

Abstract
Pathogens may impair reproduction in association or not with congenital infections. We have investigated the effect of acute infection with Trypanosoma cruzi, the protozoan agent of Chagas’ disease in Latin America, on reproduction of mice. Although mating of infected mice occurred at a normal rate, 80% of them did not become gravid. In the few gravid infected mice, implantation numbers were as in uninfected control mice, but 28% of fetuses resorbed. Such infertility and early fetal losses were significantly associated with high maternal parasitemia. The remaining fetuses presented with reduced weights and all died later in gestation or within 48 hours after birth. Several organs of these fetuses were infiltrated by polynuclear cells and presented ischemic necrosis but did not harbor T. cruzi parasites, discarding congenital infection as the cause of mortality. However, surprisingly, the deciduas were massively invaded by T. cruzi parasites, harboring 125-fold more amastigotes than the maternal heart or other placental tissues. Parasites were significantly more numerous in the placentas of dead fetuses. In addition, placentas contained inflammatory infiltrates and displayed ischemic necrosis, fibrin deposits, and vascular thromboses. These results show that acute T. cruzi infection totally impairs reproduction in mice through inducing infertility or fetal-neonatal losses in association with placental parasite invasion and ischemic necrosis.
Reproductive failures, associated or not with congenital infections, have been shown both in human and experimental infections with viruses, bacteria, and protozoa. 1,2 Besides direct effects of infection on placentas and fetuses, poor pregnancy outcome might arise from disturbances in the complex biological processes tightly regulated by hormones and cytokines involved in mammal reproduction. 3-5 Indeed, proinflammatory/type 1 cytokines such as tumor necrosis factor-α and interferon-γ, produced at high levels in response to some pathogens, have been shown to be harmful for pregnancy. 6,7
Information on the effect of infection with T. cruzi (the protozoa agent of Latin American Chagas’ disease) on pregnancy is scarce though, and according to the endemic area, such infection affects 3 to 51% of pregnant women from which 2 to 10% congenitally transmit the parasite. 8,9 Some studies suggest that Chagas’ disease might cause abortion or prematurity, whereas others fail to show such a relation. 9-11 Studies in experimental infection might reinforce our knowledge of maternofetal interactions in T. cruzi infection. Infertility and fetal growth retardation, associated or not with congenital infection, have been reported in chronic infection of mice. 12,13 In acute experimental infection, placental infection has been reported without congenital transmission, 14 despite the high circulating level of parasites, known to release proinflammatory molecules 15,16 and the systemic production of cytokines potentially deleterious for pregnancy. 17,18
The aim of the present work was to gain more insight into the relation between acute T. cruzi infection and gestation in mice, by studying the reproductive capacity, the pregnancy outcome, and the histopathology of uteroplacental units (UPUs) and fetuses in relation to maternal parasitic load. Our results show that acute infection totally impairs reproduction in the mouse, inducing infertility or fetal and perinatal losses, although congenital infection was not not observed. Infertility and early fetal losses were related to high maternal parasitemia, whereas mortality in late gestation was associated with a massive parasite invasion of the decidua and ischemic necrosis of placenta and fetuses, but not with maternal parasitemia.
Materials and Methods
Mice, T. cruzi Infection, and Mating
BALB/c mice were purchased from B&K Universal (Hull, UK) and maintained in a conventional animal house. Two-month-old females were infected by intraperitoneal or subcutaneous (in the footpad) inoculation of 100 blood trypomastigotes of the Tehuantepec strain of T. cruzi maintained in our laboratory. Parasitemia was regularly determined in tail blood as previously described. 19 The kinetics of parasitemia in gravid mice, which peaked around day 28 after infection, was similar to that of nongravid mice, as previously described. 17 The time course of gestation in relation to the parasitemia of infected mice is presented in Figure 1 ▶ . To obtain pregnancies during the ascending phase of parasitemia, female mice were mated with uninfected males during days 7 to 10 after infection. To obtain maximum fecundation rates at the time of mating, females were maintained in anestrus before mating by keeping them for 2 weeks in a different room from the males. 12 To synchronize the menstrual cycles at the time of mating, females infected 4 days previously, and uninfected age-matched controls, were placed for 2 days in cages in which the litter had been impregnated with the odor of males (ie, cages having contained males). On day 6 after infection in the afternoon, one uninfected male was added, per cage containing two females, and the presence of a vaginal plug, indicating that mating had occurred, was checked every morning for 4 days. Females showing a vaginal plug (VP+) were immediately separated from the males. The day of sighting a vaginal plug was denoted day 1 of gestation (G1). The rare VP+ mice that died during the acute phase were excluded from the study, because their gravid state was not known.
