

Abstract
Most human tumors have mutations that result in deregulation of the cdk4/cyclin-Ink4-Rb pathway. Overexpression of D-type cyclins or cdk4 and inactivation of Ink4 inhibitors are common in human tumors. Conversely, lack of cyclin D1 expression results in significant reduction in mouse skin and mammary tumor development. However, complete elimination of tumor development was not observed in these models, suggesting that other cyclin/cdk complexes play an important role in tumorigenesis. Here we described the effects of cdk4 deficiency on mouse skin proliferation and tumor development. Cdk4 deficiency resulted in a 98% reduction in the number of tumors generated through the two-stage carcinogenesis model. The absence of cdk4 did not affect normal keratinocyte proliferation and both wild-type and cdk4 knockout epidermis are equally affected after topical treatment with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA), resulting in epidermal hyperplasia. In similar fashion, cdk4 knockout keratinocytes proliferated well in an in vivo model of wound-induced proliferation. Biochemical studies in mouse epidermis showed that cdk6 activity increased twofold in cdk4-deficient mice compared to wild-type siblings. These results suggest that therapeutic approaches to inhibit cdk4 activity could provide a target to inhibit tumor development with minimal or no effect in normal tissue.
The cyclin-dependent kinases (cdks) are a family of key cell-cycle regulators that function by association with cyclins, the regulatory subunits, at specific points of the cell cycle to phosphorylate various proteins during cell cycle progression. 1,2 cdk4 and cdk6 form complexes with D-type cyclins (cyclin D1, D2, and D3) during the G1 phase of the cell cycle. 3,4 A key substrate for G1 cyclin/cdk complexes is the retinoblastoma protein, pRb. Phosphorylation of pRb, a tumor suppressor gene product, has been attributed to cyclin/cdk complexes and implicated in the regulation of proliferation in keratinocytes and other cell types. 5,6 cdk4,6/D-type cyclins complex formation is induced during the middle of the G1 phase and performs the first step of pRb phosphorylation. Then cyclin E binds and activates cdk2 at the G1/S phase transition and the second step of pRb phosphorylation is carried out on a different pRb motif. 7 Thus, phosphorylation of pRb blocks its ability to suppress the activity of S-phase promoting transcription factors such as E2F. 5,6 Phosphorylation of the C-terminal region of pRb by cdk4,6 triggers sequential intramolecular interactions that progressively block pRb functions as cells move through G1. 8,9 The first phosphorylation facilitates a second interaction that leads to phosphorylation by cdk2 and further S phase progression. 9
cdk4-Deficient mice were generated and showed normal development, although the mutant mice show defects associated with growth retardation such as testicular atrophy, insulin-deficient diabetes and perturbed corpus luteum formation. 10,11 Consistent with this observation, Rane et al have shown that cdk4 activation in a knockin mouse resulted in β-islet cell hyperplasia. 10 In addition, we reported that overexpression of cdk4 is associated with epidermal hyperplasia and hypertrophy in basal cell layers of the epidermis of cdk4 transgenic mice (K5-cdk4). 12 Consistent with those results, overexpression of cdk4 in mouse astrocytes results in an increased cell size as well as hyperploidy. 13
Amplification or translocation of cdk4 or cdk6 has been demonstrated in several sarcomas and leukemias. 14,15 In addition, cdk4 amplification and overexpression have been implicated in glioma development, in this case, mutually exclusive mutations of p16INK4a or cdk4 were observed. 1,2,13 Also, a mutation in cdk4 has been described in patients with familial melanoma 16-18 and it has recently been reported that cdk4 knockin mice harboring this point mutation (R24C) are highly susceptible to melanoma development after chemical treatment. 19
In a previous report, we have shown that cyclin D1-null mice developed a reduced number of skin tumors. 20 To investigate the contribution of cdk4 to tumor development, we have studied the effect of lack of cdk4 in mouse skin carcinogenesis and in normal keratinocyte proliferation. Here, we reported that in vivo proliferation after TPA treatment or wounding on the back of cdk4-knockout mice results in a normal proliferative response. Mechanistic studies in cdk4-deficient epidermis showed an increase in the activity of cdk6. On the other hand, after a specific carcinogenic treatment, tumor development was greatly reduced in cdk4-null and heterozygous mice, which showed a 98% and 36% reduction, respectively, in the number of squamous tumors compared with normal siblings. Thus, these results provide evidence that the putative mechanisms that compensated for the absence of cdk4 in normal skin does not allow tumor development. Thus, inhibition of cdk4 activity could provide a therapeutic target for ras-dependent tumor development.
Materials and Methods
Mice and Carcinogen Treatment
cdk4-Null mice were developed by gene targeting disruption in C57BL/6 background by Tsutsui et al 11 cdk4 +/− mice were backcrossed into the SSIN (inbred SENCAR) background. Genomic DNA was extracted from mouse-tail clips and used for PCR genotyping as was described previously. 11 Seven-week-old genetically matched cdk4−/−, cdk4+/− and cdk4+/+ mice were initiated with a single dose of 200 nmol of 7,12-dimethylbenz[a]anthracene (DMBA) (Sigma Chemical Co., St. Louis, MO) on shaved dorsal skin. Two weeks later, tumor growth was promoted by treating with 2 μg of 12-O-tetradecanoylphorbol-13-acetate (TPA) (Sigma Chemical Co.) twice a week for 20 weeks. Mice were observed twice weekly. Papillomas appeared after 6 to 7 weeks of continuous TPA treatment and were counted weekly for 20 weeks.
