present study are based on the direct examination of alterations in gene expression after HPV-16 E6/E7 transduction of proliferating keratinocytes using a microarray containing 25,000 clones. We identified three genes that are reproducibly induced in cervical epithelial cells expressing HPV-16 oncogenes. A recent study comparing gene expression in HPV-31-immortalized human foreskin keratinocytes versus primary human foreskin keratinocytes found only three genes that were up-regulated more than threefold on a microarray containing 7075 genes, although many more were up-regulated between twofold and threefold. 20 The fact that so few genes are reproducibly up-regulated during immortalization is surprising. However, it could be consistent with the hypothesis that an active genetic program is necessary for senescence, and if this program is blocked by cellular mutations or by HPV oncogene expression, immortalization results. 21 In this view, the immortalized state would be similar in gene expression to an early-passage population of normal cells, whereas a differentiated or senescent population would show more changes in gene expression. In fact, recent studies have shown that HPV-16 E6/E7 expression leads to many more changes in cellular gene expression under conditions that promote quiescence or differentiation (removal of growth factors and addition of 1.4 mmol/L of calcium) when compared with conditions that promote active proliferation. 9,10
Two of the three genes identified in our experiments, CDC2 and E2-C, were up-regulated at the earliest time examined, and this up-regulation persisted throughout immortalization. Their expression pattern was closely correlated with that of HPV-16 E6 and E7, suggesting that the elevated levels of CDC2 and E2-C were an early consequence of E6 and E7 expression. E2-C is the ubiquitin carrier protein involved in the degradation of cyclin B at the end of mitosis, 22 which results in the inactivation of CDC2 and exit from the M phase of the cell cycle. The up-regulation of CDC2 and E2-C likely reflects a faster mitotic rate of HPV-16-infected cells when compared with that of HPV-6-infected or control cells. Other recent studies using cDNA microarrays found increased expression of a number of cell cycle-regulated genes, including CDC2, CDC25A, CDC25B, cyclin A, cyclin B, cdk2, and cdk4, in cervical keratinocytes expressing HPV-16 E6/E7 when compared with expression in control cells under conditions that promote keratinocyte differentiation. 9,10 However, E2-C was not reported to be up-regulated in these experiments. The third gene identified in our studies, IGFBP-3, is up-regulated only in HPV-16-transduced cells and only at the latest time points. This indicates that E6/E7 expression alone is not sufficient to induce IGFBP-3, but that other changes associated with immortalization are likely to be necessary for IGFBP-3 induction.
Our results contrast with those from other recent studies that have examined the relationship of IGFBP-3 and HPV. Although we found an induction of IGFBP-3 in immortalized cells, Nees and colleagues 9 found that IGFBP-3 mRNA is repressed in HPV-16 E6/E7 acutely infected and immortalized cervical keratinocytes when compared with vector-only controls. In their experiments, RNA was harvested from cells under conditions that promote growth arrest. Cells were maintained for 24 hours in media without growth factors and then for 24 hours in differentiation media. In contrast, our experiments were designed to detect changes in gene expression when both control and E6/E7-transduced cells were maintained under conditions of proliferation.
Another recent study has shown an induction of IGFBP-3 mRNA in immortalized foreskin keratinocytes expressing HPV-16 E7 when compared with primary keratinocytes, but in contrast to our results, levels of IGFBP-3 protein were shown to be reduced rather than increased. 23 This study had two major differences from ours: they used foreskin keratinocytes rather than cervical cells, and they transfected the isolated HPV E7 oncogene rather than the combined E6/E7 oncogenes. These results suggest that either cell type or HPV oncogene expression could account for the observed difference. Because we have shown that an immortalized human foreskin keratinocyte cell line (Nco) expressing the HPV-18 E6/E7 genes secretes large amounts of IGFBP-3 protein (data not shown), we anticipate that the difference in IGFBP-3 stability more likely reflects the presence of the E6 oncogene rather than the cell type. It is likely, therefore, that the combined action of E6 and E7 induces a different cellular phenotype than does E7 alone.
The induction of IGFBP-3 during immortalization is somewhat surprising given previous knowledge of its regulated expression and activity. IGFBP-3 is a p53-induced gene, 24 but we have shown that IGFBP-3 is highly induced even though p53 levels are greatly reduced in our immortalized cervical cells. Apparently there are additional, strong transcriptional activators of the IGFBP-3 gene that appear in late-passage HPV-16 E6/E7 cervical cells.