Figure 1.

Time course of gestation in relation to the parasitemia of mice acutely infected with T. cruzi. Female BALB/c mice were infected at day 0. The black dotted line represents the parasitemia of gravid mice (n = 65, geometric mean ± SEM). They were mated from days 7 until 10 after infection. Areas indicate important time points of gestation. According to the day after infection at which the vaginal plug was observed, the first day of gestation (G1, gray bars) occurred before circulating parasites were detectable, implantation (G4.5, dotted bars) took place at the beginning of the parasitemic phase, the complete formation of the placenta (G9.5, oblique lined bars) occurred during the ascending phase of the parasitemia, and delivery (G20, horizontal lined bars) occurred at the peak of parasitemia. Mated mice were either killed at G9, G17, or G19, or allowed to deliver.
Determination of Viability or Time of Fetal Death
On gestation day 9 (G9), 17 (G17), or 19 (G19), some of the females were sacrificed by cervical dislocation and median laparotomy was performed. The existence of pregnancy was noted and the uterus was opened by a longitudinal incision at the anti-mesometrial side. For each pregnant mouse killed at G9, the number of implants was noted. For mice killed at G17 or G19, the number of fetuses and resorptions (quite small implants with no discernible fetus and placenta, corresponding to embryos that died before G9.5, ie, before the complete formation of a vascularized placenta 20 ) were recorded, fetuses and placentas were extracted and separately weighed, and the status of fetuses was determined. Macerated pale white fetuses, displaying stunted limb buds, observed at G17 or G19, were considered to have been dead for at least 1 week, ie, approximately at midgestation. At G19, the reddish-colored fetuses, displaying well-defined limbs, were extracted from their amniotic envelop as rapidly as possible to determine their viability by observing if movements were prompted by touching the fetus with pliers. The absence of movement indicated that the fetus had recently died. These fetuses are referred to as “immobile.” Mice that were not killed during gestation were allowed to deliver and their litter was directly numbered. Stillbirths were noted and the aspect of newborns was recorded at birth and during the days after. They were not weighed to avoid risk of cannibalism by the mother because of manipulation of newborns.
Histological Studies
In some experiments, after the uterus has been opened by a longitudinal incision, it was cut transversally between resorptions or fetuses to isolate them from each other. Each resorption or fetus, with its UPU and the adjacent uterine tissue, as well as some uterus from infected nonpregnant mice, was processed for histological examination. Briefly, each piece was immediately fixed in 10% neutral buffered formalin solution (Sigma, St. Louis, MO) for more than 24 hours, postfixed overnight, and embedded in paraffin. Serial sections of 5-μm thickness were dried at 60°C for 5 minutes and stained either with hematoxylin and eosin (H&E) or with Trichrome-Masson (for fibrosis detection) using classical procedures (all reagents from Merck KGaA, Darmstadt, Germany). Hearts of some infected gravid or not gravid-nonmated females were also processed for histological studies of tissular parasite burden after H&E staining.
Intracellular parasites were checked by microscopical examination at a magnification of ×400 of H&E-stained sections, using a grid (Van Hopplynus, Brussels, Belgium) to measure the section surface. The presence of intracellular amastigotes in the decidua was confirmed by transmission electron microscopy according to classical procedures. The search for congenital infection was performed on the whole frontal section of fetuses (dead or alive) and resorptions taken at G17 and G19. The examination of UPU, taken at G9, G17, and G19 was performed on midsagittal sections. The number of amastigote nests/mm2 of tissue (one nest corresponds to one or a small number of grouped infected cells) and the average number of amastigotes per nest were quantified.
Results
Acute Infection with T. cruzi Reduces Fecundity of Mice in Relation to Maternal Parasitemia
As shown in Table 1 ▶ , although the rates of mating (appreciated by the frequency of vaginal plugs) were similar in infected and uninfected mice, only 18 to 23% of infected females developed a gestation as compared with 63 to 82% of the noninfected mice. The route of parasite inoculation did not influence the results because no difference was observed in fecundity of mice receiving parasites either intraperitoneally or subcutaneously. The uterus of infected, mated but nongravid mice, collected 9 days after mating, ie, ∼4.5 days after implantation, presented a normal macroscopic aspect, similar to the uterus of nonmated mice, without any trace of implantation scars (data not shown).
Table 1.