To induce epidermal hyperplasia, shaved dorsal skins were treated twice a week with 2 μg of TPA for 3 weeks. Mice were sacrificed 24 hours after the last application and injected with BrdU as described below.
Kinase Assay
Mouse dorsal skins were treated with a depilatory agent for 1 minute and then washed. After the mice were sacrificed, the dorsal skin was removed, and the epidermal tissue was scraped off with a razor blade, placed into NP40 lysis buffer (50 mmol/L Tris, pH 7.5; 150 mmol/L NaCl; 50 mmol/L NaF; 0.5% NP40; 1 mmol/L Na3VO4; 1 mmol/L DTT; 1 mmol/L PMSF). The homogenate was incubated on ice for 15 minutes, homogenization was achieved with a manual homogenizer and centrifuged twice at 10,000 × g for 10 minutes at 4°C to collect the supernatant, which was used directly for the kinase assays. The protein concentration in each skin lysate was measured with the Bio-Rad protein assay system (Bio-Rad Laboratories, Richmond, CA). Three hundred micrograms of protein lysate was immunoprecipitated with antibodies against cdk4, cdk6, or cdk2. 40 μl of precoated antibody beads (Life Technologies Inc., Rockville, MD) were incubated with the lysate for 2 hours at 4°C. The beads were washed twice with NP40 lysis buffer and twice with kinase buffer (50 mmol/L HEPES, pH 7; 10 mmol/L MgCl2; 5 mmol/L MnCl2, 1 mmol/L DTT, 200 μmol/L ATP). After the last wash, 25 μl of kinase buffer and 5 μl of kinase mixture [1 μg of pRb substrate (Santa Cruz Biotechnology, Inc. Santa Cruz, CA); 10 μCi γ-32P ATP (6000 Ci/mmol) (NEN, Boston, MA); 2.5 μl kinase assay buffer] was added to each tube. The tubes were incubated for 30 minutes at 30°C in a water bath. The reaction was stopped by adding 30 μl of sodium dodecyl sulfate sample buffer (Sigma Chemical Co.) to each sample, followed by boiling for 5 minutes and electrophoresed through 10% acrylamide gel. The following antibodies were used: polyclonal antibodies cdk4 (C22), cdk6 (C21), and cdk2 (M2) (Santa Cruz Biotechnology, Inc.). Bio-image analysis was used to quantitate the expression levels of those proteins and for the kinase assay.
Ha-ras Mutation in Mouse Skin Papillomas
Ha-ras mutation was determined by restriction fragment length polymorphism (RFLP) analysis as we previously described. 21 Briefly, the Ha-ras sequence leading codon 61 was amplified by reverse transcription polymerase chain reaction and the amplification product digested with XbaI restriction enzyme (Promega Corp., Madison, WI). The mutated but not the wild-type Ha-ras allele generated two fragments when subjected to XbaI RFLP.
Immunohistochemical Staining
Epithelial cell proliferation was measured by intraperitoneal injection of BrdU (60 mg/g body weight) 30 minutes before the mice were sacrificed. BrdU incorporation was detected by immunohistochemical staining of paraffin-embedded sections with mouse anti-BrdU monoclonal antibody (Becton Dickinson Immunocytometry System; Becton Dickinson, San Jose, CA). The reaction was visualized with a biotin-conjugated anti-mouse antibody (Vector Laboratories, Inc., Burlingame, CA) and an avidin-biotin-peroxidase kit (Vectastain Elite, Vector Laboratories, Inc.) with diaminobenzidine as chromogen. The number of BrdU-positive cells and total cells were determined per 200 μm of interfollicular epithelia in each section. Ten to 14 sections of 200 μm were counted per group.
Wound-Healing Assay
Mice were anesthetized following institutionally approved protocols. The backs of the mice were shaved and sterilized with alcohol, followed by 1% iodine solution. A circular full thickness wound, approximately 8 mm in diameter, was made using a dermal biopsy punch, down to, but not through, the muscle fascia. Three mice of each genotype (cdk4−/− and cdk4+/+) were sacrificed at 3 days after wounding. The healing process was analyzed in paraffin-embedded sections, which were stained with hematoxylin and eosin (H&E) to examine general tissue.
Reproducibility of Results
Unless otherwise stated, biochemical experiments were performed three times. Results are representative of at least three experiments giving similar results.