Previous studies of the effects of exogenous IGFBP-3 on cell growth and of IGFBP-3 expression during conditions of growth stimulation and repression have shown variable results. In some cases, IGFBP-3 has a positive effect of cell growth. For example, IGFBP-3 has been shown to augment IGF-1 mitogenic responses in bovine mammary epithelial cells and in a human breast cancer cell line 25,26 and to enhance the activity of IGF-1 on cultured bovine fibroblasts. 27 In other experiments, however, IGFBP-3 inhibits the growth-enhancing activity of IGF-1, and addition of IGFBP-3 to culture media causes growth inhibition or apoptosis in several cancer cell lines. 28,29 Even in a single cell line, there is not a strict correlation between IGFBP-3 levels and cell growth. In HPV-16-immortalized cervical epithelial cells, IGFBP-3 mRNA levels increase in response to retinoic acid, a growth suppressive factor 30 but decrease in response to transforming growth factor-β, another growth suppressive factor. 31 It will be important now to determine directly the biological effect of IGFBP-3 on the growth and differentiation of normal and HPV-immortalized cervical epithelial cells.
Immortalized cervical cells have striking parallels to high-grade cervical lesions. When HPV-16- or HPV-18-immortalized cervical cells are grown in raft cultures, they form structures similar to high-grade cervical lesions, characterized by a lack of stratification and differentiation, an expansion of basal-type cells throughout the epithelium, and cellular disorganization and nuclear atypia. 14,15 In addition, high-grade cervical lesions are generally caused by persistent high-risk HPV infection. Our in situ hybridization results showing overexpression of IGFBP-3 mRNA in HGSIL patient samples supports the findings of IGFBP-3 up-regulation in immortalized cervical cells and extends these in vitro finding into relevant clinical samples.
Footnotes
Address reprint requests to Allison Berger, Ph.D., Millennium Pharmaceuticals, Inc., One Kendall Square, Bldg. 700, 3rd Floor, Cambridge, MA 02139. E-mail: aberger@mpi.com.
Supported by the National Cancer Institute (grant no. RO1CA53371) and BDGene.
References
- 1.Stoler MH: Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol 2000, 19:16-28 [DOI] [PubMed] [Google Scholar]
- 2.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R: The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 1989, 63:4417-4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fichorova RN, Rheinwald JG, Anderson DJ: Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol Reprod 1997, 57:847-855 [DOI] [PubMed] [Google Scholar]
- 4.Schlegel R, Phelps WC, Zhang Y, Barbosa M: Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. EMBO J 1988, 7:3181-3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alani RM, Munger K: Human papillomaviruses and associated malignancies. J Clin Oncol 1998, 16:330-337 [DOI] [PubMed] [Google Scholar]
- 6.zur Hausen H: Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst 2000, 92:690-698 [DOI] [PubMed] [Google Scholar]
- 7.Pei XF, Sherman L, Sun YH, Schlegel R: HPV-16 E7 protein bypasses keratinocyte growth inhibition by serum and calcium. Carcinogenesis 1998, 19:1481-1486 [DOI] [PubMed] [Google Scholar]
- 8.Steinmann KE, Pei XF, Stoppler H, Schlegel R, Schlegel R: Elevated expression and activity of mitotic regulatory proteins in human papillomavirus-immortalized keratinocytes. Oncogene 1994, 9:387-394 [PubMed] [Google Scholar]
- 9.Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD: Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical keratinocytes. J Virol 2001, 75:4283-4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nees M, Geoghegan JM, Munson P, Prabhu V, Liu Y, Androphy E, Woodworth CD: Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth-factor-β2 in cervical keratinocytes. Cancer Res 2000, 60:4289-4298 [PubMed] [Google Scholar]
- 11.Miller AD, Rosman G: Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7:980-990 [PMC free article] [PubMed] [Google Scholar]