Reproductive Capacity of T. cruzi Acutely Infected (I) and Uninfected (NI) Mice*
| Exp. | Parasite inoculation† | Mouse infection | n‡ | % VP+§ | % G+¶ |
|---|---|---|---|---|---|
| 1 | i.p. | I | 24 | 70.8 | 17.6∥ |
| NI | 16 | 68.8 | 81.8 | ||
| 2 | s.c. | I | 37 | 81.1 | 23.3** |
| NI | 23 | 82.6 | 78.9 | ||
| 3 | s.c. | I | 30 | 76.7 | 21.7†† |
| NI | 20 | 80.0 | 62.5 |
*Representative results of three experiments amongst eight.
†i.p., intraperitoneal; s.c., subcutaneous route.
‡Number of mice allowed mating in each group.
§Proportion of mice presenting a vaginal plug (VP+).
¶Proportion of VP+ mice developing gestation.
∥P < 0.005,
**P < 0.0005,
††P < 0.05, Student’s t-test, when compared to NI animals.
As shown in Figure 2 ▶ , the higher the parasitemia of the female, the higher was the proportion of infertile mice, suggesting that the reduction of fecundity was related to events associated with the maternal parasitic load.
Figure 2.

Proportion of T. cruzi acutely infected mice developing gestation in relation to the parasitemia. The presence of pregnancy and the parasitemia were determined 9 days after the occurrence of the vaginal plug. NI, age-matched noninfected mated mice; n, cumulated numbers of mice in each group. Results are expressed as means ± SEM of eight experiences.
Acute Infection with T. cruzi Induces Massive in Utero and Neonatal Mortality in Mice
Although the numbers of implantations (the sum of resorptions and fetuses) were similar in gravid infected and uninfected animals (Table 2) ▶ , a high proportion of embryos and fetuses of infected mice died during gestation, whereas the mortality rates were very low in noninfected females. Indeed, as shown in Figure 3A ▶ , 28% of embryos of infected dams resorbed in early gestation (before G9.5, Figure 3C ▶ ), 8% of fetuses died in midgestation (macerated fetuses, Figure 3D ▶ ), and 22% died in late gestation (immobile fetuses seen at G19), resulting in a cumulative mortality of 57% of fetuses at G19. In addition, fetuses still alive at G19 (ie, mobile) displayed weights strongly reduced by 40% as compared to those from noninfected mice (Figure 3E ▶ and Figure 4 ▶ ). The weight of corresponding placentas was only slightly diminished by 22% (Figure 4) ▶ , without correlation with fetal weight reductions, whereas in noninfected dams, fetal and placental weights were, as expected, significantly correlated (P = 0.0006, n = 62). This indicates asymmetrical fetal growth retardation.
Table 2.
Number of Implantations in T. cruzi Acutely Infected (I) and Uninfected (NI) Mice
| G* | Mouse infection | n† | Implantations‡ |
|---|---|---|---|
| 9 | I | 13 | 7.00 ± 0.58 |
| NI | 10 | 9.40 ± 0.54 | |
| 17 | I | 29 | 8.10 ± 0.46 |
| NI | 20 | 7.55 ± 0.37 | |
| 19 | I | 8 | 7.63 ± 0.60 |
| NI | 17 | 6.71 ± 0.41 |
*Pregnant mice were killed at different time points of gestation (G9, 17, and 19).
†Number of mice/group.
‡Number of implantations/mouse (mean ± SEM).
Figure 3.

Gestational and perinatal mortality in acutely T. cruzi-infected mice. T. cruzi-infected (filled bars) or uninfected (open bars) mice were either killed at days 17 or 19 of gestation (A) or allowed to deliver (B), and mortality of the progeny was recorded. The mortality occurring in early gestation corresponded to resorptions, that of midgestation to macerated fetuses, and that of late gestation to immobile fetuses seen at G19. The perinatal mortality comprises stillbirths and newborns that were eaten by their mothers 24 to 48 hours after birth. Results are expressed as mean ± SEM of n dams per group. C: The arrow indicates a resorption. D: Macerated fetus and its placenta. E: Alive fetuses with their placenta at G19, from an infected (left) and an uninfected (right) mouse, showing growth retardation in the fetus of infected dam.
Figure 4.

Weights of alive fetuses and their placentas from T. cruzi acutely infected mice at day 19 of gestation. Fetuses and placentas were rapidly extracted from pregnant infected (filled bars; n = 15) and noninfected (open bars; n = 62) mice killed at G19. Asterisks indicate significant differences between both groups (P < 0.005, Student’s t-test).