Results
Reduced Tumor Formation in Mouse Skin
Other laboratories and our own have shown that lack of cyclin D1 results in reduced tumor development. 20,22 However, in mouse skin loss of cyclin D1 expression results in the development of a few papillomas that still harbor Ha-ras mutation. Thus, we hypothesized that complexes others than cyclin D1/cdk4,6 participated in skin tumor development. To determine whether deficiency of cdk4 can result in a reduced number or complete elimination of papilloma development, we used the two-stage carcinogenic protocol. cdk4−/−, cdk4+/−, and cdk4+/+ mice were topically treated with a subcarcinogenic dose of genotoxic DMBA followed by topical application of the tumor promoter TPA. This treatment resulted in papilloma development, which are exophytic, hyperplastic, and benign lesions originating from a single initiated cell. Tumor appearance on the backs of cdk4+/+ mice begun at 7 weeks of promotion whereas it was delayed by 1 week in cdk4−/− and cdk4+/− (appear at 8 weeks) compared with wild-type sibling mice (Figure 1A) ▶ . The percentage of mice with papillomas (incidence) at the end of the experiment (20 weeks) was 92% (13/14) in cdk4+/+, 85% (23/27) in cdk4+/− (15% reduction) and 23% (3/13) in cdk4-null mice (77% reduction). The average number of papillomas per mouse (multiplicity) in cdk4-deficient mice was 2% of cdk4 +/+ mice and 4% of cdk4+/− mice. In addition, a gene dosage dependent effect was observed between cdk4+/− and cdk4+/+, where cdk4+/− mice showed a reduced number of tumors (64%) compared with cdk4+/+ mice (normal sibling) (Figure 1A) ▶ . Only three cdk4-deficient mice developed skin tumors (one papilloma each). The size of the tumors was checked at 15 weeks of promotion and the papillomas of cdk4 null mice showed a reduced size (1 mm3) compared with wild-type mice (5 mm3). cdk4-Deficient papillomas are well-differentiated tumors without foci of dysplasia presenting a histopathology indistinguishable from that of the cdk4+/− and cdk4+/+ (Figure 1B) ▶ . Initiation by the carcinogen DMBA induces a specific point mutation at codon 61 of the Ha-ras gene. 23-26 To analyze whether the development of cdk4 knockout papillomas also depends on Ha-ras mutations, genomic DNA of cdk4 null, heterozygous and wild-type papillomas was subjected to PCR amplification and restriction fragment length polymorphism analysis. All of the cdk4 wild-type and heterozygous papillomas carried specific mutations in the Ha-ras gene. On the other hand, analysis of two papillomas of cdk4-deficient mice showed no Ha-ras mutations at codon 61. These data suggest that lack of cdk4 abrogate ras-dependent skin tumor development, although analysis of a bigger number of cdk4−/− papillomas is necessary to confirm this hypothesis.
Figure 1.

Mouse skin tumor development in cdk4-deficient mice. A: Multiplicity and incidence of papilloma development in cdk4-deficient (KO), cdk4-Heterozygous (Het) and wild-type mice (Wt). Data are expressed as average number of papillomas per mouse (multiplicity) and percentage of mice, which developed papillomas (incidence) as a function of weeks of promotion. B: Representative paraffin-sections of papillomas at 15 weeks from cdk4 KO (−/−) and cdk4 heterozygous (+/−) mice stained with hematoxylin/eosin. cdk4-Null tumors are well-differentiated without foci of dysplasia presenting a histopathology indistinguishable from that of the cdk4+/− and cdk4+/+ (not shown).
In Vivo Keratinocyte Proliferation in cdk4-Deficient Mice
The reduced number of skin tumors in cdk4-null mice led us to hypothesize that cdk4 deficiency could result in defective keratinocyte proliferation. It is known that papillomas from the classic DMBA-TPA carcinogenesis protocol originate from clonal expansion of a cell harboring a Ha-ras mutation. 26 Thus, defective proliferation of mouse keratinocytes could result in impaired clonal expansion and further abrogation of papilloma development.
To determine whether the absence of the cdk4 gene influenced the rate of proliferation and/or the architecture of mouse skin, we analyzed the interfollicular epidermis of cdk4-null and normal sibling mice. Histochemical staining in paraffin-embedded sections showed that both cdk4-null and wild-type skin are normal with no apparent differences in the structure of follicular or interfollicular epidermis (Figure 2, A and B) ▶ . Epidermal proliferation was analyzed by BrdU incorporation as a marker of DNA synthesis. The labeling index (BrdU positive basal cells/total basal cells) of the interfollicular epidermis of both cdk4 knockout and wild-type animals did not show significant differences (wild-type, 0.052 ± 0.003, n = 14; KO, 0.060 ± 0.019, n = 10; t-test, P < 0.05). In addition, statistical analysis did not show significant differences in the number of nucleated cells between cdk4-null and wild-type sibling mice (t-test, P < 0.05).
Figure 2.

Histological and biochemical analysis of cdk4-null skin. Representative paraffin-sections of skin from wild-type (A, C) and cdk4 KO mice (B, D) were stained with hematoxylin/eosin. A and B, normal skins; C and D, hyperplastic skins induced by topical application of TPA. BrdU incorporation of paraffin sections of hyperplastic skin from wild-type (E) and cdk4-deficient (F) mice was detected with mouse monoclonal anti-BrdU antibody. G: Fresh epidermal protein lysates of cdk4-null (KO) and wild-type sibling (WT) mice were immunoprecipitated with polyclonal anti-cdk4, anti-cdk6, and anti-cdk2 antibodies and an in vitro kinase assay, with pRb as a substrate, was performed. Immunoprecipitation with normal rabbit IgG (NR) was carrying out as negative control. The level of Rb peptide phosphorylation was quantified with a densitometer and the values normalized as percentage of the maximum value. The values are the average of three independent assays.