- 12.Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch M, Sauter G, Kallioniemi O: Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998, 4:844-847 [DOI] [PubMed] [Google Scholar]
- 13.Deeds J, Cronin F, Duncan LM: Patterns of melastatin mRNA expression in melanocytic tumors. Hum Pathol 2000, 31:1346-1356 [PubMed] [Google Scholar]
- 14.Rader JS, Golub TR, Hudson JB, Patel D, Bedell MA, Laimins LA: In vitro differentiation of epithelial cells from cervical neoplasias resembles in vivo lesions. Oncogene 1990, 5:571-576 [PubMed] [Google Scholar]
- 15.Pecoraro G, Lee M, Morgan D, Defendi V: Evolution of in vitro transformation and tumorigenesis of HPV16 and HPV18 immortalized primary cervical epithelial cells. Am J Pathol 1991, 138:1-8 [PMC free article] [PubMed] [Google Scholar]
- 16.Herrero R, Hildesheim A, Bratti C, Sherman ME, Hutchinson M, Morales J, Balmaceda I, Greenberg MD, Alfaro M, Burk RD, Wacholder S, Plummer M, Schiffman M: Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst 2000, 92:464-474 [DOI] [PubMed] [Google Scholar]
- 17.Solomon D, Schiffman M, Tarone R: Comparison of three management strategies for patients with atypical squamous cells of undetermined significance: baseline results from a randomized trial. J Natl Cancer Inst 2001, 93:293-299 [DOI] [PubMed] [Google Scholar]
- 18.Klingelhutz AJ, Foster SA, McDougall JK: Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380:79-82 [DOI] [PubMed] [Google Scholar]
- 19.Stoppler H, Hartmann D-P, Sherman L, Schlegel R: The human papillomavirus type 16 E6 and E7 oncoproteins dissociate cellular telomerase activity from the maintenance of telomere length. J Biol Chem 1997, 272:13332-13337 [DOI] [PubMed] [Google Scholar]
- 20.Chang YE, Laimins LA: Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol 2000, 74:4174-4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncan EL, Reddel RR: Genetic changes associated with immortalization. A Review. Biochemistry (Moscow) 1997, 62:1263-1274 [PubMed] [Google Scholar]
- 22.Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem 1998, 67:425-479 [DOI] [PubMed] [Google Scholar]
- 23.Mannhardt B, Weinzimer SA, Wagner M, Fiedler M, Cohen P, Jansen-Durr P, Zwerschke W: Human papillomavirus type 16 E7 oncoprotein binds and inactivates growth-inhibitory insulin-like growth factor binding protein 3. Mol Cell Biol 2000, 20:6483-6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N: Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 1995, 377:646-649 [DOI] [PubMed] [Google Scholar]
- 25.Grill CJ, Cohick WS: Insulin-like growth factor binding protein-3 mediates IGF-1 action in a bovine mammary epithelial cell line independent of an IGF interaction. J Cell Physiol 2000, 183:273-283 [DOI] [PubMed] [Google Scholar]
- 26.Chen JC, Shao ZM, Sheikh MS, Hussain A, LeRoith D, Roberts CTJ, Fontana JA: Insulin-like growth factor-binding protein enhancement of insulin-like growth factor-1 (IGF-1)-mediated DNA synthesis and IGF-1 binding in a human breast carcinoma cell line. J Cell Physiol 1994, 158:69-78 [DOI] [PubMed] [Google Scholar]
- 27.Conover CA: Potentiation of insulin-like growth factor (IGF) action by IGF-binding protein-3: studies of underlying mechanism. Endocrinology 1992, 130:191-199 [DOI] [PubMed] [Google Scholar]
- 28.Rajah R, Valentinis B, Cohen P: Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-β1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem 1997, 272:12181-12188 [DOI] [PubMed] [Google Scholar]
- 29.Martin JL, Baxter RC: Oncogenic ras causes resistance to the growth inhibitor insulin-like growth factor binding protein-3 (IGFBP3) in breast cancer cells. J Biol Chem 1999, 274:16407-16411 [DOI] [PubMed] [Google Scholar]
- 30.Andreatta-Van Leyen S, Hembree JR, Eckert RL: Regulation of insulin-like growth factor 1 binding protein 3 levels by epidermal growth factor and retinoic acid in cervical epithelial cells. J Cell Physiol 1994, 160:265-274 [DOI] [PubMed] [Google Scholar]
- 31.Hembree JR, Agarwal C, Eckert RL: Epidermal growth factor suppresses insulin-like growth factor binding protein 3 levels in human papillomavirus type 16-immortalized cervical epithelial cells. Cancer Res 1994, 54:3160-3166 [PubMed] [Google Scholar]