As a result of such massive in utero mortality, the litter size at delivery of infected dams was strongly reduced to 3.57 ± 1.31, whereas it was 5.97 ± 0.36 for uninfected mice (P = 0.01). Moreover, 25% of the litter of infected dams were stillbirths (versus 3% for uninfected mice) (Figure 3B) ▶ . Although the remaining newborns were not weighed for avoiding manipulations that could enhance maternal cannibalism, they were clearly smaller than controls, and finally eaten by their mothers 24 to 48 hours after birth (Figure 3B) ▶ .
Because the infertility of infected mice was associated with the maternal parasite burden (see above), we also sought for a relation between maternal parasitemia and the rate of resorptions and fetal death. We found that the proportion of resorptions (occurring in early gestation between days 5 to 9 of gestation) in infected mice was significantly and positively correlated with the concurrent maternal parasitemia (measured at day 14 after infection; n = 38, r = 0.448, P < 0.05). However, there was no relation between the proportion of fetuses that died later during gestation (macerated and immobile fetuses) and the maternal parasitemia measured at days 21 and 24 after infection.
Fetal Mortality in Late Gestation of Mice Acutely Infected with T. cruzi Is Associated with Parasite Invasion of UPUs without Fetal Infection
To address the question of the role of parasite in the fetal mortality, we sought for intracellular amastigotes in fetal tissues of infected dams, by careful microscopic examination of histological sections of fetuses collected either at G17 (70 nonmacerated fetuses, 8 macerated fetuses, and 13 resorptions) or at G19 (9 mobile, 10 immobile and 8 macerated fetuses). Amastigote nests were never observed in such fetuses, excluding the congenital transmission of parasites as a cause of the massive mortality of fetuses.
Because parasites in the UPU might affect the local maternofetal exchanges, we also sought for amastigotes in the UPU surrounding resorptions and fetuses at G9, G17, and G19. Whereas intracellular parasites were never seen at G9 of gestation, they were found in the UPU at G17 and G19. T. cruzi amastigote nests were mainly localized in the decidua (Figure 5, A and B) ▶ , but some were also observed in chorioamniotic membrane (Figure 5D) ▶ , in the spongy zone (Figure 5E) ▶ , and more rarely in cells at the junction between the labyrinthine and the spongy zone, ie, at the maternal side of the labyrinthine. Parasites were not seen in the internal, fetal side, of the labyrinthine.
Figure 5.
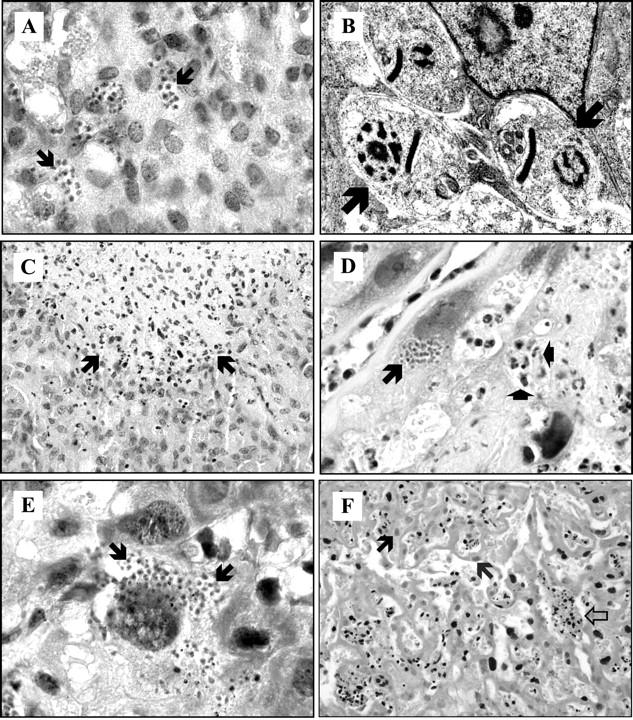
Histopathology of placenta from mice acutely infected with T. cruzi. A–C: Decidua at G17; arrows show parasite nests (A), amastigotes (B), and inflammatory infiltrates (C). D: Chorioamniotic membrane at G17; the long arrow indicates a parasite nest and the short arrows show a microabscess rich in polynuclear cells. E: Spongy zone at G17; arrows show amastigotes in a large cell. F: Labyrinthine at G19 showing necrotic cells with a pale nucleus (black arrows) and a thrombus constituted of fibrin and blood cells with black nucleus (white arrow). H&E stain; original magnifications: ×1000 (A, D, E); ×400 (C, F). Transmission electron microscopy; original magnification, ×50,000 (B).