After multiple applications of TPA on the back, both cdk4 KO and normal sibling mice developed epidermal hyperplasia with a high rate of keratinocyte proliferation in the basal cell layer (Figure 2, C and D) ▶ . A mild elevation in the rate of epidermal proliferation induced by TPA was observed in cdk4 KO epidermis compared with wild-type sibling mice (Figure 2, E and F) ▶ (labeling index, KO = 0.61 ± 0.03, n = 10, wild-type = 0.51 ± 0.02, n = 14; t-test P < 0.05), although the number of nucleated cells in interfollicular epidermis was similar (KO = 93.08 ± 9.93 nucleated cells/field; wt = 96.20 ± 0.80 nucleated cells/field; t-test P < 0.05) (Figure 2, C and D) ▶ .
A second approach was also used to determine whether keratinocytes from cdk4 knockout mice proliferate normally under a different stimulus. In this case, proliferation was induced by creating a wound on the dorsal skin of cdk4−/− and cdk4+/+ mice. The healing process involves reepithelialization, and granulation tissue formation. 27 Reepithelialization in turn involve proliferation and migration of cells from the wound edge to fill the wound site. 27 Three days after wounding, the mice were killed and the healing process was analyzed in paraffin embedded H&E-stained sections. Wound tissues showed that at day 3 wounding epidermis at wound edges had grown thicker in both wild-type and cdk4-null mice, indicating that proliferation of keratinocyte was normal (Figure 3,A and B) ▶ . The epithelial tongue (leading edges of the migrating epithelium) and epithelial thickness had formed in both cdk4 knockout and wild-type sibling mice (Figure 3, A and B) ▶ . 27,28 Analysis of keratinocyte proliferation also showed identical number of BrdU positive cells for both groups (data not shown). After incision, wound areas were measured every other day for 11 days and expressed as percent wound remaining. Again, the rate of wound healing did not show significant differences between cdk4 knockout and normal sibling mice (data not shown).
Figure 3.
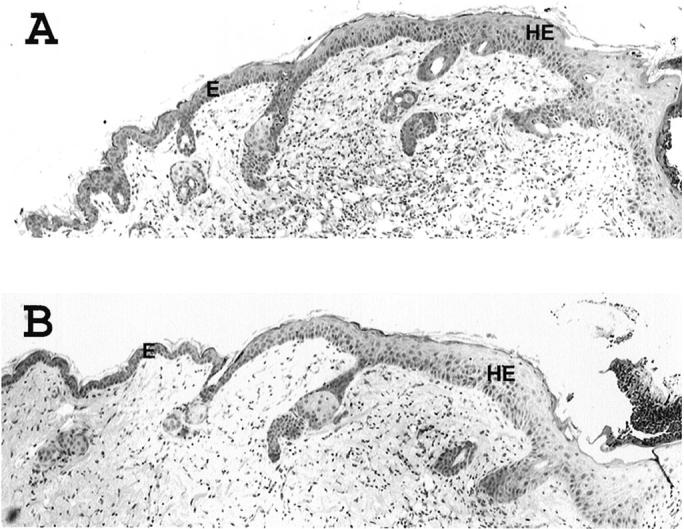
Histological analysis of skin lesion 3 days following wounding. Representative paraffin sections of hyperplastic skin from cdk4-deficienct (A) and wild-type (B) epidermis three days after wounding. Hematoxylin and eosin staining. E, epidermis; HE, hyperproliferative epithelium.
These results showed that cdk4 deficiency did not affect normal keratinocyte proliferation nor TPA or wound induced proliferation, supporting a model in which cdk4 inhibition has a strong influence on neoplastic growth with minimal or no effect on normal tissue.
Cyclin-Dependent Kinase Activities in Mouse Epidermis
Cdk4 and cdk6 activities take part in G1 phase checkpoint in mammalian cells. Thus, we investigated whether other cdk family members in mouse epidermis could compensate for the lack of cdk4. An obvious candidate was cdk6, which also binds to D-type cyclins in G1 phase. In addition, sequential phosphorylation of pRb by cdk4,6 and cdk2 led us to also analyze the kinase activity of cdk2 as a possible compensatory mechanism for the lack of cdk4.