Amastigotes were also occasionally found in the myometrial part of the uterus at G17 or G19, but not at G9, whereas parasites have not been found in the uterus of nongravid mice (data not shown).
Decidua Is a Privileged Site for Parasite Multiplication
Microscopic examination of UPU sections showed that, at G17, parasites were much more numerous in the decidua than in the other placental tissues (data not shown). It is worth noting that the decidual amastigote burden (number of amastigote nests multiplied by number of amastigotes/nest) was at this time a mean 125 times higher than in the maternal cardiac tissue (Figure 6A) ▶ . The number of nests was slightly higher in the edges than in the central part of the decidua (21.03 ± 0.90 versus 18.35 ± 1.31 nests/mm2 respectively; n = 31, P < 0.05). Interestingly, analysis of amastigote nest density in the total UPU in relation to fetal mortality showed a significant increase from alive to dead fetuses, at G17 as well as G19 (Figure 6B) ▶ .
Figure 6.

Parasite burden in maternal tissues in late gestation. T. cruzi-infected mice were killed at G17 (open bars) or G19 (filled bars), and intracellular parasites were counted in tissue sections. The state of fetuses from which placentas were obtained is indicated as follows: Mac., macerated (pale white fetuses displaying stunted limb buds); Non mac., nonmacerated (reddish-colored fetuses with well-defined limbs); Imm., immobile reddish-colored fetuses; Mob., mobile, alive, reddish-colored fetuses. Results are expressed as mean ± SEM of n samples per group. Asterisks indicate significant differences between the indicated groups (P < 0.05, Student’s t-test). ND, not determined.
Dead Fetuses and Their Placentas Suffer Massive Ischemic Necrosis in Late Gestation of Acutely Infected Mice
No abnormalities could be seen in the microarchitecture of the placenta at G9. By contrast, at G17, microabscesses rich in polynuclear cells and containing few lymphocytes were observed in deciduas, in the area between spongiotrophoblasts and the labyrinth, and in the placental membranes of all dead fetuses (Figure 5, C and D) ▶ . Microabscesses were not necessarily located close to infected cells. Fibrin deposits were not found in these placentas, even with the special Trichrome-Masson staining. At G19, such inflammatory infiltrates were associated with ischemic necrosis, numerous fibrin deposits, and vascular thromboses (Figure 5F) ▶ .
Sections of fetuses were also carefully examined at G17 and G19. Inflammatory infiltrates containing neutrophils were seen in the liver, digestive tract, and bladder, also associated with ischemic necrosis (data not shown). At G19, the pathology was more pronounced in macerated than in immobile fetuses (recently dead). In fetuses still alive at G19, no obvious histological abnormalities were found.
Discussion
This work clearly shows acute T. cruzi infection prevents reproduction in mice, in line with previously reported harmful effects on fertility and pregnancy observed in other experimental infections, as for instance leishmaniasis, 21 toxoplasmosis, 22 and bartonellosis. 23 In acute murine T. cruzi infection, we show infertility and early fetal death to be related to maternal parasitemia, whereas later fetal and neonatal mortality was associated with massive parasite invasion of decidua and ischemic necrosis of placentas and fetuses.
A lot of mated females infected with T. cruzi did not become gravid and did not show implantation scars, suggesting that the reproductive failure occurred before implantation. The reasons of such infertility are primarily unknown. However, it likely did not result from impaired mating or ovulation because mating occurred at similar rates in infected and noninfected mice, and the female mouse only copulates during estrus. 24 Harmful effects of maternal infection affected the uterus on the whole, rendering it totally unreceptive to any implantation. We may hypothesize that disturbances of the uterine cytokinic/hormonal network would be responsible for infertility. Because intracellular amastigotes were not found in the uterus of infected nongravid mice, such harmful effects could not result from local infection. By contrast, they may have been triggered by systemic events, such as a strong inflammatory response. Our observation that infertility was associated with the highest maternal parasitemia argues for a systemic effect of acute infection. Indeed, such high parasitemia is associated with the release in blood of circulating cytokines such as interferon-γ and tumor necrosis factor-α 17,25 known to be harmful for pregnancy, 6 and also likely results in the shedding of potent proinflammatory parasitic molecules. 26 Such maternal systemic inflammatory response might also contribute to induce resorptions, because a similar correlation was also observed between their proportion and the maternal parasitemia, and parasites were absent in the UPU at this early step of gestation.
However, T. cruzi amastigotes were present later in the placenta of acutely infected mice, in agreement with a previous study. 14 An original observation of our work concerns the massive invasion of decidua by T. cruzi amastigotes. The route of parasite entry is likely hematogen because amastigotes were scarce in the myometrium part of the uterus, limiting the possibility of invasion from myometrium to decidua. The occurrence of such invasion between G9 and G17 is likely related to: 1) the timing of placenta formation, which is completely vascularized at G9.5; 2) the increase of maternal parasitemia; and 3) the local release of pregnancy-associated type 2 cytokines and transforming growth factor-β, 5 antagonizing the type 1 response susceptible to control local intracellular parasite multiplication. Additionally, transforming growth factor-β, might favor the entry of trypomastigotes into host cells by activating the signaling pathway of transforming growth factor-βR required for parasite invasion. 27 The 125-fold higher parasite burden seen in decidua as compared to the heart, where there is neither type 2 imbalance nor transforming growth factor-β production, also supports this hypothesis.
The absence of congenital infection in fetuses, despite the presence of numerous intracellular parasites in the placenta and in maternal blood, points to the efficacy of the placental trophoblastic layer to block the passage of parasites from mother to fetuses, in accordance with previous observations. 14,28 The mechanisms by which the trophoblastic layer prevents fetal infection is poorly understood, although it has been suggested that lysosomal enzymes might limit T. cruzi spreading. 29,30
Fetal death in late gestation of infected dams is associated with inflammatory infiltrates with neutrophils and ischemic necrosis in several organs, showing that multiple organ failure has occurred. In the absence of fetal infection, the possibility of the transmission to fetuses of potent proinflammatory parasitic molecules 15,26 or cytokines 17,31,32 shed in maternal blood might deserve consideration as a cause of multiple organ failure. 33 However, this mechanism is probably not predominant because no correlation was observed between maternal parasitemia and the proportion of fetal losses. By contrast, placental damage, likely worsened by the local immune response generated by the presence of parasites, might drastically limit the maternofetal exchanges, leading to fetal death, as strongly suggested by the association between fetal death and placental parasite burden. The asymmetrical fetal growth retardation (ie, higher weight reduction in fetuses as compared to placentas) that we have observed in infected dams argues still more for the existence of placental dysfunctions. Indeed, asymmetrical fetal weight loss might arise from impaired placental glucose transport, known to be regulated by cytokines and to be crucial for fetal development. 34 More importantly, the ischemic necrosis, leukocyte infiltration, fibrin deposits, and vascular thrombosis certainly reduce blood flow in the placenta, leading to fetal growth retardation, hypoxia, and finally death. It is worth noting that such placental lesions are strongly similar to those recently described in the placenta of abortogenic female CBA mice mated with male DBA/2 mice. In that model, the lesions resulted from the increased local expression of a particular prothrombinase, the Fgl2, 35,36 on activation by interferon-γ and tumor necrosis factor-α, two cytokines known to be triggered by T. cruzi infection. 17,18,32 Moreover, the massive infiltration of placenta with polymorphonuclear cells, observed in our experiment, also suggests a possible involvement of complement activation by parasites 37 at the maternofetal interface as a cause of fetal demise, as recently reported. 38
In conclusion, T. cruzi acute infection deeply impairs the reproductive capacity of mice, compromising implantation and favoring placental invasion by parasites and damages, leading to fetal growth retardation and death in the absence of congenital infection. Such results might stimulate investigations on the effects of T. cruzi infection on human reproduction.
Acknowledgments
We thank Pascale Deblandre, Alain Wathelet-Depauw, Michel Vanesse, Saad Gendouz, and Rob Darrington for their technical assistance, and Gérard Chaouat (Biologie de la Relation Materno-fetale, Hôpital Antoine Béclère, Paris, France) for helpful advice and discussion of the results.
Footnotes
Address reprint requests to Carine Truyens, Laboratoire de Parasitologie, Faculté de Médecine, U.L.B., Route de Lennik 808, CP 616 B-1070 Brussels, Belgium. E-mail: ctruyens@ulb.ac.be.
Supported by grants from the Fonds National de la Recherche Scientifique Médicale (convention 3.4595.99) and the Université Libre de Bruxelles.