Epidermis of wild-type and cdk4-deficient mice was isolated and protein lysates were subjected to immunoprecipitation with cdk specific antibodies. As expected, no activity of cdk4 was observed in cdk4 knockout mice (Figure 2G) ▶ . cdk6 activity increased twofold, whereas a mild increase in cdk2 activity (1.2-fold) was observed with cdk4-null lysate compared with wild-type sibling mice (Figure 2G) ▶ . In fact, immunohistochemical analysis of normal and hyperproliferative skin from cdk4-deficient and wild-type mice with pRb site-specific phospho- residue antibodies (Ser-780 for cdk4 and cdk6; Thr-356 for cdk2) showed the same pattern of pRb phosphorylation in basal and suprabasal cell layers (data not shown). Western blot analysis of cdk6, cdk2, D-type cyclins, and p27Kip1 protein levels also revealed no significant difference between knockout and wild-type animals (data not shown). These results suggest that other cdks may compensate for the absence of cdk4 or, alternatively, phosphorylation of pRb by cdk6 is sufficient for normal epidermal proliferation. In fact, analysis of cdk complex formation showed increased binding of D-type cyclins to cdk6 and cdk2 (data not shown). Thus, it is possible that in the absence of cdk4 the available D-type cyclins bind to cdk6 and cdk2 resulting in phosphorylation of pRb protein. Other laboratories have described complex formation between D-type cyclins and cdk2, although the levels of activation of those complexes appear to be cell type specific. 29-31
Discussion
Numerous studies have implicated cdk4,6/cyclin D complexes as key regulators of the cell cycle. 32,33 The regulatory subunits of cdk4 and cdk2, D-type cyclins, and cyclin E respectively, have been implicated in the development of several kinds of human and experimental tumors. 34-39 Although much less is known about the involvement of cdks in tumorigenesis, the participation of cdk4 in the neoplastic process was suggested by amplification and/or overexpression of the cdk4 gene in human gliomas, 1,40-42 sporadic breast carcinomas, 43 lipomatous tumors, 44 and sarcomas. 45 In addition, a point mutation in cdk4 (R24C) that abrogates the binding of the product of the tumor suppressor gene, p16Ink4a, was identified in patients with familial melanoma. 18,46
In this report, we have studied how the lack of cdk4 affects neoplastic growth and normal proliferation by using cdk4-deficient mice. We found that the lack of cdk4 inhibits mouse skin tumor development. The effect of the absence of cdk4 was stronger than that observed in cyclin D1-null mice 20,47 in which papilloma multiplicity was 22% (78% reduction) of wild-type siblings whereas cdk4-deficient mice developed 2% (98% reduction) of the tumors of wild-type mice. 20 In addition, a dose effect was observed with cdk4+/− mice, which developed 64% (36% reduction) as many papillomas compared with wild-type siblings. These results suggest that cdk4/cyclin D2 or D3 complexes also have a relevant role in mouse skin tumor development and that abrogation of cdk4 impairs papilloma development. In fact, papillomas from cyclin D1 knockout mice harbor a Ha-ras mutation in codon 61 (M. L. Rodriguez-Puebla and C. J. Conti, unpublished results), whereas analysis of papillomas derived from cdk4−/− mice did not show mutations in the Ha-ras gene. Again, this result suggests that inhibition of cdk4 activity led to total inhibition of ras-dependent skin tumor development.
The fact that cdk4 knockout mice did not show any apparent defect in skin structure 10,11 led us to hypothesize that epidermal proliferation is not affected by the absence of cdk4. Using two different models of cell proliferation we have determined that in vivo keratinocyte proliferation appears normal. First, we used the same proliferative stimulus applied in the chemical carcinogenesis protocol. In this case, TPA applications to the dorsal skin showed that both cdk4-deficient and wild-type mice develop identical epidermal hyperplasia. Second, when a wound was created on the dorsal skin, the proliferative response was also identical in knockout and wild-type sibling mice. Thus, we have demonstrated that in this particular tissue absence of cdk4 affects neoplastic growth but does not inhibit proliferation of normal cells. However, we cannot rule out the possibility that in other normal cell types the absence of cdk4 has a stronger effect. In this regard, cultures of cdk4-null fibroblasts exhibits the same kinetics of proliferation as wild-type fibroblasts under conditions of continuous growth, but quiescent fibroblasts exhibited a substantial delay in S-phase entry. 11 In contrast, our would-healing experiment showed that keratinocyte and fibroblast proliferation are similar between cdk4-null and wild-type mice. In fact, the skin healing involved several processes, which depend on fibroblastic proliferation, extracellular matrix secretion and finally keratinocyte proliferation and reepithelialization. 28 Also, it is possible that tissues in continuous proliferation, such as epidermis, are less affected by the lack of cdk4 than quiescent tissues.
It is likely that the putative mechanism that compensates for the lack of cdk4 is functional in some but not in all cell types, however, this is currently unknown. In recent years there have been several proteins described which interact with cdk4 in addition to binary and ternary complexes of cdk4, D-type cyclins, and cyclin-dependent inhibitors (CKI). For example, a cdk4 association with p50Cdc37, a protein subunit of the chaperone heat shock protein 90, was recently described. 48,49 Also, a cdk4 interaction with survivin (a member of the inhibitor of apoptosis (IAP) family) was recently established as part of the caspase 3 apoptotic pathway. 50 In addition, cdk4 interacted with the muscle regulatory protein MyoD and CCAAT/enhancer binding protein α (C/EBPα) causing growth arrest. 51,52 Moreover, stable complexes between cdk4 and p16Ink4a were detected in several cell types and induction of these binary complexes caused marked inhibition of cdk2 activity by redistribution of cyclin and CKI subunits, particularly those involving p27Kip1 that bind and inhibit cdk2 activity. 53,54 These data suggest that cdk4 is present in several complexes, which are in a steady state of equilibrium. An excess or lack of cdk4 could change the equilibrium toward the formation of different complexes with a consequent impairment of the cell cycle regulation contributing to the establishment of the neoplastic phenotype. This hypothesis is supported by several reports that show cdk4 amplification in gliomas (as alternative mechanisms to p16Ink4a deletion) without amplification or overexpression of cyclin D1. 40,41 In addition, lack of cdk4 can result in the availability of D-type cyclins and further interaction with other proteins. For instance, recently it was reported that cyclin D1 binds and represses STAT3 activation, which is associated with cell transformation in src-transformed cell lines. 55 Thus, analysis of novel cdk4 and/or D-type cyclins interactions in conditions of reduced or increased overexpression will help to understand the role of those proteins in pathological conditions.