References
- 1.McDonald HM, Chambers HM: Intrauterine infection and spontaneous midgestation abortion: is the spectrum of microorganisms similar to that in preterm labor? Infect Dis Obstet Gynecol 2000, 8:220-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brabin L, Brabin BJ: Parasitic infections in women and their consequences. Adv Parasitol 1992, 31:1-81 [DOI] [PubMed] [Google Scholar]
- 3.Erlebacher A: Why isn’t the fetus rejected? Curr Opin Immunol 2001, 13:590-593 [DOI] [PubMed] [Google Scholar]
- 4.Mellor AL, Munn DH: Immunology at the maternal-fetal interface: lessons for T cell tolerance and suppression. Annu Rev Immunol 2000, 18:367-391 [DOI] [PubMed] [Google Scholar]
- 5.Clark DA, Arck PC, Chaouat G: Why did your mother reject you? Immunogenetic determinants of the response to environmental selective pressure expressed at the uterine level. Am J Reprod Immunol 1999, 41:5-22 [DOI] [PubMed] [Google Scholar]
- 6.Raghupathy R: Th1-type immunity is incompatible with successful pregnancy. Immunol Today 1997, 18:478-482 [DOI] [PubMed] [Google Scholar]
- 7.Argiles JM, Carbo N, Lopez-Soriano FJ: TNF and pregnancy: the paradigm of a complex interaction. Cytokine Growth Factor Rev 1997, 8:181-188 [DOI] [PubMed] [Google Scholar]
- 8.Brabin L: The epidemiological significance of Chagas’ disease in women. Mem Inst Oswaldo Cruz 1992, 87:73-79 [DOI] [PubMed] [Google Scholar]
- 9.Freilij H, Altcheh J, Storino R: Chagas Congénito. Enfermedad de Chagas. 1994:pp 267-278 Doyma Argentina, Division Mosby, Buenos Aires
- 10.Bittencourt AL: Brener Z Andrade ZA Barral-Netto M eds. Vertical da Doença de Chagas: Trypanosoma Cruzi e Doença de Chagas. 2000:pp 16-20 Editora Guanabara Koogan SA, Rio de Janeiro
- 11.Bittencourt AL: American trypanosomiasis (Chagas’ disease). McLeod C eds. Parasitic Infections in Pregnancy and the Newborn. 1988:pp 62-86 Oxford University Press, Oxford
- 12.Carlier Y, Rivera MT, Truyens C, Puissant F, Milaire J: Interactions between chronic murine Trypanosoma cruzi infection and pregnancy: fetal growth retardation. Am J Trop Med Hyg 1987, 37:534-540 [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez Z, Cappa SM, Mirkin GA, Solana ME, Tekiel VS: Trypanosoma cruzi pathology. Strain dependent? Medicina (B Aires) 1999, 59(Suppl 2):69-74 [PubMed] [Google Scholar]
- 14.Andrade SG: The influence of the strain of Trypanosoma cruzi in placental infections in mice. Trans R Soc Trop Med Hyg 1982, 76:123-128 [DOI] [PubMed] [Google Scholar]
- 15.Almeida IC, Camargo MM, Procopio DO, Silva LS, Mehlert A, Travassos LR, Gazzinelli RT, Ferguson MA: Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J 2000, 19:1476-1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abuin G, Couto AS, de Lederkremer RM, Casal OL, Galli C, Colli W, Alves MJ: Trypanosoma cruzi: the Tc-85 surface glycoprotein shed by trypomastigotes bears a modified glycosylphosphatidylinositol anchor. Exp Parasitol 1996, 82:290-297 [DOI] [PubMed] [Google Scholar]
- 17.Truyens C, Torrico F, Lucas R, De Baetselier P, Buurman WA, Carlier Y: The endogenous balance of soluble tumor necrosis factor receptors and tumor necrosis factor modulates cachexia and mortality in mice acutely infected with Trypanosoma cruzi. Infect Immun 1999, 67:5579-5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torrico F, Heremans H, Rivera MT, Van Marck E, Billiau A, Carlier Y: Endogenous IFN-gamma is required for resistance to acute Trypanosoma cruzi infection in mice. J Immunol 1991, 146:3626-3632 [PubMed] [Google Scholar]
- 19.Carlier Y, Rivera MT, Truyens C, Goldman M, Lambert P, Flament J, Bauwens D, Vray B: Pregnancy and humoral immune response in mice chronically infected by Trypanosoma cruzi. Infect Immun 1987, 55:2496-2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theiler K: The house mouse. Atlas of embryonic development. 1989. Springer-Verlag, New-York
- 21.Krishnan L, Guilbert LJ, Wegmann TG, Belosevic M, Mosmann TR: T helper 1 response against Leishmania major in pregnant C57BL/6 mice increases implantation failure and fetal resorptions. Correlation with increased IFN-gamma and TNF and reduced IL-10 production by placental cells. J Immunol 1996, 156:653-662 [PubMed] [Google Scholar]