In the last few years, much more effort has been directed toward inhibition of cdk2 rather than cdk4 as a potential therapeutic target. 56-58 The lack of cdk4 crystallographic data has not allowed development of specific inhibitors. Recently, however, crystallographic information using a “cdk4 mimic cdk2” protein has been obtained, showing that cdk4 has additional space in the ATP binding pocket to accommodate a large substituent such as a cdk4 selective inhibitor. 59 Thus, design of specific inhibitors for cdk4 will allow new assays to study the inhibition of neoplastic growth. Furthermore, analysis of the putative mechanism that compensate for the lack of cdk4 in cdk4-null keratinocyte should provide evidence of other potential inhibitory therapeutic targets.
In summary, we have demonstrated that lack of cdk4 results in reduced neoplastic growth in a ras-dependent tumorigenesis assay. This result correlated well with the reduce tumor development in cyclin D1-null mice using several ras-dependent tumorigenesis assays. 20,22 It appears that the cdk4/D-type cyclin complex is a preferential target for the ras oncogene. Thus, it will be interesting determine whether tumor development independent of ras mutations are also affected by the lack of cdk4; this warrants further investigation. Our results suggest that cdk4 has properties of a potential target for future therapeutic strategies.
Acknowledgments
We thank April Weiss for help with the mouse experiments; Cassie Bigbee for technical support; Dr. Suzan Fischer for helpful reading and discussion this paper; the Science Park animal facility personnel, the Science Park histology service for assistance with the immunohistochemical staining; Sharon Stockman for proofreading the paper; and Melissa Bracher for secretarial assistance.
Footnotes
Address reprint requests to Marcelo L. Rodriguez-Puebla, The University of Texas, M. D. Anderson Cancer Center, Science Park-Research Division, Smithville, TX 78957. E-mail: marcelo@rodriguez-puebla.org.
Supported by National Institutes of Health Grants CA 42157, CA 57596, and CA 90864, Institutional Grants CA 16672 to M.D. Anderson Cancer Center for the animal facility, Funds from the University Cancer Foundation at the University of Texas M. D. Anderson Cancer Center, and National Institute Environmental Health Sciences Center Grant ES07784.
References
- 1.He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day RS, James CD: CDK4 amplification is an alternative mechanism to p16 homozygous deletion in glioma cell lines. Cancer Res 1994, 54:5804-5807 [PubMed] [Google Scholar]
- 2.Ichimura K, Schmidt EE, Goike HM, Collins VP: Human glioblastomas with no alterations of the CDKN2A and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996, 13:1065-1072 [PubMed] [Google Scholar]
- 3.Sherr CJ: Mammalian G1 cyclins. Cell 1993, 73:1059-1065 [DOI] [PubMed] [Google Scholar]
- 4.Sherr CJ: D-type cyclins. Trends Biochem Sci 1995, 20:187-190 [DOI] [PubMed] [Google Scholar]
- 5.Munger K, Pieternpol JA, Pittelkow MR, Holt JT, Moses HL: Transforming growth factor B1 regulation of c-myc expression, pRb phosphorylation, and cell cycle progression in keratinocytes. Cell Growth Differ 1992, 3:291-298 [PubMed] [Google Scholar]
- 6.Nevins JR: E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258:424-429 [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa MH, Higasashi HK, Jung I, Suzuki-Takahashi M, Ikeda K, Tamai I, Kato K, Segawa E, Yoshida S, Nishimura S, Taya Y: The consensus motif for phosphorylation by cyclin D1-CDK4 is different from that for phosphorylation by cyclin A/E-CDK2. EMBO J 1996, 15:7060-7069 [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC: Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 2000, 101:79-89 [DOI] [PubMed] [Google Scholar]
- 9.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC: Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98:859-869 [DOI] [PubMed] [Google Scholar]
- 10.Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Premkumar Reddy E, Barbacid M: Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in B-islet cell hyperplasia. Nat Genet 1999, 22:44-52 [DOI] [PubMed] [Google Scholar]
- 11.Tsutsui T, Hesabi B, Moons DS, Pandolfi P, Hansel K, Koff A, Kiyokawa H: Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity. Mol Cell Biol 1999, 19:7011-7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miliani de Marval P, Gimenez-Conti I, LaCava M, Martinez L, Conti C, Rodriguez-Puebla M: Transgenic expression of CDK4 results in epidermal hyperplasia and severe dermal fibrosis. Am J Pathol 2001, 159:369-379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holland EC, Hively WP, Gallo V, Varmus HE: Modeling mutations in the G1 arrest pathway in human gliomas: overexpression of CDK4 but not loss of INK4a-ARF induces hyperploidy in cultured mouse astrocytes. Genes Dev 1998, 12:3644-3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corcoran M, Mould S, Orchard J, Ibbotson R, Chapman R, Boright A, Platt C, Tsui L, Scherer S, Oscier D: Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene 1999, 18:675-684 [DOI] [PubMed] [Google Scholar]