- 22.Stahl W, Kaneda Y, Noguchi T: Reproductive failure in mice chronically infected with Toxoplasma gondii. Parasitol Res 1994, 80:22-28 [DOI] [PubMed] [Google Scholar]
- 23.Boulouis HJ, Barrat F, Bermond D, Bernex F, Thibault D, Heller R, Fontaine JJ, Piemont Y, Chomel BB: Kinetics of Bartonella birtlesii infection in experimentally infected mice and pathogenic effect on reproductive functions. Infect Immun 2001, 69:5313-5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rugh R: Normal development of the mouse. The Mouse: Its Reproduction and Development. 1990:pp 44-101 Oxford University Press, Oxford
- 25.Laucella S, Salcedo R, Castanos-Velez E, Riarte A, de Titto EH, Patarroyo M, Orn A, Rottenberg ME: Increased expression and secretion of ICAM-1 during experimental infection with Trypanosoma cruzi. Parasite Immunol 1996, 18:227-239 [DOI] [PubMed] [Google Scholar]
- 26.Acosta-Serrano A, Almeida IC, Freitas-Junior LH, Yoshida N, Schenkman S: The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol Biochem Parasitol 2001, 114:143-150 [DOI] [PubMed] [Google Scholar]
- 27.Hall BS, Pereira MA: Dual role for transforming growth factor beta-dependent signaling in Trypanosoma cruzi infection of mammalian cells. Infect Immun 2000, 68:2077-2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delgado MA, Santos-Buch CA: Transplacental transmission and fetal parasitosis of Trypanosoma cruzi in outbred white Swiss mice. Am J Trop Med Hyg 1978, 27:1108-1115 [DOI] [PubMed] [Google Scholar]
- 29.Fretes RE, de Fabro SP: In vivo and in vitro analysis of lysosomes and acid phosphatase activity in human chagasic placentas. Exp Mol Pathol 1995, 63:153-160 [DOI] [PubMed] [Google Scholar]
- 30.Frank F, Sartori MJ, Asteggiano C, Lin S, de Fabro SP, Fretes RE: The effect of placental subfractions on Trypanosoma cruzi. Exp Mol Pathol 2000, 69:144-151 [DOI] [PubMed] [Google Scholar]
- 31.Truyens C, Angelo-Barrios A, Torrico F, Van Damme J, Heremans H, Carlier Y: Interleukin-6 (IL-6) production in mice infected with Trypanosoma cruzi: effect of its paradoxical increase by anti-IL-6 monoclonal antibody treatment on infection and acute-phase and humoral immune responses. Infect Immun 1994, 62:692-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera MT, Marques dA, Lucas R, Deman J, Truyens C, Defresne MP, De Baetselier P, Carlier Y: High tumor necrosis factor alpha (TNF-alpha) production in Trypanosoma cruzi-infected pregnant mice and increased TNF-alpha gene transcription in their offspring. Infect Immun 1995, 63:591-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karima R, Matsumoto S, Higashi H, Matsushima K: The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today 1999, 5:123-132 [DOI] [PubMed] [Google Scholar]
- 34.Kamei Y, Tsutsumi O, Yamakawa A, Oka Y, Taketani Y, Imaki J: Maternal epidermal growth factor deficiency causes fetal hypoglycemia and intrauterine growth retardation in mice: possible involvement of placental glucose transporter GLUT3 expression. Endocrinology 1999, 140:4236-4243 [DOI] [PubMed] [Google Scholar]
- 35.Clark DA, Chaouat G, Arck PC, Mittruecker HW, Levy GA: Cytokine-dependent abortion in CBA x DBA/2 mice is mediated by the procoagulant fgl2 prothrombinase [correction of prothombinase]. J Immunol 1998, 160:545-549 [PubMed] [Google Scholar]
- 36.Clark DA, Ding JW, Chaouat G, Coulam CB, August C, Levy GA: The emerging role of immunoregulation of fibrinogen-related procoagulant Fgl2 in the success or spontaneous abortion of early pregnancy in mice and humans. Am J Reprod Immunol 1999, 42:37-43 [DOI] [PubMed] [Google Scholar]
- 37.Tomlinson S, Pontes de Carvalho LC, Vandekerckhove F, Nussenzweig V: Role of sialic acid in the resistance of Trypanosoma cruzi trypomastigotes to complement. J Immunol 1994, 153:3141-3147 [PubMed] [Google Scholar]
- 38.Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H: A critical role for murine complement regulator crry in fetomaternal tolerance. Science 2000, 287:498-501 [DOI] [PubMed] [Google Scholar]