- 15.Khatib Z, Matsushime H, Valentine M: Coamplification of the Cdk4 gene with MDM2 and GL1 in human sarcomas. Cancer Res 1993, 53:5535-5541 [PubMed] [Google Scholar]
- 16.Ohta M, Nagai H, Shimizu M, Rasio D, Berd D, Mastrangelo M, Singh AD, Shields JA, Shields CJ, Croce CM: Rarity of somatic and germline mutations of the cyclin-dependent kinase 4 inhibitor gene, CDK4I, in melanoma. Cancer Res 1994, 54:5269-5272 [PubMed] [Google Scholar]
- 17.Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa-Luonnet D, Bernard J, Bressac-de Paillerets R: Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. Hum Mol Genet 1998, 7:209-216 [DOI] [PubMed] [Google Scholar]
- 18.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Büschenfelde KH, Beach D: A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269:1281-1284 [DOI] [PubMed] [Google Scholar]
- 19.Sotillo R, Garcia J, Ortega S, Martin J, Dubus P, Barbacid M, Malumbres M: Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci 2001, 98:13312-13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robles A, Rodriguez-Puebla M, Glick A, Trempus C, Hansen L, Sicinski P, Tennant R, Weinberg R, Yuspa S, Conti C: Reduced skin tumor development in cyclin D1 deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev 1998, 12:2469-2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Puebla M, LaCava M, Bolontrade M, Rusell J, Conti C: Increased expression of mutated Ha-ras during premalignant progression in SENCAR mouse skin. Mol Carcinog 1999, 26:150-156 [PubMed] [Google Scholar]
- 22.Yu Q, Geng Y, Sicinski P: Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411:1017-1021 [DOI] [PubMed] [Google Scholar]
- 23.Balmain A, Pragnell I: Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature 1983, 303:72-74 [DOI] [PubMed] [Google Scholar]
- 24.Bizub D, Wood A, Skalka A: Mutagenesis of the Ha-ras oncogene in mouse skin tumors induced by polycyclic aromatic hydrocarbons. Proc Natl Acad Sci USA 1986, 83:6048-6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roop D, Lowy D, Tambourin P, Strickland J, Harper J, Balaschak M, Spangler E, Yuspa S: An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 1986, 323:822-824 [DOI] [PubMed] [Google Scholar]
- 26.Quintanilla M, Brown K, Ramsden M, Balmain A: Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322:78-80 [DOI] [PubMed] [Google Scholar]
- 27.Martin P: Wound healing: aiming for perfect skin regeneration. Science 1997, 276:75-81 [DOI] [PubMed] [Google Scholar]
- 28.Clark R: Wound repair: overview and general considerations. Clark R eds. The Molecular and Cellular Biology of Wound Repair. 1996:pp 3-50 Plenum Press, New York
- 29.Sweeney K, Sarcevic B, Sutherland R, Musgrove E: Cyclin D2 activates Cdk2 in preference to Cdk4 in human breast epithelial cells. Oncogene 1997, 14:1329-1340 [DOI] [PubMed] [Google Scholar]
- 30.Dulic V, Drullinger L, Lees E, Reed S, Stein G: Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci USA 1993, 90:11034-11038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higashi H, Suzuki-Takahashi I, Saitoh S, Segawa K, Taya Y, Okuyama A, Nishimura S, Kitagawa M: Cyclin-dependent kinase-2 (Cdk2) forms an inactive complex with cyclin D1 since Cdk2 associated with cyclin D1 is not phosphorylated by Cdk7-cyclin-H. Eur J Biochem 1996, 237:460-467 [DOI] [PubMed] [Google Scholar]
- 32.Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J: Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol 1995, 15:2600-2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukas J, Parry D, Aagard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J: Retinoblastoma protein-dependent cell-cycle inhibition by the tumor suppressor p16. Nature 1995, 375:503-506 [DOI] [PubMed] [Google Scholar]
- 34.Arnold A, Kim H, Gaz R, Eddy R, Fukushima Y, Byers M, Shows T, Kronenberg H: Molecular cloning and chromosomal mapping of DNA rearranged with the parathyroid hormone gene in a parathyroid adenoma. J Clin Invest 1989, 83:2034-2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasogawa Y, Takamo Y, Okayasu I, Kakita A: The 5D4 abtibody (anti-cyclin D1/D2) related antigen: cytoplasmic staining is correlated to the progression of gastric cancer. Pathol Int 1998, 48:717-722 [DOI] [PubMed] [Google Scholar]
- 36.Murty V, Chaganti R: A genetic perspective of male germ cell tumors. Semin Oncol 1998, 25:133-144 [PubMed] [Google Scholar]
- 37.Houldsworth J, Reuter V, Bosl G, Chaganti R: Aberrant expression of cyclin D2 in an early event in human male germ cell tumorigenesis. Cell Growth Differ 1997, 8:292-299 [PubMed] [Google Scholar]
- 38.Delmer A, Ajchenbaum-Cymbalista F, Tang R, Ramond S, Faussat A, Marie J, Zittoun R: Overexpression of cyclin D2 in chronic B-cell malignancies. Blood 1995, 85:2870-2876 [PubMed] [Google Scholar]
- 39.Sanchez-Beato M, Camacho F, Martinez-Montero J, Saez A, Villuendas R, Sanchez-Verde L, Garcia J, Piris M: Anomalous high p27/Kip1 expression in a subset of aggressive B-cell lymphomas is associated with cyclin D3 overexpression. p27/Kip1-cyclin D3 colocalization in tumor cells. Blood 1999, 94:765-772 [PubMed] [Google Scholar]
- 40.Schmit E, Ichimura K, Reifenberger G: CdkN2 (p16/MTST1) gene deletion or Cdk4 amplification occurs in the majority of glioblastomas. Cancer Res 1994, 54:6321-6324 [PubMed] [Google Scholar]
- 41.Sonoda Y, Yoshimoto T, Sekiya T: Homozygous deletion of the MTS1/p16 and MTS2/p15 genes and amplification of the CDK4 gene in glioma. Oncogene 1995, 11:2145-2149 [PubMed] [Google Scholar]
- 42.Lam P, Di Tomaso E, Ng H, Pang J, Roussel M, Hjelm N: Expression of p19INK4d, CDK4, CDK6 in glioblastoma multiforme. Br J Neurosurg 2000, 14:28-32 [DOI] [PubMed] [Google Scholar]
- 43.An H, Beckmann MW, Reifenger G, Bender HG, Niederacher D: Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am J Pathol 1999, 154:113-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dei Tos A, Doglioni C, Piccini S, Sciot R, Furlanetto A, Boiocchi M, Dal Cin P, Maestro R, Fletcher C, Tallini G: Coordinate expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol 2000, 190:531-536 [DOI] [PubMed] [Google Scholar]
- 45.Kanoe H, Nakayama T, Murakami H, Hosaka T, Yamamoto H, Nakashima Y, Tsuboyama T, Nakamura T, Sasaki M, Toguchida J: Amplification of CDK4 gene in sarcomas: tumor specificity and relationship with the Rb mutation. Anticancer Res 1998, 18:2317-2321 [PubMed] [Google Scholar]
- 46.Zuo L: Germline mutation in the p16Ink4a binding domain of cdk4 in familial melanoma. Nat Genet 1996, 12:97-99 [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez-Puebla ML, Robles AI, Conti CJ: Ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol Carcinog 1999, 24:1-6 [PubMed] [Google Scholar]
- 48.Stepanova L, Leng X, Parker S, Harper J: Mammalian p50CDC37 is a protein kinase targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev 1999, 10:1491-1502 [DOI] [PubMed] [Google Scholar]
- 49.Dai K, Kobayashi R, Beach D: Physical interaction of mammalian CDC37 with CDK4. J Biol Chem 1996, 271:22030-22034 [DOI] [PubMed] [Google Scholar]
- 50.Suzuki A, Hayashida M, Ito T, Kawano H, Nakano T, Miura M, Akahane K, Shiraki K: Survivin initiates cell cycle entry by the competitive interactions with CDK4/p16Ink4a and CDK2/cyclin E complex activation. Oncogene 2000, 19:3225-3234 [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Goode T, Iakova P, Albrech J, Timchenko N: C/EBPα triggers proteosome-dependent degradation of cdk4 during growth arrest. EMBO J 2002, 21:930-941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J-M, Wei Q, Zhao X, Paterson B: Coupling of the cell-cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J 1999, 18:926-933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parry D, Mahony D, Wills K, Lees E: Cyclin-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol 1999, 19:1775-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McConnell B, Gregory F, Stott F, Hara E, Peters G: Induced expression of p16Ink4a inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol 1999, 19:1981-1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bienvenu F, Gascan H, Coqueret O: Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J Biol Chem 2001, 276:16840-16847 [DOI] [PubMed] [Google Scholar]
- 56.Sausville EA, Senderowicz AM: Chemical cyclin-dependent kinase inhibitors. Gutkind JS eds. Signaling Networks and Cell Cycle Control: The Molecular Basis of Cancer and Other Diseases. 2000:pp 557-567 Humana Press, Ottowa
- 57.Senderowicz AM: Small molecule modulators of cyclin-dependent kinases for cancer therapy. Oncogene 2000, 19:6600-6606 [DOI] [PubMed] [Google Scholar]
- 58.Senderowicz AM, Sausville EA: Pre-clinical and clinical development of cyclin-dependent kinase modulators. J Natl Cancer Inst 2000, 92:376-387 [DOI] [PubMed] [Google Scholar]
- 59.Ikuta M, Kamata K, Fukasawa K, Honma T, Machida T, Hirai H, Suzuki-Takahashi I, Hayama T, Nishimura S: Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J Biol Chem 2001, 276:27548-27554 [DOI] [PubMed] [Google Scholar]